

Summary
The MAP kinase p38α is essential for neuronal signaling. To better understand the molecular regulation of p38α we used atomistic and molecular techniques to determine the structural basis of p38α regulation by the two neuronal tyrosine phosphatases, PTPSL/PTPBR7 (PTPRR) and STEP (PTPN5). We show that, despite the fact that PTPSL and STEP belong to the same family of regulatory proteins, they interact with p38α differently and their distinct molecular interactions explain their different catalytic activities. While the interaction of PTPSL with p38α is similar to that of the previously described p38α:HePTP (PTPN7) complex, STEP binds and regulates p38α in an unexpected and new manner. Using NMR and small angle X-ray scattering data we generated a model of the p38α:STEP complex and define molecular differences between its resting- and active-states. Together, these results provide fundamentally new insights into molecular regulation of p38α by key regulatory proteins.
INTRODUCTION
The mitogen-activation protein kinases (MAPKs), p38, ERK and JNK, are central to evolutionarily conserved signaling pathways that are present in all eukaryotic cells. Each MAPK cascade is activated in response to a diverse array of extracellular signals and culminates in the dual-phosphorylation of a threonine and a tyrosine residue in the MAPK activation loop (Derijard et al., 1995). Dually phosphorylated MAPKs then phosphorylate both cytosolic and nuclear targets, with the localization and duration of MAPK activity determining the cellular response to the initial extracellular signal. MAPK signal duration is finely controlled by multiple phosphatases, including three kinase-interaction motif-protein tyrosine phosphatases (KIM-PTPs): PTPSL/PTPBR7 (PTPRR), STEP (PTPN5), HePTP (PTPN7, LC-PTP) (Hendriks et al., 1995; Lombroso et al., 1991; Zanke et al., 1992). KIM-PTPs control MAPK signaling by two mechanisms: 1) the KIM-PTP binds to an unphosphorylated MAPK to sequester it in the cytosol (known as the resting-state complex); or 2) the KIM-PTP dephosphorylates and inactivates a phosphorylated MAPK (known as the active-state complex) (Tarrega et al., 2002; Xu et al., 2009).
Increasing evidence suggests a crucial role for MAPK signaling in the central nervous system (CNS) and aberrant phosphorylation within the CNS is associated with a multitude of neurological disorders, including Alzheimer’s and Parkinson’s disease (Kim and Choi, 2010). Correspondingly, an additional layer of MAPK regulation is present within neurons. Two KIM-PTPs, PTPSL and STEP, are expressed within discrete regions of the CNS. PTPSL expression is highest in the cerebellum, while STEP is expressed in the hippocampus, striatum, amygdala and cortex (Chirivi et al., 2004; Lombroso et al., 1993; Van Den Maagdenberg et al., 1999).
PTPSL, STEP and the other KIM-PTP, HePTP, have a 17-residue kinase-interaction motif (KIM or D-motif) that is essential for their interaction with ERK2 and p38 (Blanco-Aparicio et al., 1999; Saxena et al., 1999). The KIM binds to a conserved binding site that is formed by the hydrophobic docking groove and an acidic patch named the common docking (CD) site (together known as the KIM-binding site or D-motif recruitment site) (Tarrega et al., 2002; Zhou et al., 2006). The KIM sequence is well conserved across the KIM-PTP family. All three KIMs contain basic residues, proposed to engage the CD site, upstream of a hydrophobic motif (L-X-L-X-φ, where φ is a methionine in STEP and PTPSL and a valine in HePTP; Figure 1A). Previously, we used nuclear magnetic resonance (NMR) spectroscopy and isothermal titration calorimetry (ITC) to define the interface of HePTPKIM binding to p38α. We determined that the hydrophobic motif occupies three hydrophobic pockets within the p38α hydrophobic docking groove, while its basic residues interact with the acidic patch (Francis et al., 2011a).
Figure 1. The mode of KIM binding is conserved within the KIM-PTP family.

(A) Sequences of STEPKIMKIS, PTPSLKIMKIS, and HePTPKIMKIS and conservation as determined using ClustalW (Larkin et al., 2007). KIM residues are colored in orange and blue. KIS residues are colored in gray. (B) Constructs used in this study. (C) 2D [1H,15N] TROSY spectrum of [2H,15N]-p38α (black) overlaid with the 2D [1H,15N] TROSY spectrum of [2H,15N]-p38α:PTPSLKIM (red; 1:8 molar ratio p38α:PTPSLKIM) (D) 2D [1H,15N] TROSY spectrum of [2H,15N]-p38α (black) overlaid with the 2D [1H,15N] TROSY spectrum of [2H,15N]-p38α:STEPKIM (red; 1:8 molar ratio p38α:STEPKIM) (E) Histograms which show the combined 1H/15N CSPs vs. p38α residue for the final titration points of each KIM-PTPKIM peptide with [2H,15N]-p38α. See also Figure S1.
C-terminal to the KIM is a region known as the kinase-specificity sequence (KIS) which is followed by a globular PTP catalytic domain (CAT) (Eswaran et al., 2006; Munoz et al., 2003; Mustelin et al., 2005). While the CAT is essential for dephosphorylation of the MAPK by the KIM-PTP, the importance of the KIS remains ambiguous, especially as the KIS sequence is divergent among the KIM-PTP family members. We previously showed that the HePTPKIS is highly flexible and contributes significantly to the interaction between HePTP and p38α, while HePTPCAT does not interact with unphosphorylated p38α. This was corroborated by small-angle x-ray scattering (SAXS) experiments that showed that the p38α:HePTP resting-state complex is elongated in solution (Francis et al., 2011a). Whether the extensive KIS interaction, limited CAT interaction and extended resting-state conformation observed in p38α:HePTP are common features of all p38α:KIM-PTP resting-state complexes is currently unknown.
Similar studies with a different MAPK, ERK2, revealed that the ERK2:HePTP resting-state complex shares several of these features. As with p38α, HePTPCAT does not interact with ERK2 in the resting-state, and the ERK2:HePTP resting-state complex is extended in solution (Piserchio et al., 2012). However, phosphorylation of ERK2 leads to a domain rearrangement in which HePTPCAT rotates by nearly 180°, the ERK2:HePTP complex compacts and the HePTP catalytic site residue moves by ~65 Å to allow for dephosphorylation of ERK2 (Francis et al., 2011b). The flexibility of the HePTPKIS and the absence of an interaction between ERK2 and HePTPCAT are critical to facilitate this large rearrangement.
Here, we combined NMR, SAXS, ITC experiments and HADDOCK to delineate the interaction of p38α with STEP and PTPSL. In combination with experiments conducted with HePTP, these studies serve as the first atomic-level characterization of MAPK regulation by an entire family of regulators – the KIM-PTP family. Our results reveal key differences between the interactions of p38α with each KIM-PTP. We show that the interaction of p38α with PTPSL is closely related to the interaction of HePTP, while the interaction of p38α with STEP is quite different and includes an unexpected contribution from STEPCAT.
RESULTS
KIM binding to p38α is conserved within the KIM-PTP family
We used NMR spectroscopy to define the p38α binding interface of the STEPKIM and PTPSLKIM peptides (constructs used in this study are illustrated in Figure 1B). Each peptide was individually titrated into [2H,15N]-p38α until binding was saturated (1:1, 1:3, 1:5, 1:8 molar ratio of p38α:peptide) and two-dimensional (2D) [1H,15N] TROSY spectra were recorded (Figure 1C, D; Supplemental Figure S1A). The fully saturated 2D [1H,15N] TROSY spectra of p38α:PTPSLKIM and p38α:STEPKIM (100 μM [2H,15N]-p38α:800 μM peptide) are highly similar (Figure 1E; Supplemental Figure S1B). All peak shifts are in fast exchange and can be readily followed with the exception of 3 peaks that map to the MAPK-specific insert (L262-Q264) and are broadened beyond detection independent of ligand (Supplemental Figure S1A). As expected, both peptides caused significant chemical shift perturbations (CSPs) for residues located in the KIM-binding site, including helix αD, helix αE, the αD–αE loop, the β7–β8 turn, loop 4 and the CD site (Figure 2). Thus, like HePTPPKIM, PTPSLKIM and STEPKIM engage all three hydrophobic pockets and interact with the acidic patch demonstrating that the mode of KIM binding to p38α is conserved within the KIM-PTP family.
Figure 2. PTPSL and STEP interaction with p38α.

Histograms showing the combined 1H/15N CSP vs. p38α residue for the following experiments: p38α with PTPSLKIM (1:8 molar ratio), PTPSLKIMKIS (1:3 molar ratio), PTPSL (1:1 ratio; SEC purified), STEPKIM (1:8 molar ratio), STEPKIMKIS (1:8 molar ratio), STEP (1:1 ratio; SEC purified), and PSChimera (1:1 ratio; SEC purified). Residues that form the p38α hydrophobic docking groove and the CD site are highlighted by blue and orange boxes, respectively. Residues with peak line-widths broadened beyond detection upon titration are colored in green. See also Figure S2.
However, there are small differences between the CSPs induced by STEPKIM and PTPSLKIM. L164 in β8 exhibits the largest CSP difference between the 2D [1H,15N] TROSY spectra of p38α:PTPSLKIM and p38α:STEPKIM (Figure 1E; Supplemental Figure S1B). The N-terminal residues of the KIM (Ile in PTPSLKIM; Met in STEPKIM) are the only residues not identical between the two peptides; therefore, any differences between the two spectra are a result of the differential interaction of this residue with p38α. Based on the structure of ERK2:HePTPKIM, this residue is immediately adjacent to the β7–β8 turn further supporting our NMR results (Zhou et al., 2006).
Although the KIM binding mode is conserved among the KIM-PTPs, the HePTPKIM sequence is only ~75% conserved when compared with PTPSLKIM and STEPKIM (Figure 1A). This leads to changes in the chemical environment for residues within the p38α KIM-binding site, which are readily observed in the 2D [1H,15N] TROSY spectra (Supplemental Figure S1C). PTPSLKIM and STEPKIM perturb 9 more peaks than HePTPKIM (Francis et al., 2011a). Compared to HePTPKIM, PTPSLKIM and STEPKIM interact differentially with several residues within the hydrophobic binding groove and loop 4, including N82, A111, L113, C119, L122, D124, D125, V127, L130, S154, E160, C162 and L164 (Figure 1E). These NMR CSP experiments correspond well with ITC measurements that showed PTPSLKIM and STEPKIM bind p38α ~2-times tighter than HePTPKIM (Table 1; Supplemental Figure S2).
Table 1.
Thermodynamic and dissociation constants for p38α:KIM-PTP domains derived from ITC experiments at 25 °C. See also Figure S2.
| Interaction | Kd (nM) | ΔH (kcal·mol−1) | TΔS (kcal·mol−1) | ΔG (kcal·mol−1) |
|---|---|---|---|---|
| p38α : STEPKIM | 3567 ± 208 | −37.7 ± 2.5 | −30.2 ± 2.5 | −7.4 ± 0.1 |
| p38α : STEPKIMKIS | 1340 ± 135 | −34.7 ± 1.4 | −26.6 ± 1.5 | −8.0 ± 0.1 |
| p38α : STEP | 577 ± 71 | −28.8 ± 2.0 | −20.3 ± 2.0 | −8.5 ± 0.1 |
| p38α : STEPF274A/F281A | 1074 ± 105 | −24.2± 0.4 | −16.0 ± 0.4 | −8.2 ± 0.1 |
| p38α : STEPF281A/I307A | 1062 ± 36 | −34.3± 1.1 | −26.2 ± 1.0 | −8.2 ± 0.1 |
| p38α : STEPM285A/I307A | 592 ± 106 | −27.6± 0.5 | −19 ± 0.3 | −8.5 ± 0.1 |
|
| ||||
| p38α : PTPSLKIM | 2840 ± 135 | −33.0 ± 1.7 | −25.4 ± 1.7 | −7.6 ± 0.1 |
| p38α : PTPSLKIMKIS | 1527 ± 23 | −38.8 ± 6.1 | −30.8 ± 6.1 | −7.9 ± 0.1 |
| p38α : PTPSL | 382 ± 65 | −30.2 ± 1.5 | −21.4 ± 1.4 | −8.8 ± 0.1 |
|
| ||||
| p38α : PSChimera | 309 ± 4 | −27.2 ± 0.7 | −18.3 ± 0.7 | −8.9 ± 0.0 |
An NMR view of the interaction between PTPSL and p38α
We expanded the NMR experiments to define the binding interface of PTPSL with p38α. In this experiment, [2H,15N]-p38α was incubated with unlabeled PTPSL, the 78 kDa p38α:PTPSL complex was purified by size exclusion chromatography (SEC) and a 2D [1H,15N] TROSY spectrum was immediately recorded (Supplemental Figure S3C).
Direct comparison of the 2D [1H,15N] TROSY spectra of free and PTPSL-bound p38α reveals CSPs of 41 peaks, 36 in fast exchange and 5 peaks with line-widths broadened beyond detection (N115, Q120, F129, L130, S154; Figure 2). Like PTPSLKIM, PTPSL perturbed residues clustered within the hydrophobic binding groove and the acidic patch of p38α (Figure 2). Two peaks, N115 (line-widths broadened beyond detection) and V127, are perturbed by full-length PTPSL but not by PTPSLKIM with additional peaks experiencing larger CSPs (G110, D112, I116, K118, Q120, Q310) (Figure 2). Correspondingly, full-length PTPSL binds ~7-times tighter to p38α than PTPSLKIM (Table 1; Supplemental Figure S2).
The majority of the additional CSPs map to a region of p38α that interacts with HePTPKIS (Francis et al., 2011a). While the PTPSLKIS and HePTPKIS sequences are only ~50% conserved, this region of p38α is thus also likely interacting with PTPSLKIS. To determine if a peptide composed of the PTPSLKIM and PTPSLKIS sequences (i.e. PTPSLKIMKIS) can recapitulate the binding of full-length PTPSL to p38α, we titrated the PTPSLKIMKIS into [2H,15N]-p38α and a series of 2D [1H,15N] TROSY spectra were recorded (Supplemental Figure S3B). Titration of a 3-fold molar excess of the PTPSLKIMKIS peptide (100 μM [2H,15N]-p38α:300 μM PTPSLKIMKIS) resulted in a nearly identical 2D [1H,15N] TROSY spectrum as that of p38α: PTPSL (Figure 2, Supplemental Figure S3H). Differences include residues N115 and S154, which are broadened beyond detection by full-length PTPSL but are either not perturbed (N115) or undergo a large CSP (S154) with PTPSLKIMKIS, respectively (Supplemental Figure S3H). Furthermore, a direct comparison of the 2D [1H,15N] TROSY spectra of p38α: PTPSLKIM with p38α: PTPSLKIMKIS shows larger CSPs for four residues (M109, K118, E160, L164), and three peaks (Q120, F129, L130) are broadened beyond detection by PTPSLKIMKIS but not by PTPSLKIM (Supplemental Figure S3H). Consistent with these results, the PTPSLKIMKIS binds p38α ~2-times tighter than PTPSLKIM (Table 1; Supplemental Figure S2).
The p38α:PTPSL resting-state complex is extended in solution
The interactions of PTPSLKIM, PTPSLKIMKIS and PTPSL with p38α are highly similar. PTPSL perturbed few residues outside of the KIM-binding site confirming that the KIM is the essential anchor for the interaction of PTPSL with p38α. Indeed, we used NMR spectroscopy to probe the interaction of p38α with PTPSLCAT, a construct lacking residues 332–360, which includes the KIM; no interactions between p38α and isolated PTPSLCAT were detected (Supplemental Figure S4A, C). This is consistent with previous results from NMR experiments which showed that the isolated HePTPCAT did not interact with either p38α or ERK2 (Francis et al., 2011a; Piserchio et al., 2012).
Consequently, we used SAXS to determine the size and shape of the p38α:PTPSL resting-state complex. The p38α:PTPSL complex was purified by SEC, and SAXS data was recorded at Beamline X9A at the National Synchrotron Light Source (Brookhaven National Laboratories). Using the Guinier approximation of five independent SAXS samples, a radius of gyration (Rg) of 33.1 ± 0.5 Å was calculated for the p38α:PTPSL complex (Table 2; Figure 3A). A maximum particle dimension (Dmax) of 105 Å was determined by analysis of the P(r) function (Table 2; Figure 3D, E). These values for Rg and Dmax are highly similar to those determined for the p38α:HePTP and ERK2:HePTP resting-state complexes and demonstrate that, under resting conditions, p38α and PTPSL adopt an extended conformation, with a rather limited interaction surface, consistent with the NMR CSP experiments.
Table 2.
SAXS analysis of p38α:PTPRR, p38α:STEP and pp38α:STEP complexes.
| p38:PTPRR | p38:STEP (Resting) | pp38:STEP (Active) | |
|---|---|---|---|
| Guinier approximation | |||
|
| |||
| Rg (Å) | 33.1 ± 0.5 | 29.9 ± 0.3 | 28.5 ± 0.3 |
|
| |||
| P(r) function calculation | |||
|
| |||
| Q-range (Å−1) | 0.013–0.303 | 0.013–0.304 | 0.014–0.297 |
| Rg (Å) | 34.1 | 29.8 | 28.2 |
| Dmax (Å) | 105 | 95 | 80 |
|
| |||
| Structure modeling | |||
|
| |||
| χ | 1.4 ± 0.1 | 1.9 ± 0.2 | 1.0 ± 0.1 |
| NSD | 1.30 ± 0.04 | 1.09 ± 0.03 | 1.00 ± 0.03 |
Figure 3. The p38α:PTPSL and p38α:STEP resting-state complex.

(A) SAXS data (I(q) vs q) of the p38α:PTPSL resting-state complex shown (black squares) with error bars (grey lines). Error bars show the experimental error based on circular averaging of the 2D solution scattering data; theoretical scattering curve from ab initio molecular envelope (red); inset, Guinier plots for samples at 2.3 mg/ml and 4.0 mg/ml. (B) SAXS data (I(q) vs q) of the p38α:STEP resting-state complex shown (black squares) with error bars (grey lines). Theoretical scattering curve from the ab initio molecular envelope (red); inset, Guinier plots for samples at 2.0 mg/ml and 4.0 mg/ml. (C) Kratky plot of the p38α:PTPSL resting-state SAXS data (green) and p38α:STEP resting-state SAXS data (blue). (D) P(r) function of the p38α:PTPSL resting-state complex (green) and p38α:STEP resting-state complex (blue). (E) p38α:PTPSL ab initio molecular envelope in two views rotated by 90° with the dimensions of the envelope. (F) The p38α:STEP ab initio molecular envelope in two views rotated by 90° with the dimensions of the envelope.
The p38α:STEP resting-state complex is compact
Equivalent SAXS measurement were performed for the p38α:STEP resting-state complex. The Guinier approximation of four independent SAXS samples was used to calculate an Rg of 29.9 ± 0.3 Å, showing that the Rg of the p38α:STEP resting-state complex is ~3.0 A smaller than that of the p38α: PTPSL and p38α:HePTP resting-state complexes (Table 2; Figure 3B). To analyze this difference in more detail, we determined the molecular envelope of the p38α:STEP resting-state complex (Table 2; Figure 3D, F). A Dmax of 95 Å was determined by analysis of the P(r) function, which is ~10–15 Å shorter than the p38α:PTPSL and p38α:HePTP complexes (Table 2; Figure 3D). Thus, the p38α:STEP resting-state complex is more compact than both the p38α:KIM-PTP and the ERK2:HePTP resting-state complexes. Therefore, unlike PTPSL and HePTP, which have limited interaction surfaces with p38α, this result suggested that STEP may interact more extensively with p38α.
STEP has interactions with p38α outside of the KIM binding pocket
To test this hypothesis, we performed NMR CSP experiments with full-length STEP and [2H,15N]-p38α. Here, [2H,15N]-p38α was incubated with unlabeled STEP, the 78 kDa p38α:STEP complex purified by SEC, and a 2D [1H,15N] TROSY spectrum recorded (Supplemental Figure S3F). Direct comparison of the 2D [1H,15N] TROSY spectra of free and STEP-bound p38α reveals CSPs of 58 peaks, 40 in fast exchange and 18 with peak line-widths that are broadened beyond detection (Figure 2). As expected, like STEPKIM, STEP perturbs residues in the p38α KIM-binding site (Figure 2). Surprisingly, STEP perturbed significantly more residues in p38α than STEPKIM and caused several additional peaks to broaden beyond detection, decreasing the total number of peaks in the 2D [1H,15N] TROSY spectrum of STEP-bound p38α (Figure 2). Direct comparison of the 2D [1H,15N] TROSY spectra of p38α:STEPKIM and p38α:STEP reveals three additional residues (V30, M109, L164) with CSPs and 18 additional peaks (D125, V127, Y132, I141, S154, D177, M179, V183, A190, Q202, R220, L222, L247, K248, S254, R256, H312, E328) that are broadened beyond detection by full-length STEP (Supplemental Figure S3I). Correspondingly, full-length STEP binds ~6-times tighter to p38α than STEPKIM (Table 1; Supplemental Figure S2). Mapping the additional perturbations reveals a previously undetected p38α interaction surface (herein referred to as the PTP-site), which connects the p38α activation loop to the MAPK-specific insert.
To corroborate our findings, we used a divide and conquer approach. To determine if the STEPKIS accounts for this additional interaction, a peptide composed of the STEPKIMKIS sequence was titrated into [2H,15N]-p38α, and a series of 2D [1H,15N] TROSY spectra were recorded (Supplemental Figure S3E). Interestingly, the p38α:STEPKIMKIS spectrum (100 μM [2H,15N]-p38α:800 μM STEPKIMKIS) is similar to that of p38α:STEPKIM at the same molar ratio (Figure 2; Supplemental Figure S3I). Direct comparison of the 2D [1H,15N] TROSY spectra of p38α:STEPKIM and p38α:STEPKIMKIS reveals differences in seven residues: V30, M109 and E160 show CSPs, while G85, D125, V127 and S154 are broadened beyond detection (Supplemental Figure S3I). Using ITC we determined that STEPKIMKIS binds ~2.5-times tighter than STEPKIM but 2-times weaker than full-length STEP (Table 1; Supplemental Figure S2). Importantly, the STEPKIMKIS did not affect residues within the new PTP-site (Figure 2). This suggests that the additional changes in the NMR spectrum must be the result of an interaction between the STEP catalytic domain (STEPCAT) and p38α.
p38α:PSChimera complex
To confirm that STEPCAT interacts directly with p38α, we generated a chimeric protein in which PTPSLKIMKIS was fused with STEPCAT (herein referred to as the PSChimera; Figure 1B). The PSChimera was expressed, purified and incubated with [2H,15N]-p38α. The p38α:PSChimera complex was then purified by SEC and a 2D [1H,15N] TROSY spectrum of the complex was recorded (Supplemental Figure S3G).
Within and around the p38α KIM binding site, the PSChimera affected the p38α 2D [1H,15N] TROSY spectrum in a PTPSL-like manner (Figure 2). Notably, additional changes in the p38α 2D [1H,15N] TROSY spectrum, which correspond to peaks that define the newly identified PTP-site, were also identified (Figure 2). Thus, the PSChimera, which has a PTPSLKIMKIS and a STEPCAT domain, interacts in a PTPSL-like manner with its KIMKIS and a STEP-like manner at the PTP-site. Minor differences between the interaction of the PSChimera and STEP with p38α at the PTP-site include Q202 and K248 that are not broadened beyond detection with PSChimera (K248 does experience a small CSP). Similar to STEP, 17 peaks in the PTP-site broaden beyond detection with PSChimera, demonstrating that the STEPCAT interaction with p38α is independent of the KIMKIS sequence. Furthermore, NMR interaction studies showed that titration of unlabeled, isolated STEPCAT (lacking residues 214–243 including the KIM) with [2H,15N]-p38α results in non-specific interactions throughout the p38α surface, – further reinforcing the critical importance of KIM anchoring for p38α regulation (Supplemental Figure S4B, C). The chimeric protein has a slightly higher binding affinity than PTPSL or STEP for p38α (Table 1). This binding affinity is the product of the PTPSLKIM, which binds the tightest of the three KIM-PTPKIM peptides, coupled with the additional binding strength afforded by the STEPCAT interaction.
Defining the p38α binding site on STEP
To define the p38α binding interface on STEP, we performed reverse NMR experiments with [2H,15N]-STEP and unlabeled p38α. We previously published the sequence-specific backbone assignments of STEPCAT (Francis et al., 2013). The 2D [1H,15N] TROSY spectrum of STEP contains 30 additional peaks when compared to that of STEPCAT, in agreement with the 31 additional N-terminal residues in STEP (Supplemental Figure S4D). All new peaks cluster around ~8.0 ppm, a chemical shift range typical for unstructured residues, suggesting that the KIM and KIS regions of STEP are unstructured in the unbound state, identical to what we have seen for HePTP (Supplemental Figure S4D) (Francis et al., 2011a). Lastly, the vast majority of peaks in the STEP 2D [1H,15N] TROSY spectrum overlap perfectly with peaks in the STEPCAT 2D [1H,15N] TROSY spectrum, showing that the flexible N-terminal residues in STEP do not interact with STEPCAT. This allowed for the transfer of the STEPCAT sequence-specific backbone assignment to STEP.
Peaks corresponding to residues 244–253, the majority of helix α0, have very different chemical shifts between STEP and STEPCAT, indicating that the presence of the flexible N-terminus (residues 214–243) alters the chemical environment of residues 244–253. This is consistent to what was observed for HePTP, in which helix α0 (residues 44–56) was also perturbed by the presence of the N-terminus, residues 15–43 (Francis et al., 2011a). Previously, it was hypothesized that helix α0 must change conformation in order for the regulatory Ser/Thr residue at the N-terminus of the helix to be phosphorylated (Mustelin et al., 2005; Szedlacsek et al., 2001). These NMR studies show that this hypothesis is correct and the pliability of helix α0 is conserved within the KIM-PTP family.
To test the effects of p38α binding on STEP, the complex between [2H,15N]-STEP and unlabeled p38α was purified by SEC, and a 2D [1H,15N] TROSY spectrum of the 78 kDa complex was immediately collected (Supplemental Figure S4E). Direct comparison of the 2D [1H,15N] TROSY spectra of free and p38α-bound STEP reveals changes in 54 peaks, 22 of which are in fast exchange and 32 with peak line-widths broadened beyond detection (Figure 4A; Supplemental Figure S4E). Included in this total are 21 of the new peaks that clustered around 8.0 ppm in the free STEP spectrum that are broadened beyond detection or overlap with other peaks in the in the 2D [1H,15N] TROSY spectrum of the p38α:STEP complex. This indicates, as expected, a strong involvement of the STEPKIM in p38α binding. Additional new peaks unperturbed by p38α binding originate from STEPKIS residues that do not contribute to p38α binding. 33 perturbed peaks are in STEPCAT, including residues in helix α-1′ (A263, E268, K269), helix α-2′ (F274, L276, E277, A278, E279, F280, F281, E282), the β4–β7 loop (N376, I377, E378, E379, M380), helix α4 (R478, T479, F482) and helix α-6 (T517, C518, Q520, H525) (Figure 4A). Other STEPCAT residues that are affected include: L254, A257, M285, N286, D294, T306, I307, G350, V353 and the catalytically important Q516 (Figure 4A).
Figure 4. STEPCAT binds to the p38α activation loop and MAPK-insert primarily through helix α-2′.

(A) Histograms showing the combined 1H/15N CSP vs. STEP residue for the interaction of p38α with [2H,15N]-STEP. (B) Alignment of the 4 lowest energy HADDOCK structures of the p38α:STEPCAT complex. p38α is colored blue and STEPCAT is colored in shades of orange. (C) Superposition of the lowest energy HADDOCK p38α:STEPCAT complex structure with the experimental SAXS molecular envelope of the p38α:STEP resting-state complex; p38α (blue), KIM (green) and STEPCAT (orange). See also Figure S4.
3D model of the p38α:STEP complex
We used the NMR data, which describe the interaction interface of STEP on p38α and vice versa, together with surface accessibility data, to restrain the docking of STEP and p38α using HADDOCK (the crystal structures of apo-p38α (1P38) and STEPCAT (2BIJ) (de Vries et al., 2007; Dominguez et al., 2003; Eswaran et al., 2006; Wang et al., 1997) were used as starting structures). While the STEPCAT structure does not include STEPKIMKIS residues (214–257), we have established – using the PSChimera – that the STEPCAT interaction occurred independent of a specific KIMKIS sequence. Consequently, only p38α residues affected by STEPCAT in both STEP and the PSChimera were used as input for the HADDOCK calculations. The lowest energy cluster produced by HADDOCK contained 76 structures with an RMSD of 0.8 Å. The four lowest energy structures from this cluster are shown in Figure 4B. In all HADDOCK models, STEP helix α-2′ binds to p38α between the activation loop and the MAPK-specific insert. The interface between the two proteins buries 2125 ± 116 Å2 of surface area. To experimentally test the validity of the model, we compared the calculated (determined using Hydropro (Ortega et al., 2011)) Rg of the lowest energy p38α:STEP HADDOCK model (Rg = 29.2 Å) with the experimentally determined Rg (Rg = 29.9 Å). The excellent consistency of the Rg’s is further supported by the excellent fit of the lowest energy p38α:STEP HADDOCK model into the molecular envelope determined for the p38α:STEP resting-state complex (NSD = 1.7; Figure 4C). Additionally, modeling the KIM peptide into the KIM-binding site of p38α (based on the ERK2:HePTPKIM crystal structure (Zhou et al., 2006)) results, as expected, a further improved model of the p38α:STEP resting-state complex (Rg = 30.1 Å; Dmax = 92 Å). This model, which is comprised of every component of the complex but the STEPKIS, superimposes even better with the p38α:STEP molecular envelope (NSD = 1.5; Figure 4C) (Kozin and Svergun, 2001).
To further verify the model of the p38α:STEP resting-state complex, we generated a number of STEP mutants that are predicted to lead to changes in binding affinity, as the mutated residues are directly involved in the p38α:STEP interaction. Indeed, mutations of residues in helix α-2′, and to a lesser extent residues C-terminal to helix α-2′, showed a reduction of the binding affinity, clearly showing that helix α-2′ is important for the interaction of STEP with p38α and further confirming the HADDOCK model (Table 1).
The p38α:STEP active-state complex
We have shown that STEPCAT, when tethered via the KIM, interacts with p38α and perturbs residues in the p38α activation loop. However, this does not imply that STEP is poised and ready to efficiently dephosphorylate p38α. In fact in the HADDOCK p38α:STEP resting-state complex model, the STEP active site cysteine is ~35 Å away from the p38α phosphorylation loop tyrosine. Therefore, a domain arrangement must occur between the resting- and active-states of the p38α:STEP complex to properly position STEPCAT for dephosphorylation of p38α.
To verify that a domain rearrangement occurs between the active- and resting-states of the p38α:STEP complex, we used SAXS to determine the size and shape of the p38α:STEP active-state complex (Akella et al., 2010; Nielsen and Schwalbe, 2011). p38α was dually phosphorylated in vitro using a constitutively active mutant of MKK6. Phosphorylated p38α was incubated with a substrate trapping mutant of STEP (C472S/T306D), the complex purified by SEC, and SAXS data recorded (Figure 5A). Using the Guinier approximation of six independent SAXS samples, an Rg of 28.5 ± 0.3 Å was calculated for the p38α:STEP active-state complex (Table 2; Figure 5A). This Rg is 1.5 Å smaller than the Rg of the resting-state complex. More strikingly, the Dmax of the active-state complex, 80 Å, is 15 Å shorter than the resting-state complex (Table 2; Figure 5C, D). These data show that the p38α:STEP active-state complex is more compact than the resting-state complex and support that a domain rearrangement occurs between the resting- and active-states of the p38α:STEP complex. Thus, although STEPCAT interacts with p38α in the resting-state, it is not properly positioned for dephosphorylation of p38α.
Figure 5. The p38α:STEP active-state complex is more compact than the resting-state complex.
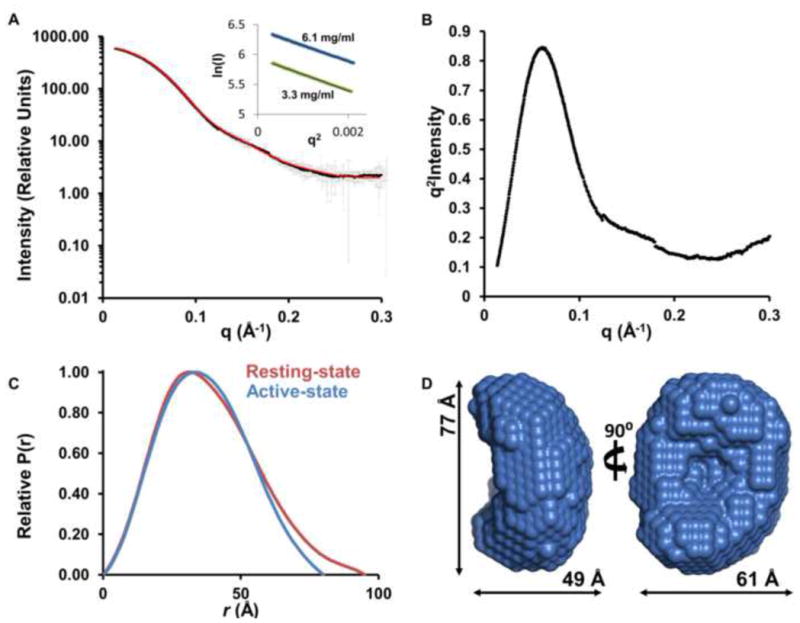
(A) Experimental SAXS data (I(q) vs q) of the p38α:STEP active-state complex shown as black squares with error bars as grey lines; theoretical scattering curve from ab initio molecular envelope (red); inset, Guinier plots for samples at 3.3 mg/ml and 6.1 mg/ml. (B) Kratky plot. (C) P(r) function of the p38α:STEP active-state complex (blue) overlaid with resting-state complex (red). (D) Averaged ab initio molecular envelope; 2 views rotated by 90° with the dimensions of the envelope.
DISCUSSION
Our current understanding of MAPK regulation by their regulatory proteins at the molecular level is extremely limited, as these essential signaling complexes have proven exceedingly difficult to investigate using traditional structural approaches. Years of efforts to crystallize MAPK:KIM-PTP complexes have not been successful, and their large molecular weights, ~80 kDa, make NMR analysis onerous. To circumvent these problems, we examined the binding of each domain of STEP and PTPSL to p38α using NMR CSP and ITC experiments and then determined the overall molecular shape of the complexes using SAXS. Using this approach, we show that, despite having similar KIM-peptide interactions and overall binding affinities with p38α, the p38α:PTPSL and p38α:STEP resting-state complexes are surprisingly different due to an additional interaction of the STEP catalytic domain with p38α. These studies, in combination with complementary studies previously performed between p38α and HePTP, provide the first detailed investigation of MAPK regulation by an entire family of regulators (Francis et al., 2011a).
Using NMR experiments, we show that the mode of KIM binding to p38α is conserved among the KIM-PTP family. Uniformly, PTPSLKIM, STEPKIM and HePTPKIM engaged residues within the three hydrophobic pockets and perturbed residues at the CD site. In contrast, crystal structures of p38α bound to KIM peptides of the activating kinases, MKK3b and MKK6, reveal a different mode of binding in which only hydrophobic pockets φA and φB are engaged and an extensive electrostatic interaction with the CD site is absent (Chang et al., 2002; Garai et al., 2012). This demonstrates a critical point in MAPK selectivity: the mechanism of KIM binding to p38α is different for each class of regulators (i.e. MAPKKs versus KIM-PTPs).
Based on NMR and SAXS data, the interaction of resting-state p38α with PTPSL is similar to that described previously between p38α and HePTP. However, the p38α:PTPSL and p38α:HePTP interactions are not identical. One key feature of the p38α:HePTP interaction, the extensive interaction between p38α and the HePTPKIS, is not observed between p38α and the PTPSLKIS (Francis et al., 2011a). Despite the limited interaction surface between p38α and PTPSL, PTPSL binds p38α nearly 6-times tighter than HePTP. Recently published in vitro phosphatase assays showed that while HePTP and PTPSL can both readily dephosphorylate p38α, PTPSL is the most efficient p38α phosphatase of all tested (Zhang et al., 2011). This is likely the product of: 1) the highest affinity interaction with p38α among the KIM-PTPs and 2) the limited interaction outside of the KIM, imposing minimal constraints on the flexibility and movement of the PTPSLCAT.
In contrast, the p38α:STEP resting-state complex is more compact than the other two p38α:KIM-PTP resting-state complexes due to an additional previously unidentified interaction between STEPCAT and p38α. In the resting-state complex, STEPCAT occludes a pocket on p38α formed between the MAPK-insert and the C-lobe that has been shown to bind allosteric signaling molecules that are known activators of the p38α cascade, such as fatty acids (Diskin et al., 2008; Nony et al., 2005; Perry et al., 2009). Binding of certain lipid-based molecules to this pocket have been shown to promote auto-phosphorylation and activation of p38α in vitro (Gills et al., 2007; Tzarum et al., 2012). STEP regulates p38α at the site of extra-synaptic glutamate receptors, where over-activation of the kinase leads to the production of pro-apoptotic signals culminating in neuronal cell death (Poddar et al., 2010). Therefore, STEP may have adapted several means to suppress p38α activity in the resting-state. First, by binding strongly to the KIM-binding site, STEP prevents upstream activating kinases from promiscuously binding and activating p38α. Second, by blocking access to the MAPK-insert pocket, through the STEPCAT interaction, STEP can prevent the binding of allosteric signaling molecules that induce auto-activation of p38α.
The orientation of STEPCAT in the p38α:STEP resting-state complex is not conducive to dephosphorylation of the tyrosine residue in the p38α phosphorylation loop and significant rotation of STEPCAT is necessary to properly position the active site for catalysis. This domain rearrangement is evident when comparing SAXS data of the resting- and active-states of the p38α:STEP complex. A study conducted by Zhang et al. showed that STEP is significantly less efficient than PTPSL and HePTP at dephosphorylating p38α. The kcat for the STEP-catalyzed dephosphorylation of p38α is ~20-fold lower than those for PTPSL or HePTP, showing that it is likely easier to move the flexibly-tethered HePTPCAT and PTPSLCAT than moving the “trapped” (i.e. p38α bound) STEPCAT (Balasu et al., 2009; Zhang et al., 2011).
Taken together, these results provide fundamental new insights into the molecular basis of p38α regulation by the neuronal KIM-PTPs. We have identified a novel means by which STEP prevents promiscuous activation of p38α by blocking access to the MAPK-insert pocket. As p38α has been implicated in the pathogenesis of numerous neurological disorders, these findings could facilitate the development of new drug therapies to treat these disorders.
EXPERIMENTAL PROCEDURES
Peptide and protein preparation
The PTPSLKIM peptide (IGLQERRGSNVSLTLDM), STEPKIM (MGLQERRGSNVSLTLDM) and STEPKIMKIS (MGLQERRGSNVSLTLDMCTPGCNEEGFGYLVSPREESAHEYLLS) were synthesized, HPLC purified and verified by mass spectrometry (MS) (>98% purity; Biosynthesis, Inc.).
Expression and purification of unphosphorylated p38α was carried out as previously described (Francis et al., 2011a). PTPSL (residues 332–655), PTPSLCAT (residues 361–655), STEP (residues 214–539), STEPCAT (residues 244–539), and PSChimera (PTPSL residues 332–373 with STEP residues 257–539; gene synthesized by DNA 2.0) were expressed and purified using the protocol previously described in detail for STEPCAT (Francis et al., 2013). STEP (residues 214–539) mutants F274A/F281A, F280A/I307A and M285A/I307A were produced by quick-change mutagenesis.
The PTPSLKIMKIS (residues 332–373) was subcloned into a derivative of the pET28 vector that includes an N-terminal His6-tag and a TEV protease recognition sequence (Peti and Page, 2007). The expression vector was transformed into BL21-(DE3) RIL E. coli cells (Invitrogen). Cultures were grown in Luria broth at 37°C with shaking at 250 rpm until reaching an optical density (OD600) between 0.6–0.8, at which point expression was induced by the addition of 1.0 mM isopropylthio-β-D-galactoside (IPTG, final concentration). The cultures were grown for an additional 3 hours at 37 °C with shaking at 250 rpm. The cells were harvested by centrifugation (6000 xg, 12 min, 4°C) and stored at −80°C until purification. All buffers and glassware used for purification were autoclaved prior to purification. For purification, cell pellets were resuspended in lysis buffer (50 mM Tris pH 8.0, 500 mM NaCl, 5 mM imidazole, 0.1% Triton X-1000, and cOmplete EDTA-free protease inhibitor cocktail tablets; Roche) and lysed by high pressure homogenization (Avestin C3 Emulsiflex). The bacterial lysate was cleared by centrifugation (35000 xg, 40 min, 4°C). After filtration, the supernatant was loaded onto a 5 mL HisTrap HP column (GE Healthcare) equilibrated with Buffer A (50 mM Tris pH 8.0, 500 mM NaCl, 5 mM imidazole). Protein was eluted from the column using a 5–500 mM imidazole gradient over 20 column volumes. Purified fractions were pooled, and the his6-tag was removed by incubation with TEV protease during overnight dialysis in 50 mM Tris pH 8.0, 500 mM NaCl at 4°C. The TEV protease, the His6-tag and uncleaved protein were removed using a second IMAC step. Final purification was achieved using size exclusion chromatography (SEC) equilibrated in 50 mM Tris pH 6.8, 500 mM NaCl. The molecular weight of the purified peptide was verified by electrospray-ionization MS.
All unlabeled, purified proteins were stored at −80°C until use at which point they were thawed and equilibrated in the appropriate buffer using SEC. All labeled proteins for NMR analysis were purified within 24 hours of data acquisition and stored on ice. PTPSLKIM, STEPKIM and STEPKIMKIS peptides were solubilized in the appropriate buffer prior to the experiment. The final protein buffer was dependent on the experiment performed. For ITC experiments, the final SEC purification was performed in ITC buffer (10 mM Tris pH 7.5, 150 mM NaCl, 0.1 mM EDTA, 0.5 mM TCEP); for NMR and SAXS experiments, the final SEC purification was performed in NMR buffer (50 mM HEPES pH 6.8, 150 mM NaCl, 5 mM dithiothreitol).
NMR sample preparation
[2H,15N]-p38α (Vogtherr et al., 2005) and [2H,15N]-STEP were expressed in M9 media supplemented with 15NH4Cl (1 g/l) in 99% D2O and purified as previously described. PTPSLKIM, PTPSLKIMKIS, STEPKIM, or STEPKIMKIS peptides were added to [2H,15N]-p38α (100 μM p38α) in the following molar excesses: PTPSLKIM (1:1 and 1:8 molar ratio, p38:peptide), PTPSLKIMKIS (1:1 and 1:3 molar ratio, p38:peptide), STEPKIM (1:1, 1:3, 1:5 and 1:8 molar ratio, p38:peptide) and STEPKIMKIS (1:1, 1:3, 1:6 and 1:8 molar ratio, p38:peptide). PTPSLCAT and STEPCAT were added to [2H,15N]-p38α (100 μM p38α) in a 2-fold molar excess (0.1 mM [2H,15N]-p38α:0.2 mM PTPSLCAT or STEPCAT). For p38α:KIM-PTP complex formation, [2H,15N]-p38α was incubated with either PTPSL, STEP or PSChimera (1:1 molar ratio) on ice for 45 min and purified using SEC (Superdex 75 26/60, NMR buffer). Each p38α:KIM-PTP complex elutes at the position expected for the p38α:KIM-PTP heterodimer. Each [2H,15N]-p38α:KIM-PTP complex was concentrated to 0.1 mM and stored on ice (<8 hours) until NMR data acquisition. For the p38α:[2H,15N]-STEP complex formation, [2H,15N]-STEP was incubated with p38α (1:1 molar ratio) on ice for 45 min and purified using SEC (Superdex 75 26/60, NMR buffer). The p38α:[2H,15N]-STEP complex was concentrated to 0.1 mM and stored on ice (<2 hours) until NMR data acquisition. A 2D [1H,15N]-TROSY spectrum was recorded for all samples described above. All NMR experiments were performed at 298 K on a Bruker Advance 800 MHz spectrometer equipped with a TCI HCN z-gradient cryoprobe. NMR samples were prepared in NMR buffer in 90% H2O/10% D2O. The NMR spectra were processed with Topspin 2.1/3.0/3.1 (Bruker, Billerica, MA) and analyzed using CARA (http://cara.nmr.ch).
Chemical shift perturbation studies
The p38α:STEPKIM and p38α: PTPSLKIM peptide titration series (1:1, 1:3, 1:5 and 1:8 ratio p38α:STEPKIM) were used to follow chemical shift changes in p38α upon peptide or protein binding. Nearly all peaks showed fast exchange and thus could be easily tracked. The observed trajectory of each peak was applied to the analysis of the KIMKIS peptides (PTPSLKIMKIS and STEPKIMKIS) and the full-length complexes (PTPSL, STEP, and PSChimera) allowing for high certainty of peak assignments of p38α in the bound conformations. All chemical shift differences (Δδ) were calculated using
To allow for direct comparison across all CSP experiments, we used identical cut-off CSPs as in the HePTP NMR experiments; small CSP 0.07–0.103 ppm, medium CSP 0.104–0.131 ppm; large CSP ≥0.132 (Francis et al., 2011a).
HADDOCK Calculations
HADDOCK was used to dock p38α and STEP using ambiguous NMR-derived restraints. p38α (PBDID 1P38) and STEP (PBDID 2BIJ) were used as starting input. Active residues were defined as those that undergo a CSP and have high solvent accessibility in the unbound protein structure (p38α residues: 177, 179, 183, 222, 248, 254, 256; STEP residues: 263, 268, 269, 274, 277, 278, 279, 281, 282, 285, 286, 294, 306, 307, 377, 378, 379, 516, 517, 518). Passive residues are those have a CSP but have low solvent accessibility (p38α residues: 190, 195, 247, 255, 328; STEP residues: 276, 280, 350, 353, 376, 380, 478, 479, 482, 520, 525). CSPs were the only experimental restraint type used in the HADDOCK calculation and default settings were used for all docking steps, as used by the easy interface of the HADDOCK webserver.
Small angle X-ray scattering
For the resting-state p38α: PTPSL and p38α:STEP complexes, purified, unphosphorylated p38α was incubated in a 1:1 molar ratio with purified PTPSL331–655 and/or purified STEP214–539 on ice for 30 min and the complexes purified using SEC (Superdex 75 26/60, NMR buffer). To form the active-state complex of p38α:STEP, purified, dually-phosphorylated p38α was incubated in a 1:1 molar ratio with purified STEP214–539 T306D/C472S on ice for 30 min and the complex was purified using SEC (Superdex 75 26/60, NMR buffer). For p38α:PTPSL, data was collected on samples at 0.4, 0.8, 1.6, 2.3 and 4.0 mg/ml. For resting-state p38α:STEP, data was collected on samples at 1.0, 2.0 and 4.0 mg/ml. For active-state p38α:STEP, data was collected on samples at 0.8, 3.3 and 6.1 mg/ml. All samples were prepared within 48 hours of data acquisition and stored on ice at 4°C. All samples were filtered (0.02 μM filter, Whatman) immediately prior to data collection. 20 μl of sample was continuously flowed through a 1 mm diameter capillary and exposed to an X-ray beam for 30 s (p38α:PTPSL), 60 s (active-state p38α:STEP), or 180 s (resting-state p38α:STEP). Normalization for beam intensity, buffer subtraction and merging of the data from both detectors were carried out using PRIMUS (Konarev et al., 2003). A Guinier approximation, I(q) = I(0)exp(−q2Rg2/3), where a plot of ln[I(q)] and q2 is linear for q<1.3/Rg, was performed on at least four independent scattering trials and averaged to determine the radius of gyration. The linearity of the Guinier region and the intensity at zero scattering angle, I(0), were used to validate that all samples were monodisperse in solution. I(0)/c, where c is concentration, was consistent for all measurements for a single complex. GNOM (Svergun, 1992) was used to determine the pair-distribution function, P(r), for each complex. Twenty-four envelopes were generated for each complex using GASBOR (Svergun et al., 2001) and were aligned and averaged using the DAMAVER program suite (Volkov and Svergun, 2003).
Supplementary Material
Highlights.
The p38α:PTPSL interaction is confined mainly to the KIM-binding site.
The p38α:PTPSL complex adopts an extended conformation.
The STEP catalytic domain interacts with a novel p38α binding site.
The p38α:STEP resting- and active-states have distinct conformations.
Acknowledgments
The authors thank Drs. L. Yang and M. Allaire (National Synchrotron Light Source, NSLS) for their support at NSLS beamline X9. We thank Dr. Paul Lombroso, Yale University, for providing PTPN5 DNA. This research was supported by grant RSG-08-067-01-LIB from the American Cancer Society to R.P. and by R01GM100910 from the National Institute of Health to W.P. Use of NSLS at Brookhaven National Laboratory was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under contract no. DE-AC02-98CH10886. 800 MHz NMR data were recorded at Brandeis University; the instrument was purchased with support from NIH S10-RR017269.
Footnotes
Supplemental Information includes figures and Supplemental Experimental Procedures and can be found with this article online at http://
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akella R, Min X, Wu Q, Gardner KH, Goldsmith EJ. The third conformation of p38alpha MAP kinase observed in phosphorylated p38alpha and in solution. Structure. 2010;18:1571–1578. doi: 10.1016/j.str.2010.09.015. [DOI] [PubMed] [Google Scholar]
- Balasu MC, Spiridon LN, Miron S, Craescu CT, Scheidig AJ, Petrescu AJ, Szedlacsek SE. Interface analysis of the complex between ERK2 and PTP-SL. PLoS One. 2009;4:e5432. doi: 10.1371/journal.pone.0005432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Aparicio C, Torres J, Pulido R. A novel regulatory mechanism of MAP kinases activation and nuclear translocation mediated by PKA and the PTP-SL tyrosine phosphatase. J Cell Biol. 1999;147:1129–1136. doi: 10.1083/jcb.147.6.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CI, Xu BE, Akella R, Cobb MH, Goldsmith EJ. Crystal structures of MAP kinase p38 complexed to the docking sites on its nuclear substrate MEF2A and activator MKK3b. Mol Cell. 2002;9:1241–1249. doi: 10.1016/s1097-2765(02)00525-7. [DOI] [PubMed] [Google Scholar]
- Chirivi RG, Dilaver G, van de Vorstenbosch R, Wanschers B, Schepens J, Croes H, Fransen J, Hendriks W. Characterization of multiple transcripts and isoforms derived from the mouse protein tyrosine phosphatase gene Ptprr. Genes Cells. 2004;9:919–933. doi: 10.1111/j.1365-2443.2004.00773.x. [DOI] [PubMed] [Google Scholar]
- de Vries SJ, van Dijk AD, Krzeminski M, van Dijk M, Thureau A, Hsu V, Wassenaar T, Bonvin AM. HADDOCK versus HADDOCK: new features and performance of HADDOCK2.0 on the CAPRI targets. Proteins. 2007;69:726–733. doi: 10.1002/prot.21723. [DOI] [PubMed] [Google Scholar]
- Derijard B, Raingeaud J, Barrett T, Wu IH, Han J, Ulevitch RJ, Davis RJ. Independent human MAP-kinase signal transduction pathways defined by MEK and MKK isoforms. Science. 1995;267:682–685. doi: 10.1126/science.7839144. [DOI] [PubMed] [Google Scholar]
- Diskin R, Engelberg D, Livnah O. A novel lipid binding site formed by the MAP kinase insert in p38 alpha. J Mol Biol. 2008;375:70–79. doi: 10.1016/j.jmb.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Dominguez C, Boelens R, Bonvin AM. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J Am Chem Soc. 2003;125:1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- Eswaran J, von Kries JP, Marsden B, Longman E, Debreczeni JE, Ugochukwu E, Turnbull A, Lee WH, Knapp S, Barr AJ. Crystal structures and inhibitor identification for PTPN5, PTPRR and PTPN7: a family of human MAPK-specific protein tyrosine phosphatases. Biochem J. 2006;395:483–491. doi: 10.1042/BJ20051931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis DM, Page R, Peti W. Sequence-specific backbone 1H, 13C and 15N assignments of the 34 kDa catalytic domain of PTPN5 (STEP) Biomol NMR Assign . 2013 doi: 10.1007/s12104-013-9480-8. Submitted. [DOI] [PubMed] [Google Scholar]
- Francis DM, Rozycki B, Koveal D, Hummer G, Page R, Peti W. Structural basis of p38alpha regulation by hematopoietic tyrosine phosphatase. Nat Chem Biol. 2011a;7:916–924. doi: 10.1038/nchembio.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis DM, Rozycki B, Tortajada A, Hummer G, Peti W, Page R. Resting and active states of the ERK2:HePTP complex. J Am Chem Soc. 2011b;133:17138–17141. doi: 10.1021/ja2075136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garai A, Zeke A, Gogl G, Toro I, Fordos F, Blankenburg H, Barkai T, Varga J, Alexa A, Emig D, et al. Specificity of linear motifs that bind to a common mitogen-activated protein kinase docking groove. Sci Signal. 2012;5:ra74. doi: 10.1126/scisignal.2003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gills JJ, Castillo SS, Zhang C, Petukhov PA, Memmott RM, Hollingshead M, Warfel N, Han J, Kozikowski AP, Dennis PA. Phosphatidylinositol ether lipid analogues that inhibit AKT also independently activate the stress kinase, p38alpha, through MKK3/6-independent and -dependent mechanisms. J Biol Chem. 2007;282:27020–27029. doi: 10.1074/jbc.M701108200. [DOI] [PubMed] [Google Scholar]
- Hendriks W, Schepens J, Brugman C, Zeeuwen P, Wieringa B. A novel receptor-type protein tyrosine phosphatase with a single catalytic domain is specifically expressed in mouse brain. Biochem J. 1995;305(Pt 2):499–504. doi: 10.1042/bj3050499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 2010;1802:396–405. doi: 10.1016/j.bbadis.2009.12.009. [DOI] [PubMed] [Google Scholar]
- Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. Journal of Applied Crystallography. 2003;36:1277–1282. [Google Scholar]
- Kozin MB, Svergun DI. Automated matching of high- and low-resolution structural models. Journal of Applied Crystallography. 2001;34:33–41. [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Lombroso PJ, Murdoch G, Lerner M. Molecular characterization of a protein-tyrosine-phosphatase enriched in striatum. Proc Natl Acad Sci U S A. 1991;88:7242–7246. doi: 10.1073/pnas.88.16.7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombroso PJ, Naegele JR, Sharma E, Lerner M. A protein tyrosine phosphatase expressed within dopaminoceptive neurons of the basal ganglia and related structures. J Neurosci. 1993;13:3064–3074. doi: 10.1523/JNEUROSCI.13-07-03064.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz JJ, Tarrega C, Blanco-Aparicio C, Pulido R. Differential interaction of the tyrosine phosphatases PTP-SL, STEP and HePTP with the mitogen-activated protein kinases ERK1/2 and p38alpha is determined by a kinase specificity sequence and influenced by reducing agents. Biochem J. 2003;372:193–201. doi: 10.1042/BJ20021941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustelin T, Tautz L, Page R. Structure of the hematopoietic tyrosine phosphatase (HePTP) catalytic domain: structure of a KIM phosphatase with phosphate bound at the active site. J Mol Biol. 2005;354:150–163. doi: 10.1016/j.jmb.2005.09.049. [DOI] [PubMed] [Google Scholar]
- Nielsen G, Schwalbe H. NMR spectroscopic investigations of the activated p38α mitogen-activated protein kinase. Chembiochem. 2011;12:2599–2607. doi: 10.1002/cbic.201100527. [DOI] [PubMed] [Google Scholar]
- Nony PA, Kennett SB, Glasgow WC, Olden K, Roberts JD. 15S-Lipoxygenase-2 mediates arachidonic acid-stimulated adhesion of human breast carcinoma cells through the activation of TAK1, MKK6, and p38 MAPK. J Biol Chem. 2005;280:31413–31419. doi: 10.1074/jbc.M500418200. [DOI] [PubMed] [Google Scholar]
- Ortega A, Amoros D, Garcia de la Torre J. Prediction of hydrodynamic and other solution properties of rigid proteins from atomic- and residue-level models. Biophys J. 2011;101:892–898. doi: 10.1016/j.bpj.2011.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry JJ, Harris RM, Moiani D, Olson AJ, Tainer JA. p38alpha MAP kinase C-terminal domain binding pocket characterized by crystallographic and computational analyses. J Mol Biol. 2009;391:1–11. doi: 10.1016/j.jmb.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peti W, Page R. Strategies to maximize heterologous protein expression in Escherichia coli with minimal cost. Protein Expr Purif. 2007;51:1–10. doi: 10.1016/j.pep.2006.06.024. [DOI] [PubMed] [Google Scholar]
- Piserchio A, Francis DM, Koveal D, Dalby KN, Page R, Peti W, Ghose R. Docking Interactions of Hematopoietic Tyrosine Phosphatase with MAP Kinases ERK2 and p38alpha. Biochemistry. 2012;51:8047–8049. doi: 10.1021/bi3012725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poddar R, Deb I, Mukherjee S, Paul S. NR2B-NMDA receptor mediated modulation of the tyrosine phosphatase STEP regulates glutamate induced neuronal cell death. Journal of neurochemistry. 2010;115:1350–1362. doi: 10.1111/j.1471-4159.2010.07035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena M, Williams S, Tasken K, Mustelin T. Crosstalk between cAMP-dependent kinase and MAP kinase through a protein tyrosine phosphatase. Nat Cell Biol. 1999;1:305–311. doi: 10.1038/13024. [DOI] [PubMed] [Google Scholar]
- Svergun D. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. Journal of Applied Crystallography. 1992;25:495–503. [Google Scholar]
- Svergun DI, Petoukhov MV, Koch MH. Determination of domain structure of proteins from X-ray solution scattering. Biophys J. 2001;80:2946–2953. doi: 10.1016/S0006-3495(01)76260-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szedlacsek SE, Aricescu AR, Fulga TA, Renault L, Scheidig AJ. Crystal structure of PTP-SL/PTPBR7 catalytic domain: implications for MAP kinase regulation. J Mol Biol. 2001;311:557–568. doi: 10.1006/jmbi.2001.4890. [DOI] [PubMed] [Google Scholar]
- Tarrega C, Blanco-Aparicio C, Munoz JJ, Pulido R. Two clusters of residues at the docking groove of mitogen-activated protein kinases differentially mediate their functional interaction with the tyrosine phosphatases PTP-SL and STEP. J Biol Chem. 2002;277:2629–2636. doi: 10.1074/jbc.M108874200. [DOI] [PubMed] [Google Scholar]
- Tzarum N, Eisenberg-Domovich Y, Gills JJ, Dennis PA, Livnah O. Lipid molecules induce p38alpha activation via a novel molecular switch. J Mol Biol. 2012;424:339–353. doi: 10.1016/j.jmb.2012.10.007. [DOI] [PubMed] [Google Scholar]
- Van Den Maagdenberg AM, Bachner D, Schepens JT, Peters W, Fransen JA, Wieringa B, Hendriks WJ. The mouse Ptprr gene encodes two protein tyrosine phosphatases, PTP-SL and PTPBR7, that display distinct patterns of expression during neural development. The European journal of neuroscience. 1999;11:3832–3844. doi: 10.1046/j.1460-9568.1999.00802.x. [DOI] [PubMed] [Google Scholar]
- Vogtherr M, Saxena K, Grimme S, Betz M, Schieborr U, Pescatore B, Langer T, Schwalbe H. NMR backbone assignment of the mitogen-activated protein (MAP) kinase p38. J Biomol NMR. 2005;32:175. doi: 10.1007/s10858-005-2449-x. [DOI] [PubMed] [Google Scholar]
- Volkov VV, Svergun DI. Uniqueness of ab initio shape determination in small-angle scattering. Journal of Applied Crystallography. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Harkins PC, Ulevitch RJ, Han J, Cobb MH, Goldsmith EJ. The structure of mitogen-activated protein kinase p38 at 2.1-A resolution. Proc Natl Acad Sci U S A. 1997;94:2327–2332. doi: 10.1073/pnas.94.6.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Kurup P, Zhang Y, Goebel-Goody SM, Wu PH, Hawasli AH, Baum ML, Bibb JA, Lombroso PJ. Extrasynaptic NMDA receptors couple preferentially to excitotoxicity via calpain-mediated cleavage of STEP. J Neurosci. 2009;29:9330–9343. doi: 10.1523/JNEUROSCI.2212-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanke B, Suzuki H, Kishihara K, Mizzen L, Minden M, Pawson A, Mak TW. Cloning and expression of an inducible lymphoid-specific, protein tyrosine phosphatase (HePTPase) Eur J Immunol. 1992;22:235–239. doi: 10.1002/eji.1830220134. [DOI] [PubMed] [Google Scholar]
- Zhang YY, Wu JW, Wang ZX. Mitogen-activated protein kinase (MAPK) phosphatase 3-mediated cross-talk between MAPKs ERK2 and p38alpha. J Biol Chem. 2011;286:16150–16162. doi: 10.1074/jbc.M110.203786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Sun L, Humphreys J, Goldsmith EJ. Docking interactions induce exposure of activation loop in the MAP kinase ERK2. Structure. 2006;14:1011–1019. doi: 10.1016/j.str.2006.04.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.