

Abstract
The fragment indole-6-carboxylic acid (1F1), previously identified as a flap site binder in a fragment-based screen against HIV protease (PR), has been co-crystallized with pepstatin-inhibited PR and with apo-PR. Another fragment, 3-indolepropionic acid (1F1-N), predicted by AutoDock calculations and confirmed in a novel ‘inhibition of nucleation’ crystallization assay, exploits the same interactions in the flap site in two crystal structures. Both 1F1 and 1F1-N bind to the closed form of apo-PR and to pepstatin:PR. In solution, 1F1 and 1F1-N raise the Tm of apo-PR by 3.5–5 °C as assayed by differential scanning fluorimetry (DSF), and show equivalent low-micromolar binding constants to both apo-PR and pepstatin:PR, assayed by backscattering interferometry (BSI). The observed signal intensities in BSI are greater for each fragment upon binding to apo-PR than to pepstatin-bound PR, consistent with greater conformational change in the former binding event. Together, these data indicate that fragment binding in the flap site favors a closed conformation of HIV PR.
Keywords: HIV protease inhibitor, flap mobility, fragment screen, combination therapy, drug resistance
Introduction
HIV is no longer the rapid death sentence it once was due to the advent of drug therapies including HAART (highly-active antiretroviral therapy). However, drug resistance is an increasing problem. It is estimated that 14% of new HIV infections in the United States occur with strains of HIV that are already resistant to one or more of the components of HAART, of which 4.5% represents drug resistance to protease inhibitors(1). While new therapeutics, vaccines, and/or microbicides being developed to prevent HIV infection may significantly decrease the incidence of this devastating disease in the future, such therapies do not address the problem of resistance for the 33 million people(2) currently living with HIV.
Of the components in the HAART cocktail, HIV protease (PR) inhibitors have been shown to have the best dose-response behavior(3). However, HIV often develops resistance to PR inhibitors both through mutations in the protease and mutations in processing sites on the gag polyprotein(4). Eight of the nine FDA approved active site inhibitors are based on the same hydroxyethylene core, with only tipranavir being different. Only darunavir has a different resistance profile compared to the other approved PR inhibitors, and the differences are slight(5). To overcome resistance, drugs with novel mechanisms of action are required.
Allosteric inhibitors are an attractive alternative to active site inhibitors, particularly for systems prone to developing resistance mutations. Combinations of allosteric inhibitors and active site inhibitors can be even more effective, such as the combination of non-nucleoside reverse transcriptase inhibitors (NNRTIs) and active site nucleoside analog reverse transcriptase inhibitors (NRTIs) for HIV-1. When administered alone, HIV rapidly develops resistance to NRTIs. However, when a combination of an NRTI, such as azidothymidine (AZT), and an allosteric NNRTI is administered, the evolution of resistance to both drugs is suppressed(6). An advantage of an allosteric inhibitor is that it will not be subject to the same cross-resistance pressures shared by all active-site inhibitors(7). This may be the case for darunavir, a PR inhibitor that binds both to the active site and a secondary site near the flap site of HIV-1 protease. Darunavir remains effective against PR mutants that are resistant to other protease inhibitors, perhaps because of the alternative binding site on the protein surface for the S-enantiomer(8–10), in combination with the strong interactions of darunavir with the PR main chain and favorable occupation of the PR ‘substrate envelope’(11, 12). In addition, the dynamic biomechanical relationships that allosteric inhibitors could exploit raise the genetic barrier for mutations that maintain sufficient catalytic efficiency for virus replication(13). Co-administration of allosteric and active-site inhibitors to decrease the evolution of resistance has been demonstrated both in vitro and ex vivo in the case of ABL kinase, a target for cancer chemotherapy(14, 15). Hence, an allosteric inhibitor to HIV PR could restore the efficacy of active site inhibitors against multi-drug-resistant mutants and, in combination with an active site inhibitor, potentially lessen the evolution of resistance.
A previous fragment screen utilizing X-ray crystallography identified a novel binding site on the surface of HIV PR, termed the flap site; two of the 384 fragments screened bound in this site(16). This site holds much interest as a potential allosteric site for HIV PR, as suggested by the conformational changes observed in the reported crystal structures with compounds 1F1 (indole-6-carboxylic acid) and 2F4 (2-acetyl-benzothiophene), co-crystallized with TL-3-inhibited PR(16). Here, we report crystal structures with 1F1 bound to both apo-PR and pepstatin:PR, revealing each to be in the closed conformation. Further, we present crystal structures with a 1F1 derivative, 1F1-N (3-indolepropionic acid), bound to the same site in both apo-PR and pepstatin:PR, again with each in the closed conformation. 1F1-N binding was predicted from in silico docking calculations against a library of related fragments, and selected for co-crystallization based on a novel ‘inhibition of nucleation’ crystallization screen. 1F1-N recapitulates each of three principle interactions with PR observed for 1F1, even though it binds in a unique orientation. Interaction of 1F1 and 1F1-N with apo-PR and pepstatin:PR in solution was confirmed using differential scanning fluorimetry (DSF) as well as back-scattering interferometry (BSI). BSI establishes Kd values for each compound and reveals a greater extent of conformational change when the fragments bind to apo-PR compared to pepstatin:PR.
RESULTS AND DISCUSSION
AutoDock and inhibition of nucleation assay
As a first step to expand the 1F1 fragment hit(16) into a larger molecule, a library of 2,499 commercially available compounds with similar structural features was selected and virtually screened against the crystallographic 1F1 binding site using AutoDock 4.2(17), as described in Methods. From a list of the top 45 hits, 22 were selected for further screening (Supplemental Table 2). Initially, these compounds were screened for co-crystallization under the same conditions that yielded co-crystals with 1F1 bound to PR:pepstatin, but no hits were obtained. An ‘inhibition of nucleation’ screen was employed to identify compounds affecting the crystallization behavior of HIV PR, indicating potential binding in the flap site or elsewhere on the protease.
The first “inhibition of nucleation” screen was based on the growth of the semi-open conformation of apo NL4-3 PR in P41212 crystals grown in the presence of Mg2+ (e.g. PDB_id: 2PC0(18)). These crystals nucleate overnight, and crystal growth is compatible with 10% ethanol or methanol, but not 10% DMSO. It was observed that these crystals do not nucleate in the presence of 20 mM of 1F1 dissolved in methanol (Supplemental Figure 1). Further trials demonstrated dose-dependence of crystal formation (Supplemental Figure 2). In addition to 1F1, 1F1-F (4-iodo-1H-indole-6-carboxylic acid), 1F1-N (3-indolepropionic acid) and 1F1-U (2-(6-chloro-9H-carbazol-2-yl)propanoic acid) inhibited nucleation of the P41212 form at 20 mM or saturation. In contrast, similar compounds such as 1F1-A (2,3-dimethyl-indole-6-carboxylic acid) do not inhibit the nucleation of these crystals (Supplemental Table 2), suggesting that the effect is specific for fragments that bind specifically to PR. We postulate that binding in the flap site selects for HIV PR in a closed form, which is incompatible with the semi-open PR conformation in the P41212 lattice(18).
A second “inhibition of nucleation” assay was developed to overcome the non-compatibility of the P41212 crystal form with DMSO, as several of the compounds of interest were only soluble in DMSO. For this assay, P6122 crystals of TL-3-inhibited HIV PR were used (e.g. PDB_id: 3KFP(16)). The P6122 crystal form grows readily under a number of conditions, including those containing 10% DMSO, but these crystals shatter and dissolve if incubated with 1F1 and will not grow in its presence (Supplemental Figure 3). In this assay, 1F1-E (3-(carboxymethyl)-1H-indole-2,6-dicarboxylic acid), 1F1-H (3-(carboxymethyl)-2-methyl-1H-indole-6-carboxylic acid), and 1F1-N inhibited crystal growth completely (Supplemental Table 2). Although the inhibition of nucleation assays are not quantitative and may not be specific for interactions only in the flap site, they allowed further co-crystallization trials to focus on a small subset of fragments. Only 1F1 and 1F1-N inhibited nucleation in both assays, and co-crystals were obtained with 1F1-N bound to apo-PR and pepstatin:PR.
PR:pepstatin with 1F1
1F1 was co-crystallized with pepstatin-inhibited HIV PR. Chunky prismatic P212121 crystals grew under several conditions, and a structure to 1.1 Å resolution was obtained (Table 1; Supplemental Figure 1; Figure 1). Both the crystal form and the 1F1 binding site are similar to the published TL-3-inhibited HIV PR structure (PDB_id: 3KFR(16)). One minor difference is in the conformation of Arg57. In 3KFR, Arg57 is 100% flipped to form a hydrogen bonding interaction with the carboxylic acid moiety of 1F1, whereas in the pepstatin-bound structure, Arg57 is only ~50% flipped to bind to 1F1, with the other occupied conformation maintaining the usual hydrogen bonding interaction with Glu35.
Table 1.
Crystals and data collection.
| Protease/Inhibitor | Compound | Space Group | Cell Dimensions | Morphology | Crystallization Conditions | Flaps | Resolution Data Source |
|---|---|---|---|---|---|---|---|
| NL4-3 TL-3 (3KFR) |
1F1 | P212121 | 28.72 65.57 92.36 |
Blocks | 0.5 M KSCN, 0.1 M MES- HCL, pH 5.8, 10% DMSO | Closed | 1.3 Å SRRLa |
| NL4-3 pepstatin (4EJD) |
1F1 | P212121 | 28.82 65.63 92.93 |
Blocks | 0.2 M KBr, 0.2M KSCN, 0.1M NaCacodylate pH 6.5, 3% PGA-LM, 10% DMSO, 3% MPD | Closed | 1.1 Å SSRL |
| AR-PR (apo) (4EJ8) |
1F1 | P212121 | 28.95 66.53 91.71 |
Long needles | 0.1M Tris pH 7.5, 28% PEG 4K, 10% DMSO | Invertedb | 2.5 Å Bruker |
| NL4-3 pepstatin (4EJK) |
1F1-N | P212121 | 28.65 65.79 92.05 |
Rods | 1.0 M NaFormate, 0.1 M NaOAc pH 4.6, 10% DMSO | Closed | 1.8 Å Bruker |
| AR-PR (apo) (4EJL) |
1F1-N | P212121 | 28.82 65.91 92.51 |
Small rods | 0.8 M NaFormate, 0.1 M NaOAc pH 5.5, 15% PEG4K | Closed | 2.0 Å Bruker |
Reference (16)
Inverted = closed conformation of the flaps with reversed “handedness”
Figure 1.
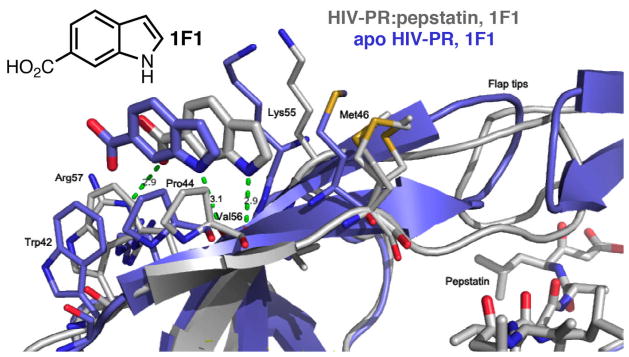
Structure of HIV PR:pepstatin bound to 1F1 (gray) compared to structure of apo HIV PR bound to 1F1 (blue). Note the inverted orientation of the flap tips and the shift of the binding site for the apo-PR complex. Met46 is disordered in the PR:pepstatin structure and adopts three conformations with partial occupancy.
Apo-PR with 1F1
Co-crystallization of 1F1 with autolysis-resistant apo-PR (mutations relative to wild-type are Q7K, L33I, L63I, C67A, C95A(21)) yielded rod-shaped P212121 crystals, and a structure was obtained at 2.5 Å (Table 1; Supplemental Table 1; Figure 1). This structure of apo-PR with 1F1 is unusual in that the flaps are closed and the active site is not occupied by a peptide or peptidomimetic inhibitor; it is empty aside from two DMSO solvent molecules, one glycerol, and ten ordered water molecules (Figure 2; compare Supplemental Figure 8a). Another unusual feature of this structure is that the flaps are closed in a conformation with opposite “handedness” compared to the standard closed conformation of HIV PR. Residues 43–58 of each monomer shift and the stacking at the tips of the flaps is inverted (Figure 1). A closed conformation of HIV PR with inverted flap tips was first observed in MD simulations(22), and this conformation of the flaps is commonly observed in semi-open structures of apo-PR(18), but this is the first time it has been described for a crystal structure with closed flaps. These results suggest that apo-PR in solution adopts a variety of conformations with both conformations of the flap tips represented, and either conformation can be trapped in a crystal lattice. The 1F1 binding site is shifted ~2.5 Å due to a shift in the flap relative to the structures with pepstatin and TL-3 inhibited PR, but the key interactions of the fragment are maintained (Figure 1). In this crystal form, Arg57 does not flip, but remains hydrogen bonded to Glu35.
Figure 2.

1F1 and active site electron density for apo-PR with 1F1 bound. Note the absence of peptide-like density in the active site (compare Supplemental Figure 8).
Pepstatin:PR with 1F1-N
The structure of 1F1-N bound to pepstatin:PR was obtained from rod-shaped P212121 crystals to 1.8Å (Table 1, Supplemental Table 1, Figure 3) and is very similar to the structure of 1F1 bound to pepstatin:PR with respect to the protein (RMSD 0.19Å, Supplemental Table 3), but interestingly, the binding mode of 1F1-N is “flipped,” with the orientation of the indole ring inverted relative to the binding of 1F1, as predicted by AutoDock (Figure 3; Supplemental Figure 5). This places the carboxylic acid groups in the same general orientation relative to the protein for both 1F1 and 1F1-N, and preserves all of the key interactions PR. Hydrophobic interactions with Trp42, Pro 44, Met 46, and Lys55 are maintained; the indole nitrogen forms a hydrogen bond with the carbonyl of Val56; and an electrostatic interaction occurs with Arg57. The observed binding mode of the indole ring corresponds closely to that predicted by AutoDock (Fig. S5a), while two alternate conformations with comparable cluster size are predicted for the propionic acid side chain: one with the carboxyl group oriented toward Lys55, and one with the carboxyl group interacting with Arg57 (Supplemental Figure 5b). The latter pose is similar to that observed in the crystal structure (Supplemental Figure 5a), with the carboxylic acid moiety at a distance of 4.9 Å from the guanidinium group of Arg57.
Figure 3.

HIV PR:pepstatin bound to 1F1(gray) superposed onto HIV PR:pepstatin bound to 1F1-N (magenta). Note the flipped binding mode of the indole ring but maintenance of the hydrogen bond from the indole nitrogen to the backbone of Val56.
Apo-PR with 1F1-N
The structure of 1F1-N bound to autolysis-resistant apo-PR was obtained from rod-like P212121 crystals to 2.0 Å (Table 1, Supplemental Table 1, Figure 4) and is similar to the structures of 1F1 and 1F1-N bound to pepstatin:PR (RMSD ~0.8 Å, Supplemental Table 3); i.e., the flaps are in a closed conformation. As in the structure of apo-PR with 1F1, the active site is occupied only by a handful of solvent molecules (Supplemental Figure 4). However, here the flaps have the usual “handedness,” unlike the structure of apo-PR with 1F1 in which the inner and outer flaps are switched. 1F1-N occupies a virtually identical position in the co-crystal structures with apo-PR and pepstatin:PR (Figure 4).
Figure 4.

Structure of HIV PR:pepstatin bound to 1F1-N (magenta) compared to structure of apo HIV PR bound to 1F1-N (navy).
Protease Conformation
Flap mobility in HIV PR has been examined by NMR, double electron-electron resonance (DEER), pulse-EPR spectroscopy with nitroxide spin-labels, and MD simulations(23–31). These studies indicate that individual flaps in the native protein sample several different conformations, including closed, open or closed with inverted flaps, semi-open, and wide-open, with the majority of the protein (60–80% depending on subtype/mutations(28)) being in the semi-open state. DEER profiles are significantly shifted to favor the closed state with the addition of active-site PR inhibitors(29). Of 270 structures of HIV protease in the PDB, the only apo structures in the closed conformation are of tethered dimers(32–36). The only other known apo-PR crystal structures either have flaps in an open or semi-open conformation stabilized by lattice contacts, or the flaps are disordered(18). The structures of apo-PR with 1F1 and 1F1-N bound are notable for being the first crystal structures of apo-PR with closed flaps (see Supplemental Figure 8; further discussion in Supporting Information), and suggest that the binding of compounds in the flap site favors a closed flap conformation of the protease in solution.
Comparison of the pepstatin-bound structures of PR:Acetyl-pepstatin (5HVP(19)), PR:pepstatin:1F1, and PR:pepstatin:1F1-N reveals nearly identical interactions with pepstatin in the active site. The most significant differences among these structures are observed in the conformation of the weakly-ordered 34–41 loop (‘ear’) on chain B when the fragment binds in the flap site of chain A. Additionally, the side chain of Lys55 in the A chain, which adopts a conformation blocking the flap site, is displaced when fragments are bound (Supplemental Figure 6).
An additional unusual feature of these structures is crystallization in the P212121 space group with unit cell dimensions of ~28, 65, 92 Å. Twelve structures of HIV PR are deposited in the PDB in this space group (4FAE, 4FAF, 3UFN, 3S53, 3S56, 3SO9, 2HS1, 2HS2, 1Z8C, 1NHO, 3KFR, 3KFS). Nine of these are complexes with flap site binders(8, 16, 37). Inspection of the electron density maps of the remaining three indicates unmodeled or poorly modeled electron density in the flap site (for further discussion, see Supporting Information). Thus, crystallization of HIV PR in the P212121 crystal form is an indicator of binding in the flap site in that the bound conformation is unique and favors this particular packing arrangement.
Differential scanning fluorimetry
To confirm that the interactions of 1F1 and 1F1-N observed in crystals also occur in solution, DSF curves were measured with varying concentrations of 1F1 and 1F1-N with autolysis-resistant apo-PR. In both cases, the compounds increased the thermal stability of HIV PR in a concentration-dependent manner, indicating protein-ligand binding(38, 39). At 5 mM, 1F1 increases the melting temperature of apo HIV PR by 5°C, while 1F1-N increases the melting temperature of apo HIV PR by 3.5°C (Figure 5). As a control, DSF experiments were performed on selected compounds from the 22-member library, and stabilizing temperature shifts were not observed (Supplemental Figure 7a). However, DSF was not a useful tool to narrow down potential binding because the background fluorescence of a number of the compounds was too high to obtain reliable results. For example, two fragments that inhibited crystal nucleation, 1F1-E and 1F1-F, exhibited levels of fluorescence that overwhelmed the DSF signal at the required millimolar concentrations (Supplemental Figure 7b). The larger shift caused by 1F1 is consistent with the hydrogen bonds with Arg57 observed in the 1F1 complexes compared to the longer range electrostatic interaction in the 1F1-N complexes. Thermal stabilization via fragments interacting with residues in the flap site is consistent with MD simulations which identify conserved, noncovalent interactions that couple secondary structure elements within the tertiary structure of the PR dimer(40).
Figure 5.
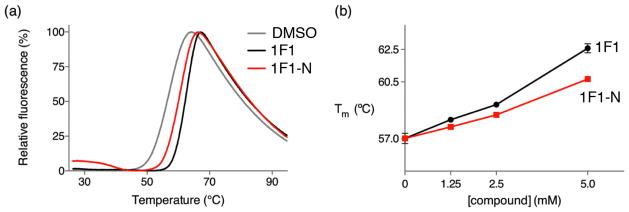
Differential scanning fluorimetry of apo, autolysis resistant HIV PR in the presence and absence of fragments showing that 1F1 and 1F1-N stabilize the protease in solution, consistent with binding in the site observed in crystal structures and flap closure. (a) The presence of 5 mM 1F1 and 1F1-N increases the melting temperature of protease, as indicated by shifts in the DSF curves. (b) 1F1 and 1F1-N stabilize protease in a dose-dependent manner.
Back-Scattering Interferometry
To further investigate the binding of 1F1 and 1F1-N to HIV PR in solution, back-scattering interferometry (BSI) was performed on samples of apo-PR and pepstatin:PR with both compounds. BSI detects changes in refractive index upon binding that can be used to determine Kd from dose-response measurements(41–45). The signal detected in a BSI measurement is the maximal refractive index change upon binding of a solute to a dilute protein solution. Both 1F1 and 1F1-N were tested with apo-PR and pepstatin:PR (Figure 6). The Kd values for both fragments are in the low μM range and within experimental error both with and without pepstatin. The results indicate that fragment binding can occur when pepstatin is bound and the flap is constrained to a closed conformation. Further, the results show that fragments bind equally well to apo-PR in which the flaps can adopt a range of conformations. Interestingly, the maximal change in phase change, which is a measure of the extent of overall conformational change, was greater in the absence of pepstatin, indicating a larger change in conformation of the protein upon compound binding(46). The difference in effective concentrations (mM for DSF and μM for BSI) reflects the nature of the different assays. It is worth noting that the DSF assay was performed with 5% DMSO, while BSI had 1% DMSO. Stabilization of protein to thermal-induced motion requires a much greater degree of site occupancy, so DSF reports on saturating concentrations. In contrast, BSI signals are generated in proportion to binding site occupancy, giving a true Kd value for a given compound; however, it is also possible that additional binding sites other than the crystallographic binding site are occupied under these conditions. Note that the label-free nature of BSI is necessary for such small molecules as 1F1 and 1F1-N: the addition of any label other than a radioisotope would significantly change the properties of the compounds.
Figure 6.

Back-scattering interferometry for (a) AR-PR:pepstatin + 1F1, (b) apo-PR + 1F1, (c) AR- PR:pepstatin + 1F1-N, and (d) apo-PR + 1F1-N. Note that all Kd values are equal within experimental error, but the phase change is significantly greater for compound binding to apo-PR.
HIV Protease Activity Assay
Inhibition of PR activity by 1F1 and 1F1-N were tested using a fluorogenic peptide substrate. The fluorescence of 1F1 overwhelms the signal of the assay at high μM concentrations. No significant inhibition of PR was observed for 1F1-N up to 1mM; nor was a significant increase observed for the inhibition of PR by pepstatin in the presence of 1F1-N (Supplemental Figure 8). However, our long-term goal is to develop larger, higher affinity compounds from these fragment-like starting points. Growing these fragments will enable additional contacts with the protein, increasing their potential for inhibition. Since it is also possible that the 1F1 binding site is involved in protein-protein interactions, larger derivatives may show effects in more sophisticated assays using full-length Gag domains.
Fragment-optimization and future directions
The common structural features arising from different binding modes of 1F1 and 1F1-N can be exploited to design larger molecules. The hydrophobic core of the fragments is stabilized by hydrophobic and van der Waals interactions with Trp42, Pro44 and Lys55 side chains, while its orientation depends on the hydrogen bond acceptor-donor pattern with Val56 and Arg57, respectively (Figure 7). This binding mode places the molecules along a groove roughly delimited by Met46 and Pro79, defining two main directions where molecules can be grown to further stabilize current interactions and possibly extend the compound’s interaction network.
Figure 7.
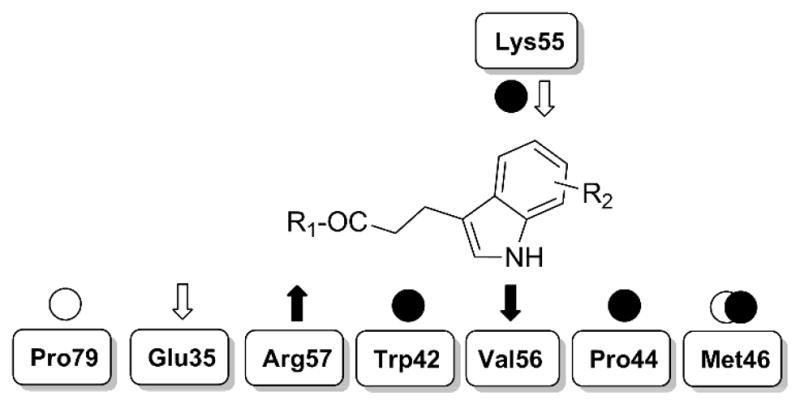
Schematic representation of essential interactions identified in the crystallographic structures and potentially exploitable ones. Experimental interactions are tagged with filled arrows and circles; interactions exploitable by chemical derivatives are shown with empty arrows and circles. Arrows = h-bonds; circles = vdW interactions.
Both fragments establish a single hydrogen bond with Arg57, suggesting that the carboxyl moiety could be esterified while keeping the key hydrogen bond. Larger groups could reach and engage the cavity enclosed by Arg57, Glu35 and Pro79. An opportune hydrogen bond donor here could also help establish a directional interaction with Glu35, contributing to destabilization of its salt bridge with Arg57. On the other side of the groove, hydrogen bonding with Val56 orients the fragment’s aromatic rings so they interact with Pro44, Lys55 and Met46. Substituents on the rings could be used to extend contacts with these residues. In particular, a hydrogen bond acceptor group could interact with the amino group of Lys55, strengthening the binding. Additionally, hydrophobic groups could further extend the molecules toward Met46, increasing close contacts and stabilizing its side chain.
Such modifications could improve overall binding, and might lead to a compound with observable protease inhibition activity. Also, since Pro79 is fairly close to the active site (i.e., approximately 10 Å away from one of the benzyl groups of TL-3), molecular hybrids linking 1F1-site binders and active site inhibitors could be designed to test the hypothesis of cooperative binding.
Conclusions
Allosteric regulation of HIV protease activity is a novel potential way to limit the development of drug-resistance. We show here that indole-carboxylate small molecules occupy an external site on the protease in both the solid state (by X-ray crystallography) and in solution (by measurements of conformational stabilization and direct site occupancy). Future work will focus on expanding these molecules into larger compounds with observable inhibition of PR.
MATERIALS AND METHODS
Expression and purification
Cloning of wild type NL4-3 HIV-1 PR was described previously(47). The autolysis-resistant HIV-1 protease (Q7K, L33I, L63I, C67A, C95A)(21), used for apo structures, was constructed using multiple rounds of the QuikChange site-directed mutagenesis protocol. For both variants, protein expression was induced in E. Coli. Inclusion bodies were isolated by centrifugation, solubilized, and purified by ion-exchange chromatography and FPLC. PR was dialyzed against 10 mM sodium acetate buffer, pH 5.2, 0.1% 2-mercaptoethanol prior to filtration and concentration to 3–5mg/mL. The purified PR was separated using SDS-PAGE, and purity was verified by Western blot using rabbit antiserum against HIV PR. For further details, see Supporting Information.
In Silico Docking
The structural features of 1F1 were used to search for potential derivatives to be screened by molecular docking. The ZINC online database (version 11)(49) was filtered for compounds containing both the benzindole ring and at least one carboxylic acid group. The results of this first filtering round were refined by rejecting molecules that do not have the properties of a fragment hit(MW:<=300; HB donors: <=3; HB acceptors: <=6), leading to a set of 2499 commercially available compounds. Finally, this set was prepared with AutoDock|Raccoon (available online at http://autodock.scripps.edu/resources/raccoon) and docked with AutoDock v4.2(17) on the HIV protease monomer bound to 1F1 (PDB_id: 3KFR, chain B). Docking results were ranked and filtered by using 1F1 docking score (−5.57 Kcal mol-1) and ligand efficiency (−0.43) as references. Ligands with docking score of −4.50 Kcal mol-1 or better were visually inspected to obtain list of the top 45 hits, and 22 of them were purchased and submitted to crystallization-based screening. For further details, see Supporting Information.
Crystallization and data collection
Crystallization was performed by vapor-diffusion in 96-well or 24-well plates at 18°C. For all co-crystallization experiments, protein samples were prepared by adding 10% of a 200 mM solution of the compound of interest in DMSO to the protein sample. For the inhibited forms of PR, pepstatin or TL-3 were added prior to the addition of compound. Screens of apo-PR were always conducted using the autolysis-resistant variant. Samples were centrifuged to remove any insoluble inhibitor or compound prior to crystallization. Crystallization conditions were obtained using commercially available 96-well screens. Conditions for each of the crystal forms are summarized in Table 1. For further details of cryoprotection, data collection, and data processing, see Supporting Information.
Differential Scanning Fluorimetry
Differential scanning fluorimetry (DSF) measurements were taken using a LightCycler 480 Real-Time PCR System (Roche) using 465 nm excitation and 580 nm emission wavelengths. The temperature was increased from 25°C to 95°C at a rate of 0.06°C/s, with 10 acquisitions/°C.
Samples were loaded into white 96-well plates. Each well contained a total volume of 20 μL consisting of 4 μM apo-PR, 10 mM HEPES at pH 7.5, 150 mM NaCl, 20X SYPRO Orange (Invitrogen), and 5% DMSO. Melting temperature calculations were carried out using the LightCycler 480 Protein Melting Software (Roche).
Backscattering Interferometry
All experiments were performed in Dulbecco’s Phosphate Buffered Saline (pH 7.4) with 1% (v/v) DMSO. Samples of apo-PR (2 μM) were incubated in the presence and absence of fixed concentrations of pepstatin (4.8 μM) and varying concentrations of small molecule ligands (1F1, 1F1-N, and tryptophan) for 1 hour at 20°C. Samples were then deposited into the channels of a microfluidic chip for analysis using a custom-built backscattering interferometer (BSI). Microfluidic chip surfaces were regenerated between uses by brief rinsing with a 3:2 solution of methanol and deionized water. The microfluidic devices were maintained at 25°C using a feedback-controlled peltier system. The high contrast interference fringes produced by each sample were generated using a fiber-coupled HeNe laser and recorded on a CCD camera. Measurements were analyzed using a combination of in-house software, Microsoft Excel and OriginPro.
Supplementary Material
Acknowledgments
This research was supported by Program Project grant P01 GM083658-05 from the National Institutes of Health, National Institute for General Medical Sciences. T. Tiefenbrunn was funded by the Molecular Basis of Viral Pathogenesis Training Grant, 2T32AI007354. Assistance provided by the support staff at the Stanford Synchrotron Radiation Lightsource (SSRL) for data collection is greatly appreciated. We thank the Scripps High-Performance Computing for the support provided to Garibaldi HPC Linux cluster. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program (P41RR001209), and the National Institute of General Medical Sciences.
Footnotes
ACCESSION NUMBERS: Coordinates and structure factors have been deposited in the Protein Data Bank with accession numbers 4EJ8, 4EJD, 4EJK, and 4EJL.
This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Wheeler WH, Ziebell RA, Zabina H, Pieniazek D, Prejean J, Bodnar UR, Mahle KC, Heneine W, Johnson JA, Hall HI. Prevalence of transmitted drug resistance associated mutations and HIV-1 subtypes in new HIV-1 diagnoses, U.S.-2006. AIDS. 2010;24:1203–1212. doi: 10.1097/QAD.0b013e3283388742. [DOI] [PubMed] [Google Scholar]
- 2.UNAIDS report on the global AIDS epidemic 2010. Joint United Nations Programme on HIV/AIDS (UNAIDS); Geneva: 2010. [Google Scholar]
- 3.Shen L, Siliciano RF. Viral reservoirs, residual viremia, and the potential of highly active antiretroviral therapy to eradicate HIV infection. J Allergy Clin Immunol. 2008;122:22–28. doi: 10.1016/j.jaci.2008.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ali A, Bandaranayake RM, Cai Y, King NM, Kolli M, Mittal S, Murzycki JF, Nalam MN, Nalivaika EA, Ozen A, Prabu-Jeyabalan MM, Thayer K, Schiffer CA. Molecular Basis for Drug Resistance in HIV-1 Protease. Viruses. 2011;2:2509–2535. doi: 10.3390/v2112509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wensing AM, van Maarseveen NM, Nijhuis M. Fifteen years of HIV Protease Inhibitors: raising the barrier to resistance. Antiviral Res. 2009;85:59–74. doi: 10.1016/j.antiviral.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 6.De Clercq E. Perspectives of non-nucleoside reverse transcriptase inhibitors (NNRTIs) in the therapy of HIV-1 infection. Il Farmaco. 1999;54:26–45. doi: 10.1016/s0014-827x(98)00103-7. [DOI] [PubMed] [Google Scholar]
- 7.Lee GM, Craik CS. Trapping moving targets with small molecules. Science. 2009;324:213–215. doi: 10.1126/science.1169378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kovalevsky AY, Liu F, Leshchenko S, Ghosh AK, Louis JM, Harrison RW, Weber IT. Ultra-high resolution crystal structure of HIV-1 protease mutant reveals two binding sites for clinical inhibitor TMC114. J Mol Biol. 2006;363:161–173. doi: 10.1016/j.jmb.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kovalevsky AY, Ghosh AK, Weber IT. Solution kinetics measurements suggest HIV-1 protease has two binding sites for darunavir and amprenavir. J Med Chem. 2008;51:6599–6603. doi: 10.1021/jm800283k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kovalevsky AY, Louis JM, Aniana A, Ghosh AK, Weber IT. Structural evidence for effectiveness of darunavir and two related antiviral inhibitors against HIV-2 protease. J Mol Biol. 2008;384:178–192. doi: 10.1016/j.jmb.2008.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prabu-Jeyabalan M, Nalivaika E, Schiffer CA. Substrate shape determines specificity of recognition for HIV-1 protease: analysis of crystal structures of six substrate complexes. Structure. 2002;10:369–381. doi: 10.1016/s0969-2126(02)00720-7. [DOI] [PubMed] [Google Scholar]
- 12.Nalam MN, Ali A, Altman MD, Reddy GS, Chellappan S, Kairys V, Ozen A, Cao H, Gilson MK, Tidor B, Rana TM, Schiffer CA. Evaluating the substrate-envelope hypothesis: structural analysis of novel HIV-1 protease inhibitors designed to be robust against drug resistance. J Virol. 2010;84:5368–5378. doi: 10.1128/JVI.02531-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perryman AL, Lin JH, McCammon JA. HIV-1 protease molecular dynamics of a wild-type and of the V82F/I84V mutant: Possible contributions to drug resistance and a potential new target site for drugs. Protein Science. 2004;13:1108–1123. doi: 10.1110/ps.03468904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Azam M, Powers JT, Einhorn W, Huang WS, Shakespeare WC, Zhu X, Dalgarno D, Clackson T, Sawyer TK, Daley GQ. AP24163 inhibits the gatekeeper mutant of BCR-ABL and suppresses in vitro resistance. Chem Biol Drug Des. 2010;75:223–227. doi: 10.1111/j.1747-0285.2009.00911.x. [DOI] [PubMed] [Google Scholar]
- 15.Zhang J, Adrian FJ, Jahnke W, Cowan-Jacob SW, Li AG, Iacob RE, Sim T, Powers J, Dierks C, Sun F, Guo GR, Ding Q, Okram B, Choi Y, Wojciechowski A, Deng X, Liu G, Fendrich G, Strauss A, Vajpai N, Grzesiek S, Tuntland T, Liu Y, Bursulaya B, Azam M, Manley PW, Engen JR, Daley GQ, Warmuth M, Gray NS. Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature. 2010;463:501–506. doi: 10.1038/nature08675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perryman AL, Zhang Q, Soutter HH, Rosenfeld R, McRee DE, Olson AJ, Elder JE, Stout CD. Fragment-based screen against HIV protease. Chem Biol Drug Des. 2010;75:257–268. doi: 10.1111/j.1747-0285.2009.00943.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heaslet H, Rosenfeld R, Giffin M, Lin Y-C, Tam K, Torbett BE, Elder JH, McRee DE, Stout CD. Conformational flexibility in the flap domains of ligand-free HIV protease. Acta Crystallographica Section D. 2007;63:866–875. doi: 10.1107/S0907444907029125. [DOI] [PubMed] [Google Scholar]
- 19.Fitzgerald PM, McKeever BM, VanMiddlesworth JF, Springer JP, Heimbach JC, Leu CT, Herber WK, Dixon RA, Darke PL. Crystallographic analysis of a complex between human immunodeficiency virus type 1 protease and acetyl-pepstatin at 2.0-A resolution. J Biol Chem. 1990;265:14209–14219. [PubMed] [Google Scholar]
- 20.Heaslet H, Kutilek V, Morris GM, Lin YC, Elder JH, Torbett BE, Stout CD. Structural insights into the mechanisms of drug resistance in HIV-1 protease NL4–3. J Mol Biol. 2006;356:967–981. doi: 10.1016/j.jmb.2005.11.094. [DOI] [PubMed] [Google Scholar]
- 21.Mildner AM, Rothrock DJ, Leone JW, Bannow CA, Lull JM, Reardon IM, Sarcich JL, Howe WJ, Tomich CS, Smith CW, et al. The HIV-1 protease as enzyme and substrate: mutagenesis of autolysis sites and generation of a stable mutant with retained kinetic properties. Biochemistry. 1994;33:9405–9413. doi: 10.1021/bi00198a005. [DOI] [PubMed] [Google Scholar]
- 22.Scott WR, Schiffer CA. Curling of flap tips in HIV-1 protease as a mechanism for substrate entry and tolerance of drug resistance. Structure. 2000;8:1259–1265. doi: 10.1016/s0969-2126(00)00537-2. [DOI] [PubMed] [Google Scholar]
- 23.Katoh E, Louis JM, Yamazaki T, Gronenborn AM, Torchia DA, Ishima R. A solution NMR study of the binding kinetics and the internal dynamics of an HIV-1 protease-substrate complex. Protein Science. 2003;12:1376–1385. doi: 10.1110/ps.0300703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Freedberg DI, Ishima R, Jacob J, Wang YX, Kustanovich I, Louis JM, Torchia DA. Rapid structural fluctuations of the free HIV protease flaps in solution: Relationship to crystal structures and comparison with predictions of dynamics calculations. Protein Science. 2002;11:221–232. doi: 10.1110/ps.33202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ishima R, Freedberg DI, Wang YX, Louis JM, Torchia DA. Flap opening and dimer-interface flexibility in the free and inhibitor-bound HIV protease, and their implications for function. Structure with Folding & Design. 1999;7:1047–1055. doi: 10.1016/s0969-2126(99)80172-5. [DOI] [PubMed] [Google Scholar]
- 26.Galiano L, Bonora M, Fanucci GE. Interflap distances in HIV-1 protease determined by pulsed EPR measurements. J Am Chem Soc. 2007;129:11004–11005. doi: 10.1021/ja073684k. [DOI] [PubMed] [Google Scholar]
- 27.Ding F, Layten M, Simmerling C. Solution structure of HIV-1 protease flaps probed by comparison of molecular dynamics simulation ensembles and EPR experiments. J Am Chem Soc. 2008;130:7184–7185. doi: 10.1021/ja800893d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kear JL, Blackburn ME, Veloro AM, Dunn BM, Fanucci GE. Subtype polymorphisms among HIV-1 protease variants confer altered flap conformations and flexibility. J Am Chem Soc. 2009;131:14650–14651. doi: 10.1021/ja907088a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blackburn ME, Veloro AM, Fanucci GE. Monitoring inhibitor-induced conformational population shifts in HIV-1 protease by pulsed EPR spectroscopy. Biochemistry. 2009;48:8765–8767. doi: 10.1021/bi901201q. [DOI] [PubMed] [Google Scholar]
- 30.Torbeev VY, Raghuraman H, Hamelberg D, Tonelli M, Westler WM, Perozo E, Kent SB. Protein conformational dynamics in the mechanism of HIV-1 protease catalysis. Proc Natl Acad Sci U S A. 2011;108:20982–20987. doi: 10.1073/pnas.1111202108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Torbeev VY, Raghuraman H, Mandal K, Senapati S, Perozo E, Kent SB. Dynamics of “flap” structures in three HIV-1 protease/inhibitor complexes probed by total chemical synthesis and pulse-EPR spectroscopy. J Am Chem Soc. 2009;131:884–885. doi: 10.1021/ja806526z. [DOI] [PubMed] [Google Scholar]
- 32.Pillai B, Kannan KK, Hosur MV. 1.9 A x-ray study shows closed flap conformation in crystals of tethered HIV-1 PR. Proteins. 2001;43:57–64. doi: 10.1002/1097-0134(20010401)43:1<57::aid-prot1017>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 33.Das A, Mahale S, Prashar V, Bihani S, Ferrer JL, Hosur MV. X-ray Snapshot of HIV-1 Protease in Action: Observation of Tetrahedral Intermediate and Short Ionic Hydrogen Bond SIHB with Catalytic Aspartate. Journal of the American Chemical Society. 2010;132:6366–6373. doi: 10.1021/ja100002b. [DOI] [PubMed] [Google Scholar]
- 34.Kumar M, Kannan KK, Hosur MV, Bhavesh NS, Chatterjee A, Mittal R, Hosur RV. Effects of remote mutation on the autolysis of HIV-1 PR: X-ray and NMR investigations. Biochemical and Biophysical Research Communications. 2002;294:395–401. doi: 10.1016/S0006-291X(02)00482-5. [DOI] [PubMed] [Google Scholar]
- 35.Mittal S, Cai Y, Nalam MNL, Bolon DNA, Schiffer CA. Hydrophobic Core Flexibility Modulates Enzyme Activity in HIV-1 Protease. Journal of the American Chemical Society. 2012;134:4163–4168. doi: 10.1021/ja2095766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bihani SC, Das A, Prashar V, Ferrer JL, Hosur MV. Resistance mechanism revealed by crystal structures of unliganded nelfinavir-resistant HIV-1 protease non-active site mutants N88D and N88S. Biochemical and Biophysical Research Communications. 2009;389:295–300. doi: 10.1016/j.bbrc.2009.08.138. [DOI] [PubMed] [Google Scholar]
- 37.Brynda J, Rezacova P, Fabry M, Horejsi M, Stouracova R, Sedlacek J, Soucek M, Hradilek M, Lepsik M, Konvalinka J. A Phenylnorstatine Inhibitor Binding to HIV-1 Protease: Äâ Geometry, Protonation, and Subsite àíPocket Interactions Analyzed at Atomic Resolution. Journal of Medicinal Chemistry. 2004;47:2030–2036. doi: 10.1021/jm031105q. [DOI] [PubMed] [Google Scholar]
- 38.Vedadi M, Niesen FH, Allali-Hassani A, Fedorov OY, Finerty PJ, Jr, Wasney GA, Yeung R, Arrowsmith C, Ball LJ, Berglund H, Hui R, Marsden BD, Nordlund P, Sundstrom M, Weigelt J, Edwards AM. Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc Natl Acad Sci U S A. 2006;103:15835–15840. doi: 10.1073/pnas.0605224103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protocols. 2007;2:2212–2221. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- 40.Rücker P, Horn A, Meiselbach H, Sticht H. A comparative study of HIV-1 and HTLV-I protease structure and dynamics reveals a conserved residue interaction network. Journal of Molecular Modeling. 2011;17:2693–2705. doi: 10.1007/s00894-011-0971-1. [DOI] [PubMed] [Google Scholar]
- 41.Bornhop DJ, Latham JC, Kussrow A, Markov DA, Jones RD, Sorensen HS. Free-solution, label-free molecular interactions studied by back-scattering interferometry. Science. 2007;317:1732–1736. doi: 10.1126/science.1146559. [DOI] [PubMed] [Google Scholar]
- 42.Latham JC, Stein RA, Bornhop DJ, McHaourab HS. Free-solution label-free detection of alpha-crystallin chaperone interactions by back-scattering interferometry. Anal Chem. 2009;81:1865–1871. doi: 10.1021/ac802327h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baksh MM, Kussrow AK, Mileni M, Finn MG, Bornhop DJ. Label-free quantification of membrane-ligand interactions using backscattering interferometry. Nat Biotechnol. 2011;29:357–360. doi: 10.1038/nbt.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kussrow A, Baksh MM, Bornhop DJ, Finn MG. Universal sensing by transduction of antibody binding with backscattering interferometry. Chembiochem. 2011;12:367–370. doi: 10.1002/cbic.201000671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kussrow A, Enders CS, Bornhop DJ. Interferometric methods for label-free molecular interaction studies. Anal Chem. 2011;84:779–792. doi: 10.1021/ac202812h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Olmsted IR, Xiao Y, Cho M, Csordas AT, Sheehan JH, Meiler J, Soh HT, Bornhop DJ. Measurement of aptamer-protein interactions with backscattering interferometry. Anal Chem. 2011;83:8867–8870. doi: 10.1021/ac202823m. [DOI] [PubMed] [Google Scholar]
- 47.Buhler B, Lin YC, Morris G, Olson AJ, Wong CH, Richman DD, Elder JH, Torbett BE. Viral evolution in response to the broad-based retroviral protease inhibitor TL-3. J Virol. 2001;75:9502–9508. doi: 10.1128/JVI.75.19.9502-9508.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee T, Laco GS, Torbett BE, Fox HS, Lerner DL, Elder JH, Wong CH. Analysis of the S3 and S3’ subsite specificities of feline immunodeficiency virus (FIV) protease: development of a broad-based protease inhibitor efficacious against FIV, SIV, and HIV in vitro and ex vivo. Proc Natl Acad Sci U S A. 1998;95:939–944. doi: 10.1073/pnas.95.3.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Irwin JJ, Shoichet BK. ZINC--a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Congreve M, Carr R, Murray C, Jhoti H. A ‘rule of three’ for fragment-based lead discovery? Drug Discov Today. 2003;8:876–877. doi: 10.1016/s1359-6446(03)02831-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.