

Abstract
The use of ultraviolet photodissociation (UVPD) for the activation and dissociation of peptide anions is evaluated for broader coverage of the proteome. To facilitate interpretation and assignment of the resulting UVPD mass spectra of peptide anions, the MassMatrix database search algorithm was modified to allow automated analysis of negative polarity MS/MS spectra. The new UVPD algorithms were developed based on the MassMatrix database search engine by adding specific fragmentation pathways for UVPD. The new UVPD fragmentation pathways in MassMatrix were rigorously and statistically optimized using two large data sets with high mass accuracy and high mass resolution for both MS1 and MS2 data acquired on an Orbitrap mass spectrometer for complex Halobacterium and HeLa proteome samples. Negative mode UVPD led to the identification of 3663 and 2350 peptides for the Halo and HeLa tryptic digests, respectively, corresponding to 655 and 645 peptides that were unique when compared with electron transfer dissociation (ETD), higher energy collision-induced dissociation, and collision-induced dissociation results for the same digests analyzed in the positive mode. In sum, 805 and 619 proteins were identified via UVPD for the Halobacterium and HeLa samples, respectively, with 49 and 50 unique proteins identified in contrast to the more conventional MS/MS methods. The algorithm also features automated charge determination for low mass accuracy data, precursor filtering (including intact charge-reduced peaks), and the ability to combine both positive and negative MS/MS spectra into a single search, and it is freely open to the public. The accuracy and specificity of the MassMatrix UVPD search algorithm was also assessed for low resolution, low mass accuracy data on a linear ion trap. Analysis of a known mixture of three mitogen-activated kinases yielded similar sequence coverage percentages for UVPD of peptide anions versus conventional collision-induced dissociation of peptide cations, and when these methods were combined into a single search, an increase of up to 13% sequence coverage was observed for the kinases. The ability to sequence peptide anions and cations in alternating scans in the same chromatographic run was also demonstrated. Because ETD has a significant bias toward identifying highly basic peptides, negative UVPD was used to improve the identification of the more acidic peptides in conjunction with positive ETD for the more basic species. In this case, tryptic peptides from the cytosolic section of HeLa cells were analyzed by polarity switching nanoLC-MS/MS utilizing ETD for cation sequencing and UVPD for anion sequencing. Relative to searching using ETD alone, positive/negative polarity switching significantly improved sequence coverages across identified proteins, resulting in a 33% increase in unique peptide identifications and more than twice the number of peptide spectral matches.
The advent of new high-performance tandem mass spectrometers equipped with the most versatile collision- and electron-based activation methods and ever more powerful database search algorithms has catalyzed tremendous progress in the field of proteomics (1–4). Despite these advances in instrumentation and methodologies, there are few methods that fully exploit the information available from the acidic proteome or acidic regions of proteins. Typical high-throughput, bottom-up workflows consist of the chromatographic separation of complex mixtures of digested proteins followed by online mass spectrometry (MS) and MSn analysis. This bottom-up approach remains the most popular strategy for protein identification, biomarker discovery, quantitative proteomics, and elucidation of post-translational modifications. To date, proteome characterization via mass spectrometry has overwhelmingly focused on the analysis of peptide cations (5), resulting in an inherent bias toward basic peptides that easily ionize under acidic mobile phase conditions and positive polarity MS settings. Given that ∼50% of peptides/proteins are naturally acidic (6) and that many of the most important post-translational modifications (e.g. phosphorylation, acetylation, sulfonation, etc.) significantly decrease the isoelectric points of peptides (7, 8), there is a compelling need for better analytical methodologies for characterization of the acidic proteome.
A principal reason for the shortage of methods for peptide anion characterization is the lack of MS/MS techniques suitable for the efficient and predictable dissociation of peptide anions. Although there are a growing array of new ion activation methods for the dissociation of peptides, most have been developed for the analysis of positively charged peptides. Collision-induced dissociation (CID)1 of peptide anions, for example, often yields unpredictable or uninformative fragmentation behavior, with spectra dominated by neutral losses from both precursor and product ions (9), resulting in insufficient peptide sequence information. The two most promising new electron-based methods, electron-capture dissociation and electron-transfer dissociation (ETD), are applicable only to positively charged ions, not to anions (10–13). Because of the known inadequacy of CID and the lack of feasibility of electron-capture dissociation and ETD for peptide anion sequencing, several alternative MSn methods have been developed recently. Electron detachment dissociation using high-energy electrons to induce backbone cleavages was developed for peptide anions (14, 15). Another new technique, negative ETD, entails reactions of radical cation reagents with peptide anions to promote electron transfer from the peptide to the reagent that causes radical-directed dissociation (16, 17). Activated-electron photodetachment dissociation, an MS3 technique, uses UV irradiation to produce intact peptide radical anions, which are then collisionally activated (18, 19). Although they represent inroads in the characterization of peptide anions, these methods also suffer from several significant shortcomings. Electron detachment dissociation and activated-electron photodetachment dissociation are both low-efficiency methods that require long averaging cycles and activation times that range from half a second to multiple seconds, impeding the integration of these methods with chromatographic timescales (14–19). In addition, the fragmentation patterns frequently yield many high-abundance neutral losses from product ions, which clutter the spectra (14–17), and few sequence ions (14, 18, 19). Recently, we reported the use of 193-nm photons (ultraviolet photodissociation (UVPD)) for peptide anion activation, which was shown to yield rich and predictable fragmentation patterns with high sequence coverage on a fast liquid chromatographic timeline (20). This method showed promise for a range of peptide charge states (i.e. from 3- to 1-), as well as for both unmodified and phosphorylated species.
Several widely used or commercial database searching techniques are available for automated “bottom-up” analysis of peptide cations; SEQUEST (21), MASCOT (22), OMSSA (23), X! Tandem (24), and MASPIC (25) are all popular choices and yield comparable results (26). MassMatrix (27), a recently introduced searching algorithm, uses a mass accuracy sensitive probability-based scoring scheme for both the total number of matched product ions and the total abundance of matched products. This searching method also utilizes LC retention times to filter false positive peptide matches (28) and has been shown to yield results comparable to or better than those obtained with SEQUEST, MASCOT, OMSSA, and X! Tandem (29). Despite the ongoing innovation in automated peptide cation analysis, there is a lack of publically available methods for automated peptide anion analysis.
In this work, we have modified the mass accuracy sensitive probabilistic MassMatrix algorithms to allow database searching of negative polarity MS/MS spectra. The algorithm is specific to the fragmentation behavior generated from 193-nm UVPD of peptide anions. The UVPD pathways in MassMatrix were rigorously and statistically optimized using two large data sets with high mass accuracy and high mass resolution for both MS1 and MS2 data acquired on an Orbitrap mass spectrometer for complex HeLa and Halo proteome samples. For low mass accuracy/low mass resolution data, we also incorporated a charge-state-filtering algorithm that identifies the charge state of each MS/MS spectrum based on the fragmentation patterns prior to searching. MassMatrix not only can analyze both positive and negative polarity LC-MS/MS files separately, but also can combine files from different polarities and different dissociation methods into a single search, thus maximizing the information content for a given proteomics experiment. The explicit incorporation of mass accuracy in the scores for the UVPD MS/MS spectra of peptide anions increases peptide assignments and identifications. Finally, we showcase the utility of integrating MassMatrix searching with positive/negative polarity MS/MS switching (i.e. data-dependent positive ETD and negative UVPD during a single proteomic LC-MS/MS run). MassMatrix is available to the public as a free search engine online.
EXPERIMENTAL PROCEDURES
Materials
HPLC buffer components and proteomics-grade trypsin were obtained from Sigma-Aldrich (St. Louis, MO). All other reagents and solvents were obtained from Fisher Scientific (Fairlawn, NJ). An equimolar mixture of mitogen-activated protein kinases (MAPKs), including c-Jun N-terminal kinase 2, mitogen-activated protein kinase 1, and p38 mitogen-activated kinase 14, were prepared as described elsewhere (20). HeLa cells were obtained from American Type Culture Collection (CCL-2) (Manassas, VA) and were lysed, reduced, alkylated, and digested as reported previously (30). Halobacterium salinarum NRC-1 was purchased from the American Type Culture Collection. The bacteria were grown in the recommended medium (American Type Culture Collection medium 2185). HeLa cells were suspended in low-salt buffer (10 mm Tris-HCl, 10 mm KCl, 1.5 mm MgCl2, pH 8.0) to swell prior to dounce homogenization to lyse the cells. The whole cell lysate was centrifuged at 1000 × g to clarify the soluble lysate and to remove the insoluble pellet.
Sample Preparation
Proteins from Halobacterium and HeLa lysates were digested overnight with trypsin using a FASP protein digestion kit (Protein Discovery, San Diego, CA). Prior to digestion, proteins were reduced in 5 mm dithiothreitol for 30 min at 55 °C, and this was followed by alkylation in 15 mm iodoacetamide at room temperature in the dark for 30 min. Trypsin was added in a 1:20 enzyme-to-protein ratio, and the solution was buffered at pH 7.5 in 50 mm ammonium bicarbonate.
Mass Spectrometry, Liquid Chromatography, Ultraviolet Photodissociation, and Electron Transfer Dissociation
All mass spectrometric measurements were carried out on a Thermo Fisher Scientific LTQ XL (San Jose, CA) linear ion trap mass spectrometer or a Thermo Fisher Scientific Orbitrap Elite (Thermo Fisher Scientific, Bremen, Germany) mass spectrometer. The LTQ mass spectrometer was equipped with a Coherent (Santa Clara, CA) ExciStar XS excimer laser operated at 193 nm via a setup similar to that previously described (30, 31). Liquid chromatography was carried out using a Dionex UltiMate 3000 system (Sunnyvale, CA) with an Agilent ZORBAX 300SB-C18 column (Santa Clara, CA) (150 × 75 μm, 3.5-μm particle size) at flow rate of 300 nl/min. The nano-electrospray ionization (ESI) setup consisted of a conductive mini microfilter assembly from IDEX Health and Science (Oak Harbor, WA) coupled to a New Objectives uncoated PicoTip® nanoESI emitter (Woburn, MA), which was operated at an ESI voltage of 2 kV. A 60-min linear gradient from 5% eluent B to 40% eluent B was used for the first set of MAPK samples, in which eluent A was 10 mm piperidine in water and eluent B was 10 mm piperidine in acetonitrile. A 210-min linear gradient from 5% eluent B to 35% eluent B was used for the HeLa samples and the second set of MAPK samples, in which eluent A was 0.1% acetic acid in water and eluent B was 0.1% acetic acid in acetonitrile. The mitogen-activated kinase mixture was injected at approximately one picomole of tryptic digest per protein. For the HeLa sample, ∼2 μg of total lysate was injected. Data-dependent nanoLC-MS/UVPD was performed as follows: the first event was the full negative mass scan (m/z range of 400–2000), which was followed by five UVPD events on the five most abundant ions from the first event. A series of eight 1-mJ UV pulses (applied during a 16-ms activation period) was used per MS/MS scan with a q-value set at 0.1. Data-dependent nanoLC-MS/CID was performed as follows: the first event was the full positive mass scan (m/z range of 400–2000), which was followed by 10 CID events on the 10 most abundant ions from the first event. For CID, a normalized collision energy of 35%, a q-value of 0.25, and a collision duration of 30 ms were used. Polarity switching data-dependent nanoLC-MS/MS was performed as follows: the first event was the full negative mass scan (m/z range of 400–2000), which was followed by five UVPD events on the five most abundant ions from the first event; the seventh event was the full positive mass scan (m/z range of 400–2000), and that was followed by five ETD events on the five most abundant ions from the seventh event. The maximum ion injection time was 100 ms for both full scan and MS/MS events. A dynamic exclusion duration of 50 s was used with a list size of 500 allowed m/z values and a single repeat count. ETD experiments used fluoranthene as the reagent anion and a reaction time of 100 ms. CID, ETD, and UVPD spectra were the average of 3, 5, and 10 microscans, respectively. UVPD spectra from polarity switching experiments were the average of five microscans. To allow both positive ETD and negative UVPD to be performed in the same nanoLC-MS/MS run, a 1.8-mm hole was drilled in the ETD ion volume to create a direct path for the excimer laser beam from the optical window to the ion trap through the intervening ETD source.
The Halobacterium and HeLa samples were analyzed on a Thermo Scientific Orbitrap Elite (Thermo Fisher Scientific, Bremen, Germany) outfitted with a 193-nm excimer laser (same Coherent ExciStar XS excimer as used in the procedures described above) and modified to allow UV activation in the higher energy collision-induced dissociation (HCD) cell of the instrument. Photodissociation was implemented in a manner similar to that described previously (32, 33). LC separations were performed using water (A) and acetonitrile (B) mobile phases containing 0.05% acetic acid (pH ∼ 4) on a Dionex Ultimate 3000 configured for pre-concentration with a loading solvent of 98% water, 2% acetonitrile, and 0.05% acetic acid. Trap columns (35 mm × 0.1 mm) and analytical columns with an integrated emitter (150 cm × 0.075 cm) were packed in house using IntegraFrit and PicoFrit capillaries (New Objective, Woburn, MA) and 3 μm Michrom Magic C18 packing material. Approximately 3 μg of digest was loaded onto the trap at 5 μl/min for 3 min. For the Halobacterium digests, the solvent composition remained at 3% B for 2 min before increasing to 7% B over 10 min, then to 20% B over 180 min, and finally to 30% B over 25 min. The LC separations of the HeLa digests started with 2 min running at 3% B followed by an increase to 7% B in 20 min, a further increase to 17% B in 150 min, and a final gradient to 35% B in 45 min. For nanospray, 1.7 kV was applied at a liquid voltage junction located pre-column. One microscan was used for MS and MSn scans in all experiments. Ion trap automatic gain control targets were 30,000 for full MS and 10,000 for MSn, and Fourier transform automatic gain control targets were 1,000,000 for both full MS and MSn. The maximum ion time was 100 ms for MS and MSn.
All data-dependent nanoLC-MS methods on the Orbitrap involved a Fourier transform full mass survey scan (m/z range of 400–2000) at a resolution of 120,000 followed by a series of MS/MS scans on the top 10 most abundant ions from scan event 1. The minimum signal required for MS2 selection was 4000, and the isolation width was fixed at 2. Dynamic exclusion was enabled for 45 s with a repeat count of 1 and a list size of 500 m/z values. For CID, a normalized collision energy of 35 was used with activation q 0.25 and activation time 10 ms. For HCD, the Fourier transform resolution was 15,000, the normalized collision energy was 30, and the activation time was 0.1 ms. ETD was performed using fluoranthene reagent ions and an activation time of 100 ms. For negative UVPD, a normalized collision energy of 1 was used to transfer ions into the HCD cell, where two 2-mJ pulses were delivered over a period of 4 ms for activation. Product ions from UVPD were detected in the Orbitrap at a resolution of 15,000.
Data Analysis
The MassMatrix UVPD search algorithm was developed based on the MassMatrix search algorithms for CID and ETD by adding new fragmentation pathways for UVPD. MassMatrix has also been adapted to search MS data from negative mode and mixed positive/negative mode LC-MS/MS equipped with one or more fragmentation methods. The scoring models involved in the new UVPD algorithm are the same as those used for CID as described elsewhere (27, 28, 34). Three independent statistical scores, pp, pp2, and pptag, from the scoring models are used mainly to evaluate the quality of peptide-spectrum matches in MassMatrix. MassMatrix also features a precursor filtering algorithm (including intact charge-reduced peaks) to improve database search results for UVPD. For low mass accuracy and low mass resolution UVPD tandem MS data, an automated charge deconvolution algorithm was incorporated in MassMatrix to resolve precursor charge states. Database search results from MassMatrix are reported in HTML files. Protein and peptide match lists are also exported to tables presented in .CSV text file format in MassMatrix (version 2.3.4 or later) (29).
The RAW data files collected on the mass spectrometer were converted to mzXML files by use of MassMatrix data conversion tools (v3.9). For high mass accuracy and high mass resolution Orbitrap data, isotope distributions for the precursor ions of the MS/MS spectra were deconvoluted to obtain the charge states and monoisotopic m/z values of the precursor ions during the data conversion. All data were searched using an in-house MassMatrix web server (v2.4.2). The search parameters in MassMatrix were set as follows: (i) enzyme: trypsin; (ii) missed cleavages: 2; (iii) modifications: fixed iodoacetamide derivative of cysteine and variable oxidation of methionine; (iv) precursor ion mass tolerances: 20 ppm for Orbitrap data and 2.0 Da for LTQ data; (v) product ion mass tolerances: 0.02 for HCD and UVPD tandem MS data on Orbitrap, and 0.6 Da for ion trap (CID, ETD, and UVPD on LTQ) data; (vi) maximum number of modifications allowed for each peptide: 2; (vii) peptide length: 6 to 40 amino acid residues; (viii) score thresholds of 5.3 and 1.3 for the pp/pp2 and pptag scores, respectively. Phosphorylation of serine, threonine, and tyrosine was set as an additional variable side-chain modification for MAPK samples. The UniProt-SwissProt Human database (release 2011 06, 20,236 entries) was used for MAPK and HeLa data sets, and the Halobacterium sp nrc1 was used for Halo data sets. Peptide and protein identifications were both filtered at a 1% false discovery rate (FDR). Peptides that were shared by multiple proteins were counted only once. Counted proteins that shared some common peptides had to have at least one unique peptide during protein-level FDR calculation.
RESULTS AND DISCUSSION
Recently, we investigated the utility of using 193-nm UVPD to sequence both singly and multiply charged peptide anions (20). This method generated rich and predictable fragmentation patterns, with a-/x-type products being the most prolific and abundant sequence ions, often yielding 100% sequence coverage. Representative examples of the UVPD spectra are shown in supplemental Figs. S1 and S2. A series of UVPD spectra for three peptides from the HeLa cell digest with precursor charge states of 1-, 2-, and 3- are shown in supplemental Fig. S1. Supplemental Fig. S2 shows an example of a UVPD mass spectrum of one peptide from the LC-MS analysis of the Halobacterium lysate. Abundant a and x ions that afford 100% sequence coverage were observed following a single UV pulse. The promise of this UVPD technique for acidic proteome characterization, as highlighted in our initial experiments, motivated us to create an automated searching platform specific to UVPD. Thus, we modified the MassMatrix algorithm to accept anion spectra and to incorporate UVPD product ion specificity. This MassMatrix package can also simultaneously search MS/MS spectra from multiple ion activation methods (CID, UVPD, ETD/electron-capture dissociation, negative ETD, etc.), as well as ions of both polarities (+/−), to maximize proteome coverage.
To test and validate MassMatrix searching of UVPD spectra, we utilized three different sample types. The first two samples were complex tryptic peptide mixtures from the cytosolic portion of HeLa cervical cancer cells and Halobacterium and were used to generate large peptide datasets for the optimization of the search algorithm and comparison between UVPD and other conventional fragmentation methods. The predicted proteome of Halobacterium sp. NRC-1, an archaea that flourishes in a high-salt environment, is highly acidic. It has been used as a model system for probing prokaryotic adaptation to extreme conditions. With a mean isoelectric point of 4.9, Halobacterium is an ideal organism for evaluating the negative polarity UVPD strategy for acidic peptides and proteins. The third sample was a mixture of three MAPKs. These proteins in effect regulate all cellular activity and thus are thought to play key roles in cancer development (35). Conveniently, the MAPKs utilized herein also yield many acidic peptides after trypsin digestion, making them a useful model for validating the negative-mode UVPD (NUVPD) method.
Optimization of Fragmentation Pathways of UVPD for MassMatrix Database Search
Fragmentation patterns of UVPD for peptide anions are relatively complicated compared with CID or ETD fragmentation of peptide cations. Manual inspection of the UVPD peptide anion tandem mass spectra showed that a and x ions were the two main ion series created by UVPD fragmentation (20). However, other ions, including some y, z, and a/x ions with hydrogen migration, also contribute significantly to the tandem mass spectra generated via UVPD of peptide anions. We have rigorously optimized the fragmentation pathways for UVPD. Supplemental Scheme S1 shows the workflow diagram of the optimization strategy for development of the in silico algorithm for UVPD. The workflow used an iterative process to fully optimize the pathways for UVPD anions.
We first started with the initial fragmentation pathways for all peptides that considered only a and x ions based on the basic knowledge of UVPD fragmentation.
We then built a search algorithm based on the common pathways and searched two large data sets from the complex Halo and HeLa proteome samples. Although the search algorithm was based on non-optimized pathways, it successfully identified high-quality peptide matches at a low FDR of 1%. The FDR estimation was rigorously calculated based on the gold-standard target-decoy search strategy (36).
Based on these high-quality peptide matches with very low FDRs, we performed a statistical analysis to elucidate the pathways in more detail.
Based on the statistical results, the initial pathways were further refined and optimized.
We then repeated steps 2, 3, and 4 based on the refined pathways until the statistical results were consistent with the pathways that were used to generate the peptides for statistical analysis.
To recap, starting with a base algorithm that included only a/x ions, a subset of high-quality peptide spectral matches using a 1% FDR was defined. Those same high-caliber UVPD spectra were then re-inspected for other product ions that could consistently be ascribed to other types of fragments derived from in silico–generated pathways, such as b, c, y, z, CO2 losses, and hydrogen migrations. Identification of the additional types of fragment ions allowed the original UVPD spectra to be iteratively searched and the to be algorithm trained. In this way, the algorithm was refined and expanded to include a greater array of fragment ions consistently generated upon UVPD of peptide anions, and the spectral re-inspection process triggered by strong a/x matches ensured confidence that other non-a/x fragment ions in those same spectra would in fact be meaningful diagnostic ions worthy of assignment and tabulation. This iterative procedure ultimately led to the identification of the predominant or favored pathways upon UVPD of peptide anions, including a/x, a+1/x+1, and z+1, plus a few charge-state-dependent pathways, such as a+2/x+2 for doubly and triply charged anions and several observed consistently only for singly charged ions (c, y−1, and y). These types of fragment ions are shown graphically as heat maps in Fig. 1 and are discussed in more detail later.
Fig. 1.
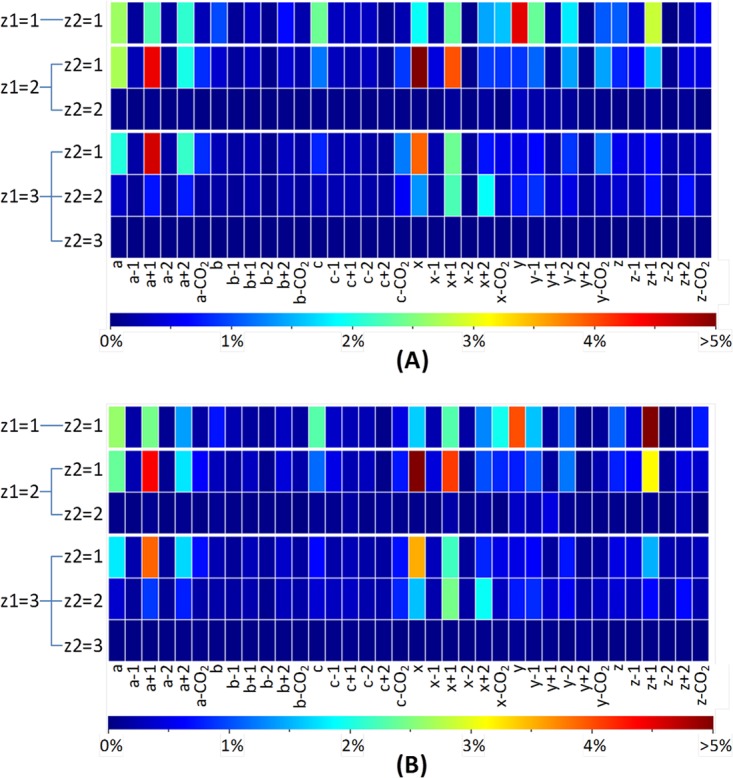
The fragmentation behavior of tryptic HeLa and Halo peptide anions as determined via UVPD from the optimized search algorithm in MassMatrix: ion intensities of various product ions normalized by total ion intensities of the spectrum (after precursor and intact charge-reduced peak filtering) for tryptic peptide matches with FDR < 1% from (A) Halo and (B) HeLa proteome samples. For the Halo proteome sample, 14,802 peptide spectral matches (PSMs) from 3663 unique peptides were identified. Among them, 726 PSMs from 328 unique peptides were singly charged, 12,112 PSMs from 3121 unique peptides were doubly charged, and 1964 PSMs from 617 unique peptides were triply charged. For the HeLa proteome sample, there were 7886 PSMs from 2349 unique peptides identified. Among them, 232 PSMs from 121 unique peptides were singly charged, 5972 PSMs from 1991 unique peptides were doubly charged, and 1682 PSMs from 568 unique peptides were triply charged. Only one representative PSM for each unique peptide with a given charge state was used in plotting the heat maps.
For both the Halo and HeLa proteome samples used in the pathway optimization, nanoLC-MS/UVPD was undertaken using a mobile phase system consisting of 0.05% acetic acid in water and 0.05% acetic acid in acetonitrile. Because the use of acetic acid as a mobile phase modifier/ion-pairing agent results in an acidic mobile phase that limits the overall charging of peptide anions, the charge state distribution is more narrowly focused and decreases the overlap between peptide species that co-elute and have the same m/z value. Interestingly, no significant differences in overall ion abundances were observed relative to those obtained using more basic mobile phases; only the charge state distributions were different (data not shown). Furthermore, the use of an acidic mobile phase system for negative ESI-MS analysis means that no special equipment or columns are needed (i.e. the same chromatographic system and conditions can be used for both positive- and negative-mode proteome analysis). Others have noted the ability to generate negative ions from acidic solutions (37, 38), so this phenomenon is not unprecedented.
In the 193-nm UVPD mass spectra of peptide anions, some of the most prevalent product ions arise from sequential electron detachment events that create charge-reduced, radical species. In addition, both precursor ions and charge-reduced radical species undergo neutral losses of CO2. These products give no direct information about the peptide sequence. From the statistical results of the initial searches considering only a and x ions (as well as all the optimized pathways), we confirmed that most of the peptide UVPD spectra were dominated by intact precursor ions, charge-reduced precursors, and radical precursor species with neutral losses of CO2. For the Halo peptides, these precursor species contributed 74.4%, 63.4%, and 63.5% of the total ion abundance of a UVPD mass spectrum on average for the 1-, 2-, and 3- charge states, respectively. For the HeLa peptides, the contributions from the non-dissociated precursors were 69.5%, 59.0%, and 57.4% for the 1-, 2-, and 3- charge states, respectively. These precursor species give no direct information about the peptide sequence and can significantly lower the specificity of a database search. Therefore, we developed an automated filtering algorithm to filter these ions prior to database searching. The subsequent optimization of the UVPD pathways was then based on the filtering of the precursor species.
To gain unbiased and statistically relevant insight into the UVPD fragmentation behavior of peptide anions, we utilized UVPD patterns from a large population of tryptic peptides from HeLa and Halobacterium cells and built an automated in-house program written in Python to measure the frequencies and intensities of various product ions during statistical analysis in the optimization. The ion intensities defined by each product ion normalized by the total spectral ion intensity are plotted in heat maps for the Halo and HeLa data sets (Fig. 1). The heat maps illustrate the overall importance of various product ions for UVPD fragmentation. This statistical result was considered consistent with the pathways used to generate the search results when all the product ions used in the pathways were the ones of greatest contributions in the heat maps. The pathways were refined iteratively until consistent results were achieved. The heat maps of the final optimized pathways for the Halo and HeLa data sets are shown in Fig. 1 and are categorized by precursor charge state (1- through 3-) and distribution of sequence ions a, b, c, x, y, and z. The product ions considered in the final optimized pathways are listed in Table I. The patterns of the heat maps of Halo and HeLa data are very similar and give identical optimized pathways. Basically, a/x, a+1/x+1, and z+1 ions were observed with significant contribution to the UVPD spectra for all 1-, 2-, and 3- peptides. a+2/x+2 ions were observed with significance for those peptides with 2- and 3- charge states. c, y−1, and y ions were observed with significance for singly charged (1-) peptides only.
Table I. UVPD product ions with charge state selectivity that were incorporated into the MassMatrix algorithm.
| Product ion | a/x | a+1/x+1 | z+1 | a+2 | x+2 | c | y−1 | y |
| Charge state selectivity | All | All | All | 2-/3- | 2-/3- | 1- | 1- | 1- |
To test the benefits of this pathway optimization (i.e. automated precursor filtering (including intact charge-reduced peaks)) and using UVPD-specific, complementary product ions in addition to the a/x series, receiver operating characteristic (ROC) curves were generated from these MassMatrix search results of the tryptic Halobacterium and HeLa peptide anions analyzed via nanoLC-MS/UVPD as shown in Fig. 2. ROC curves were created by plotting the number of true positives against the number of false positives as the score thresholds decreased in the search results. An ideal database search should return all true positives with scores higher than all false positives and an ROC curve with a right angle. An ROC curve shifted toward the left indicates higher specificity (i.e. a better false positive rate for a given number of matches). A curve shifted toward the top indicates higher sensitivity (i.e. more true positives at a given false positive rate). It can be seen in Fig. 2 that both the sensitivities and the specificities of the searches were improved significantly after pathway optimization, or filtering of precursor species and incorporating, in addition to a/x ions, more product ions of significance. All the subsequent results for negative UVPD are from the search algorithm utilizing the optimized pathways. This MassMatrix UVPD search algorithm with the optimized pathways is publically available online (version 2.4.4 or later).
Fig. 2.
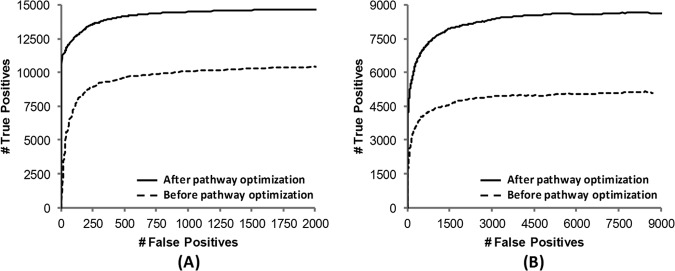
ROC curves of MassMatrix search results for UVPD of (A) Halo and (B) HeLa tryptic digests. Searches were performed before pathway optimization using only the a/x ion series without any filtering and again after pathway optimization using all product ions listed in Table I and applying precursor/charge-reduced peak filtering and CO2 neutral loss filtering.
Comparisons of Peptide and Protein Identifications by High Mass Accuracy NUVPD, ETD, CID, and HCD
Tryptic digests of the Halobacterium and HeLa cell proteins were analyzed in triplicate on the Orbitrap mass spectrometer via nanoLC-MS/MS using NUVPD (−), ETD (+), CID (+), and HCD (+) (raw files, protein match lists, and peptide match lists may be accessed from the Peptide Atlas data repository using the information provided in supplemental Tables S1 and S3). Among these sets of results, the MS/MS product ions obtained upon NUVPD (−) and HCD (+) were analyzed in the Orbitrap analyzer with high mass accuracy and high mass resolution. The product ions generated via ETD (+) and CID (+) were analyzed in the linear ion trap with low mass accuracy and low mass resolution. The results are summarized in Tables II and III in terms of the numbers of peptides and proteins identified by each MS/MS method, as well as via Venn diagrams (Figs. 3 and 4) that illustrate the intersection in the identifications obtained with the various MS/MS methods. With respect to the total number of peptides identified via each method, 3663 and 2350 unique peptides were identified via NUVPD for the Halo and HeLa digests, respectively. This represents fewer total unique peptides than identified via the CID or HCD methods (6088 to 10,121 peptides). However, 655 and 645 peptides were uniquely identified via NUVPD for the Halobacterium and HeLa digests, respectively, confirming that UVPD analysis of peptide anions results in the mapping of new regions of proteins. In terms of the corresponding protein maps derived from the peptide identifications, the UVPD results led to the identification of 805 and 659 proteins for the Halobacterium and HeLa samples, again corresponding to fewer total protein identifications than CID or HCD, but 49 and 50 proteins not identified via ETD, CID, or HCD were uniquely found through NUVPD for the Halobacterium and HeLa proteomes, respectively (see Figs. 3 and 4). The ability to identify ∼50 new proteins in a proteome is a significant advance and is attributed to the ability of UVPD to generate informative MS/MS spectra from peptides that ionize efficiently in the negative mode, ones that might not ionize effectively in the positive mode, or give uninformative fragmentation patterns in the positive mode. This is especially true for the acidic proteome of Halobacterium, as we were able to identify 1181 total proteins by combining UVPD with other traditional MS/MS methods (Fig. 4)—the most proteins identified to date for this organism (to the best of our knowledge) (39, 40). The 1181 identified proteins covered 45% of the 2630 predicted proteins from Halobacterium NRC-1 (41) and were identified without the use of labor-intensive sample prefractionation techniques.
Table II. The number of unique peptides identified via various fragmentation methods with FDR < 1%.
| UVPD (−) | ETD (+) | CID (+) | HCD (+) | |
|---|---|---|---|---|
| Halo | 3663 | 1220 | 6088 | 7335 |
| HeLa | 2350 | 1228 | 9716 | 10,121 |
Table III. The number of proteins identified via various fragmentation methods with FDR < 1%.
| UVPD (−) | ETD (+) | CID (+) | HCD (+) | |
|---|---|---|---|---|
| Halo | 805 | 431 | 996 | 1051 |
| HeLa | 659 | 381 | 1469 | 1656 |
Fig. 3.
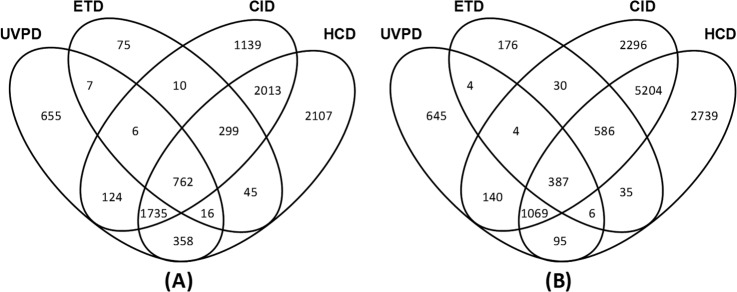
The number of unique peptides identified via various MS/MS methods with FDR < 1% and Venn diagrams showing overlap of peptide identifications for (A) Halobacterium and (B) HeLa tryptic digests.
Fig. 4.
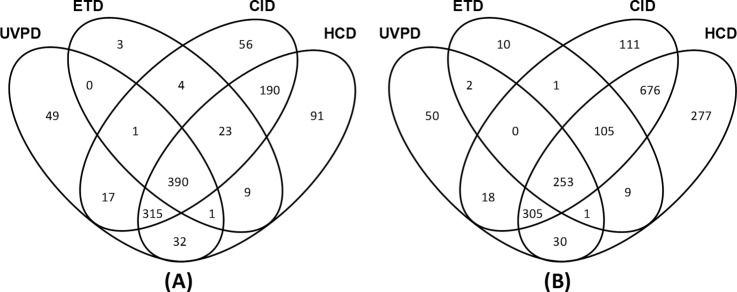
The number of unique proteins identified via various MS/MS methods with FDR < 1% and Venn diagrams showing overlap of protein identifications for (A) Halobacterium and (B) HeLa tryptic digests.
The distributions of the pI values of the peptides identified via each of the MS/MS methods are summarized in Fig. 5 as shown by the probability density as a function of pI. Although the general distributions parallel each other for all four activation methods, the probability density is significantly enhanced at the lowest pI values for the UVPD dataset. This result reflects the greater ionization efficiencies of the more acidic peptides in the negative ESI mode, leading to the production of diagnostic fragmentation patterns upon UVPD that facilitate identification. We anticipate that the enhancement would be even greater using a high-pH mobile phase that would significantly improve ionization in the negative mode, and implementation of this strategy is underway. The amino acid composition of the peptides identified via UVPD, CID/HCD, and ETD was also determined and is summarized in bar-graph form in supplemental Fig. S3. No significant trends were discerned.
Fig. 5.
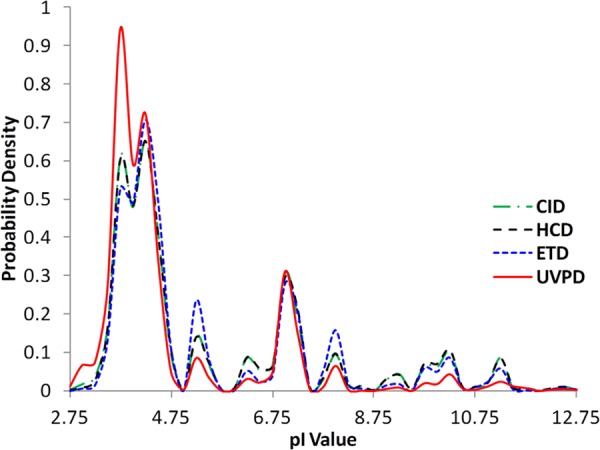
Distributions of pI values of peptides identified via different MS/MS methods for merged data from both Halo and HeLa proteome samples.
Automated Precursor Charge Identification for Low Mass Accuracy UVPD Data
For low mass accuracy and low mass resolution tandem MS data, the charge states of peptide precursors usually cannot be resolved using precursor isotopic components, and thus multiple charge states of a precursor must be searched. Although the charge-reduced precursor ions in UVPD give no direct information about the peptide sequence, their presence provides evidence about the precursor charge state of the given peptide. Using this information, we have developed a charge deconvolution algorithm that automatically identifies the charge state. For each UVPD MS/MS, the eight most abundant ions are identified such that each is sufficiently distinctive in m/z to represent a different ion. Using the precursor m/z value of the spectrum, a list of offset values is created such that each offset value represents a possible m/z value for the ion peaks representing the charge-reduced radical species from electron detachment. Each m/z value depends on both a presumed precursor charge and the number of additional charge reduction events, for which
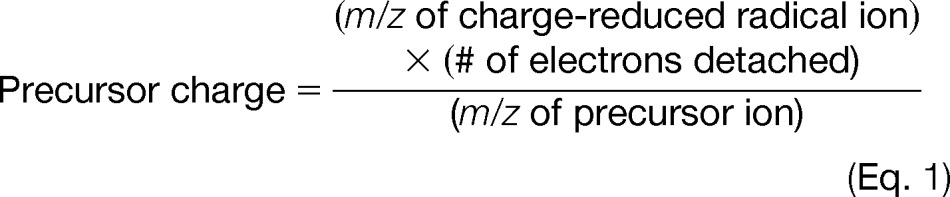 |
and for each precursor charge state (n) there will be a total of n − 1 charge-reduction events. To identify the most likely precursor charge of a spectrum, the most abundant ion peaks in the mass spectra are compared with the calculated theoretical ion peak values and their CO2-loss counterparts via a matched-peak count system. The tolerance for matching peaks is set at 0.15% of the precursor m/z value. A score is maintained for each possible charge, and when there is a match, scoring increases. This increase is determined such that ions of the same type are ranked by abundance in the spectrum. When two charge states share the same score, the lower absolute charge state is chosen. For example, ion peaks from charge reduction of the precursor (without dissociation) will have the same m/z for 2- precursors with one electron detachment and 4- precursors with two electron detachments. Thus, in this case, the lower charge state is selected. Furthermore, compensation is made for precursors with large m/z values whose charge-reduced analogs might have m/z values outside the detectable m/z range. This precursor charge state assignment for low mass accuracy and low mass resolution UVPD data has been incorporated into the MassMatrix searching algorithm. The algorithm enables MassMatrix to search only the interpreted charge state for each low mass accuracy UVPD tandem mass spectrum, which both increases searching selectivity and reduces searching time. This algorithm is similar to other charge-state-determining computational tools used for positive-mode ETD (42–45) but is specific to the unique fragmentation behavior of negative-mode UVPD.
As an example demonstrating the charge state determination method, nanoLC-MS/UVPD using a mobile phase gradient comprising 10 mm piperidine in water and 10 mm piperidine in acetonitrile was used to analyze the MAPK mixture and HeLa tryptic peptides. Because the high basicity of piperidine increases the overall charging of peptide anions, the accuracy of the charge assignment can be tested over a wider range of precursor charges when using this basic mobile phase. Supplemental Fig. S4A illustrates the accuracy of the automated charge state assignment algorithm for precursor charges 1- through 5- verified by means of manual inspection. MS/MS spectra with poor signal-to-noise were not included in the figure, as it is anticipated that they likely would not result in both accurate charge-state assignment and true positive searching results. Supplemental Fig. S4B illustrates the accuracy of the automated charge-state assignment algorithm for precursor charges 1- through 3- verified by means of MassMatrix database search. The systematic verification of the algorithm using a MassMatrix database search was achieved by comparing the charge states of the peptide spectral matches (PSMs) at FDR < 1% from the MassMatrix database search with the charge determination algorithm disabled to the charge states of the same MS/MS spectra predicted by the charge determination algorithm. As seen in supplemental Fig. S4, the accuracy ranged from 86% to 100%, illustrating a high level of reliability for the algorithm. Interestingly, the majority of the incorrect charge state assignments from spectra of good quality arose from the dissociation of multiple peptide species with different charges but having the same m/z value (i.e. ones that co-elute during LC separation). The accuracy of the charge state algorithm was tested by undertaking a search using high mass accuracy Orbitrap/UVPD data sets for which the charge states were alternately assigned by the mass spectrometer software (using precursor isotopes) versus the custom algorithm created for low mass accuracy spectra. In terms of proteins matched, the charge state assignments based on the custom (low mass accuracy) algorithm led to 91% of the same protein matches as the high-accuracy isotope-based algorithm for the HeLa data sets and 98% of the same protein matches for the Halobacterium data sets. Translating this to the peptide level, the total number of peptide spectral matches obtained for the Halobacterium data set using the custom (low mass accuracy) charge state assignment was 92% of the total number derived from the isotope-based charge-assignment method. Examination of the charge states of the peptides indicated that 75% of the missed charge state assignments were for the 3- charge state, and 25% were for the 2- charge state.
Verifying MassMatrix Interpretation of Peptide Anion Populations by Means of Low-resolution UVPD
The accuracy and specificity of the MassMatrix UVPD search algorithm was also assessed for low-resolution, low mass accuracy UVPD on a linear ion trap using a tryptic mixture of three MAPK proteins analyzed via nanoLC-MS/MS. As mentioned previously, the MAPKs yield many acidic peptides after trypsin digestion, making them a useful model for method verification. An acidic mobile phase system (acetonitrile/water/acetic acid) was again used for this aspect of the study, and the resulting base peak chromatogram for one of the runs is shown in supplemental Fig. S5. A search was conducted against the UniProt-SwissProt Human database using MassMatrix, and the top three hits were the accurate identification of the three MAPKs (MAPK 14, MAPK 1, and MAPK 9). The sequence coverage for MAPK 14, MAPK 1, and MAPK 9 was 52%, 36%, and 28% at FDR < 1%. When comparing the same MAPK mixture by positive-mode CID, the sequence coverage for MAPK 14, MAPK 1, and MAPK 9 was 47%, 49%, and 43% at FDR < 1%. Interestingly, only 3899 MS/MS spectra were collected for UVPD, whereas 15,760 MS/MS spectra were collected for CID. This decrease in collection rate for UVPD is due to the decrease in signal intensity inherent in the LTQ instrumentation for negative-mode analysis relative to positive mode; thus, more averages are needed per MS/MS spectrum, injection times are increased, and the overall sensitivity is lower. In order to take advantage of data from both positive and negative analysis, the new MassMatrix setup is capable of mixing and matching information from various positive- and negative-mode MS/MS techniques and combining the data into a single search, which can improve the overall data quality. When the CID and UVPD nanoLC-MS/MS runs for the MAPK mixture were combined, the sequence coverage improved to 52%, 49%, and 45% for MAPK 14, MAPK 1, and MAPK 9, respectively (raw files, peptide match lists, and protein match lists may be accessed from the Peptide Atlas data repository using the information provided in supplemental Tables S2 and S3).
Positive/Negative Polarity Switching for Simultaneous Sequencing of both Anion and Cation Peptides
The strategy of accumulating MS/MS data for both positive and negative ions via alternating scans in a single LC-MS/MS run has been demonstrated previously for screening drugs in biological matrices, ultimately yielding high sensitivity and structural information (46). Such dual-polarity alternating MS/MS methods have not been routinely exploited for peptide sequencing, for several reasons. As mentioned above, CID studies of peptide anions do not reliably produce highly diagnostic spectra, instead promoting uninformative and unpredictable neutral losses. The electron-based activation methods are likewise not readily amenable to dual-polarity studies, requiring either unique chemical reagents or different instrument specifications to generate electron-donating versus electron-accepting reagent ions (12, 16, 17). In contrast, UVPD requires no re-tuning or re-optimization when switching between positive and negative ion modes and can be easily coupled to a variety of standard positive-mode dissociation techniques (CID, ETD, etc.). Also, the new MassMatrix algorithm described herein offers the ability to automate the sequencing of both peptide cations and anions in single LC-MS/MS runs. The success of these polarity-switching experiments will depend on the combination of MS/MS methods used, as well as the particular sample being analyzed (acidic versus basic peptide populations). In terms of positive-mode MS/MS, ETD has been shown to yield high peptide sequence coverage and has demonstrated significant success for characterizing acidic post-translational modifications such as phosphorylation (47); however, this technique has a significant bias toward the identification of highly basic peptides (48). We anticipated that negative UVPD could complement ETD to improve the identification of the more acidic peptides in complex mixtures via a polarity-switching LC-MS/MS strategy. To test our hypothesis, tryptic peptides from the cytosolic fraction of HeLa cells were analyzed via polarity-switching nanoLC-MS/MS utilizing ETD for cation sequencing and UVPD for anion sequencing. A mobile phase system using acetic acid was again used, and the extracted negative and positive polarity base peak chromatograms are illustrated in supplemental Figs. S6A and S6B, respectively. Supplemental Figs. S6C and S6D show examples of UVPD and ETD MS/MS spectra from the polarity-switching experiment that were used by MassMatrix to successfully identify HeLa tryptic peptides (e.g. the more acidic GDLGIEIPAEK from pyruvate kinase isozyme and the more basic KQSLGELIGTLNAAK from triosephosphate isomerase). The raw MassMatrix output for this experiment is shown in supplemental Table S1.
To investigate the extent to which UVPD aided ETD in improving proteome analysis, MassMatrix was used to search ETD spectra alone and then when combined with negative UVPD spectra for the LC-MS data shown in supplemental Fig. S7. Supplemental Fig. S7A illustrates the sequence coverage of proteins identified via positive ETD only versus that of proteins identified via positive ETD plus negative UVPD. As seen in supplemental Fig. S7A, a notable increase in sequence coverage was observed when negative UVPD was combined with positive ETD for more than 150 identified proteins. Also, the number of post-translational modifications increased from 669 to 1271 at a FDR < 1%, and the number of unique peptide identifications increased from 515 to 687 in favor of the combined MassMatrix UVPD(−)/ETD(+) search, as seen in supplemental Fig. S7B (raw data, peptide match lists, and protein match lists may be accessed from the Peptide Atlas data repository using the information provided in supplemental Tables S2 and S3). These findings establish a compelling benchmark for obtaining high sequence information of both acidic and basic peptides via alternating positive/negative MS/MS scans in the same chromatographic run.
CONCLUSION AND OUTLOOK
We have presented a modified MassMatrix algorithm that allows database searching of negative polarity MS/MS data and which has been adapted for the highly diagnostic fragmentation arising from UVPD of peptide anions. Owing to the fact that MassMatrix accepts the common mzXML file format (as well as mzData and MGF), essentially any LC-MS/MS data set can be processed, regardless of the instrument make or model. The UVPD pathways in MassMatrix were statistically optimized using two large data sets with high mass accuracy and high mass resolution for both MS1 and MS2 data acquired on an Orbitrap mass spectrometer for complex HeLa and Halo proteome samples. Filtering precursor species and inclusion of the additional diagnostic product ions (in addition to a/x ions) from UVPD give rise to higher sensitivity and specificity and lower false discovery rates in database searches. UVPD of peptide anions from tryptic digests of Halobacterium or HeLa cell lysates led to the identification of more unique peptides and proteins not found by means of conventional CID, HCD, or ETD in the positive mode, underscoring the potential strategic benefits of extending sequence coverage by utilizing both positive and negative modes. The benefit of UVPD was especially highlighted in the analysis of the acidic proteome of Halobacterium, as 45% of the predicted proteins were identified by combining the negative-mode UVPD method with traditional positive-mode MS/MS techniques (without sample prefractionation). UVPD alone identified 68% of the 1181 proteins found for the Halobacterium samples. Moreover, UVPD of peptide anions in combination with ETD of peptide cations in alternating scans affords a substantial increase in sequence coverage.
Supplementary Material
Acknowledgments
J.A.M. acknowledges the ACS division of Analytical Chemistry and The Society for Analytical Chemists of Pittsburgh for support.
Footnotes
* Funding from the NIH (R21GM099028) is acknowledged. K.D. acknowledges funding from the Welch Foundation (F-1390) and NIH (GM59802). J.S.B. acknowledges funding from the Welch Foundation (F-1155). MST acknowledges funding from the NIH (NIAID AI064184).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are::
- CID
- collision-induced dissociation
- ESI
- electrospray ionization
- ETD
- electron-transfer dissociation
- FDR
- false discovery rate
- HCD
- higher energy collision-induced dissociation
- MAPK
- mitogen-activated protein kinase
- NUVPD
- negative-mode ultraviolet photodissociation
- PSM
- peptide spectral match
- ROC
- receiver operating characteristic
- UVPD
- ultraviolet photodissociation.
REFERENCES
- 1. Griffiths W. J., Wang Y. (2009) Mass spectrometry: from proteomics to metabolomics and lipidomics. Chem. Soc. Rev. 38, 1882–1896 [DOI] [PubMed] [Google Scholar]
- 2. Mann M., Hendrickson R. C., Pandey A. (2001) Analysis of proteins and proteomes by mass spectrometry. Annu. Rev. Biochem. 70, 437–473 [DOI] [PubMed] [Google Scholar]
- 3. Mitchell P. (2010) Proteomics retrenches. Nat. Biotechnol. 28, 665–670 [DOI] [PubMed] [Google Scholar]
- 4. Duncan M. W., Aebersold R., Caprioli R. M. (2010) The pros and cons of peptide-centric proteomics. Nat. Biotechnol. 28, 659–664 [DOI] [PubMed] [Google Scholar]
- 5. de Godoy L. M. F., Olsen J. V., Cox J., Nielsen M. L., Hubner N. C., Froehlich F., Walther T. C., Mann M. (2008) Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature 455, 1251–1254 [DOI] [PubMed] [Google Scholar]
- 6. Kiraga J., Mackiewicz P., Mackiewicz D., Kowalczuk M., Biecek P., Polak N., Smolarczyk K., Dudek M. R., Cebrat S. (2007) The relationships between the isoelectric point and: length of proteins, taxonomy and ecology of organisms. BMC Genomics 8, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dalpathado D. S., Desaire H. (2008) Glycopeptide analysis by mass spectrometry. Analyst 133, 731–738 [DOI] [PubMed] [Google Scholar]
- 8. Lee K. A., Means G. D., Patterson S. D. (2006) Phosphoproteomics: challenges and opportunities. Curr. Proteomics 3, 249–257 [Google Scholar]
- 9. Ewing N. P., Cassady C. J. (2001) Dissociation of multiply charged negative ions for hirudin (54–65), fibrinopeptide B, and insulin A (oxidized). J. Am. Soc. Mass Spectrom. 12, 105–116 [DOI] [PubMed] [Google Scholar]
- 10. Zubarev R. A. (2003) Electron-capture dissociation tandem mass spectrometry. Curr. Opin. Biotechnol. 15, 12–16 [DOI] [PubMed] [Google Scholar]
- 11. Zubarev R. A., Zubarev A. R., Savitski M. M. (2008) Electron capture/transfer versus collisionally activated/induced dissociations: solo or duet? J. Am. Soc. Mass Spectrom. 19, 753–761 [DOI] [PubMed] [Google Scholar]
- 12. Syka J. E. P., Coon J. J., Schroeder M. J., Shabanowitz J., Hunt D. F. (2004) Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 101, 9528–9533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Coon J. J. (2009) Collisions or electrons? Protein sequence analysis in the 21st century. Anal. Chem. 81, 3208–3215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kjeldsen F., Silivra O. A., Ivonin I. A., Haselmann K. F., Gorshkov M., Zubarev R. A. (2005) Cα-C backbone fragmentation dominates in electron detachment dissociation of gas-phase polypeptide polyanions. Chem. Eur. J. 11, 1803–1812 [DOI] [PubMed] [Google Scholar]
- 15. Kjeldsen F., Horning O. B., Jensen S. S., Giessing A. M. B., Jensen O. N. (2008) Towards liquid chromatography time-scale peptide sequencing and characterization of post-translational modifications in the negative-ion mode using electron detachment dissociation tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 19, 1156–1162 [DOI] [PubMed] [Google Scholar]
- 16. Coon J. J., Shabanowitz J., Hunt D. F., Syka J. E. P. (2005) Electron transfer dissociation of peptide anions. J. Am. Soc. Mass Spectrom. 16, 880–882 [DOI] [PubMed] [Google Scholar]
- 17. Huzarska M., Ugalde I., Kaplan D. A., Hartmer R., Easterling M. L., Polfer N. C. (2010) Negative electron transfer dissociation of deprotonated phosphopeptide anions: choice of radical cation reagent and competition between electron and proton transfer. Anal. Chem. 82, 2873–2878 [DOI] [PubMed] [Google Scholar]
- 18. Antoine R., Joly L., Tabarin T., Broyer M., Dugourd P., Lemoine J. (2007) Photo-induced formation of radical anion peptides. Electron photodetachment dissociation experiments. Rapid Commun. Mass. Spectrom. 21, 265–268 [DOI] [PubMed] [Google Scholar]
- 19. Larraillet V., Vorobyev A., Brunet C., Lemoine J., Tsybin Y. O., Antoine R., Dugourd P. (2010) Comparative dissociation of peptide polyanions by electron impact and photo-induced electron detachment. J. Am. Soc. Mass Spectrom. 21, 670–680 [DOI] [PubMed] [Google Scholar]
- 20. Madsen J. A., Kaoud T. S., Dalby K. N., Brodbelt J. S. (2011) 193-nm photodissociation of singly and multiply charged peptide anions for acidic proteome characterization. Proteomics 11, 1329–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Eng J. K., McCormack A. L., Yates J. R., III (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 22. Perkins D. N., Pappin D. J. C., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 23. Geer L. Y., Markey S. P., Kowalak J. A., Wagner L., Xu M., Maynard D. M., Yang X., Shi W., Bryant S. H. (2004) Open mass spectrometry search algorithm. J. Proteome Res. 3, 958–964 [DOI] [PubMed] [Google Scholar]
- 24. Craig R., Beavis R. C. (2004) TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20, 1466–1467 [DOI] [PubMed] [Google Scholar]
- 25. Narasimhan C., Tabb D. L., VerBerkmoes N. C., Thompson M. R., Hettich R. L., Uberbacher E. C. (2005) MASPIC: intensity-based tandem mass spectrometry scoring scheme that improves peptide identification at high confidence. Anal. Chem. 77, 7581–7593 [DOI] [PubMed] [Google Scholar]
- 26. Balgley B. M., Laudeman T., Yang L., Song T., Lee C. S. (2007) Comparative evaluation of tandem MS search algorithms using a target-decoy search strategy. Mol. Cell. Proteomics 6, 1599–1608 [DOI] [PubMed] [Google Scholar]
- 27. Xu H., Freitas Michael A. (2007) A mass accuracy sensitive probability based scoring algorithm for database searching of tandem mass spectrometry data. BMC Bioinformatics 8, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xu H., Yang L., Freitas Michael A. (2008) A robust linear regression based algorithm for automated evaluation of peptide identifications from shotgun proteomics by use of reversed-phase liquid chromatography retention time. BMC Bioinformatics 9, 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xu H., Freitas M. A. (2009) MassMatrix: a database search program for rapid characterization of proteins and peptides from tandem mass spectrometry data. Proteomics 9, 1548–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Madsen J. A., Boutz D. R., Brodbelt J. S. (2010) Ultrafast ultraviolet photodissociation at 193 nm and its applicability to proteomic workflows. J. Proteome Res. 9, 4205–4214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gardner M. W., Vasicek L. A., Shabbir S., Anslyn E. V., Brodbelt J. S. (2008) Chromogenic cross-linker for the characterization of protein structure by infrared multiphoton dissociation mass spectrometry. Anal. Chem. 80, 4807–4819 [DOI] [PubMed] [Google Scholar]
- 32. Vasicek L. A., Ledvina A. R., Shaw J., Griep-Raming J., Westphall M. S., Coon J. J., Brodbelt J. S. (2011) Implementing photodissociation in an Orbitrap mass spectrometer. J. Am. Soc. Mass Spectrom. 22, 1105–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Han S.-W., Lee S.-W., Bahar O., Schwessinger B., Robinson M. R., Shaw J. B., Madsen J. A., Brodbelt J. S., Ronald P. C. (2012) Tyrosine sulfation in a Gram-negative bacterium. Nat. Commun. 3, 1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu H., Freitas M. A. (2008) Monte Carlo simulation-based algorithms for analysis of shotgun proteomic data. J. Proteome Res. 7, 2605–2615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sebolt-Leopold J. S., Herrera R. (2004) Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat. Rev. Cancer 4, 937–947 [DOI] [PubMed] [Google Scholar]
- 36. Huttlin E. L., Hegeman A. D., Harms A. C., Sussman M. R. (2007) Prediction of error associated with false-positive rate determination for peptide identification in large-scale proteomics experiments using a combined reverse and forward peptide sequence database strategy. J. Proteome Res. 6, 392–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kelly M. A., Vestling M. M., Fenselau C. C., Smith P. B. (1992) Electrospray analysis of proteins: a comparison of positive-ion and negative-ion mass spectra at high and low pH. Org. Mass Spectrom. 27, 1143–1147 [Google Scholar]
- 38. Mansoori B. A., Volmer D. A., Boyd R. K. (1997) Wrong-way-round electrospray ionization of amino acids. Rapid Commun. Mass Spectrom. 11, 1120–1130 [Google Scholar]
- 39. Goo Y. A., Yi E. C., Baliga N. S., Tao W. A., Pan M., Aebersold R., Goodlett D. R., Hood L., Ng W. V. (2003) Proteomic analysis of an extreme halophilic archaeon, Halobacterium sp. NRC-1. Mol. Cell. Proteomics 2, 506–524 [DOI] [PubMed] [Google Scholar]
- 40. Gan R. R., Yi E. C., Chiu Y., Lee H., Kao Y.-C. P., Wu T. H., Aebersold R., Goodle D. R., Ng W. V. (2006) Proteome analysis of Halobacterium sp. NRC-1 facilitated by the biomodule analysis tool BMSorter. Mol. Cell. Proteomics 5, 987–997 [DOI] [PubMed] [Google Scholar]
- 41. Ng W. V., Kennedy S. P., Mahairas G. G., Berquist B., Pan M., Shukla H. D., Lasky S. R., Baliga N. S., Thorsson V., Sbrogna J., Ng Wailap Victor, Kennedy Sean P., Mahairas Gregory G., Berquist Brian, Pan Min, Shukla Hem Dutt, Lasky Stephen R., Baliga Nitin S., Thorsson Vesteinn, Sbrogna Jennifer, Swartzell Steven, Weir Douglas, Hall John, Dahl Timothy A., Welti Russell, Goo Young Ah, Leithauser Brent, Keller Kim, Cruz Randy, Danson Michael J., Hough David W., Maddocks Deborah G., Jablonski Peter E., Krebs Mark P., Angevine Christine M., Dale Heather, Isenbarger Thomas A., Peck Ronald F., Pohlschroder Mechthild, Spudich John L., Jung Kwang-Hwan, Alam Maqsudul, Freitas Tracey, Hou Shaobin, Daniels Charles J., Dennis Patrick P., Omer Arina D., Ebhardt Holger, Lowe Todd M., Liang Ping, Riley Monica, Hood Leroy, DasSarma Shiladitya. (2000) Genome sequence of Halobacterium species NRC-1. Proc. Natl. Acad. Sci. U.S.A. 97, 12176–12181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Baker P. R., Medzihradszky K. F., Chalkley R. J. (2010) Improving software performance for peptide electron transfer dissociation data analysis by implementation of charge state- and sequence-dependent scoring. Mol. Cell. Proteomics 9, 1795–1803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sridhara V., Bai D. L., Chi A., Shabanowitz J., Hunt D. F., Bryant S. H., Geer L. Y. (2012) Increasing peptide identifications and decreasing search times for ETD spectra by pre-processing and calculation of parent precursor charge. Proteome Sci. 10, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sharma V., Eng J. K., Feldman S., von Haller P. D., MacCoss M. J., Noble W. S. (2010) Precursor charge state prediction for electron transfer dissociation tandem mass spectra. J. Proteome Res. 9, 5438–5444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Carvalho P. C., Cociorva D., Wong C. C. L., Carvalho M. d. G. d. C., Barbosa V. C., Yates J. R. (2009) Charge prediction machine: tool for inferring precursor charge states of electron transfer dissociation tandem mass spectra. Anal. Chem. 81, 1996–2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Marquet P., Saint-Marcoux F., Gamble T. N., Leblanc J. C. Y. (2003) Comparison of a preliminary procedure for the general unknown screening of drugs and toxic compounds using a quadrupole-linear ion-trap mass spectrometer with a liquid chromatography-mass spectrometry reference technique. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 789, 9–18 [DOI] [PubMed] [Google Scholar]
- 47. Molina H., Horn D. M., Tang N., Mathivanan S., Pandey A. (2007) Global proteomic profiling of phosphopeptides using electron transfer dissociation tandem mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 104, 2199–2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kjeldsen F., Giessing A. M. B., Ingrell C. R., Jensen O. N. (2007) Peptide sequencing and characterization of post-translational modifications by enhanced ion-charging and liquid chromatography electron-transfer dissociation tandem mass spectrometry. Anal. Chem. 79, 9243–9252 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.