

Abstract
Granulibacter bethesdensis is a Gram-negative pathogen in patients with Chronic Granulomatous Disease (CGD), a deficiency in the phagocyte NADPH oxidase. Repeated isolation of genetically identical strains from the same patient over years, and prolonged waxing and waning seropositivity in some subjects, raises the possibility of long-term persistence. G. bethesdensis resists killing by serum, CGD polymorphonuclear leukocytes (PMN), and antimicrobial peptides, indicating resistance to non-oxidative killing mechanisms. While G. bethesdensis extends the survival of PMN, persistent intracellular bacterial survival might rely on longer-lived macrophages and their precursor monocytes. Therefore, we examined phagocytic killing by primary human monocytes and monocyte-derived macrophages (MDM). Cells from both normal and CGD subjects internalized G. bethesdensis similarly. G. bethesdensis stimulated superoxide production in normal monocytes, but to a lesser degree than in normal PMN. Normal but not CGD monocytes and MDM killed G. bethesdensis and required in vitro treatment with interferon-γ (IFN-γ) to maintain this killing effect. Although in vitro IFN-γ did not enhance G. bethesdensis killing in CGD monocytes, it restricted growth in proportion to CGD PMN residual superoxide production, providing a potential method to identify patients responsive to IFN-γ therapy. In IFN-γ-treated CGD MDM, G. bethesdensis persisted for the duration of the study (7 days) without decreasing viability of the host cells. These results indicate that G. bethesdensis is highly resistant to oxygen-independent microbicides of myeloid cells, requires an intact NADPH oxidase for clearance, and can persist long-term in CGD mononuclear phagocytes, likely relating to the persistence of this microorganism in infected CGD patients.
Introduction
Granulibacter bethesdensis, a member of the Acetobacteraceae family, is a human pathogen that causes infection in patients with Chronic Granulomatous Disease (CGD), a deficiency in the NADPH oxidase (1, 2). This Gram-negative, rod-shaped bacterium has been isolated from at least 6 CGD patients in the United States and Spain (3–5). G. bethesdensis can cause recurrent infections in a CGD patient over several years that may be the result of reinfection with different bacterial strains or reactivation of the same strain (3, 4). This organism displays a high degree of resistance to host defenses as evidenced by isolation from spleens of wild-type mice 76 days after infection and from excised lymph nodes of a CGD patient 5 months after the onset of symptoms and unresponsiveness to antimicrobial treatment (3). The increasing numbers of clinical infections caused by G. bethesdensis as well as other species of acetic acid bacteria (6–12) underscore the need for a better understanding of the microbial pathogenesis of the Acetobacteraceae family.
Previous work has demonstrated that G. bethesdensis is serum-resistant and internalized by human peripheral blood polymorphonuclear leukocytes (PMN) in a complement-dependent manner (13). CGD PMN were incapable of killing G. bethesdensis, suggesting that this organism is resistant to non-oxidative killing mechanisms. PMN are short-lived cells that undergo apoptosis and efferocytosis by macrophages for inflammation resolution (14). Delayed PMN apoptosis and defective macrophage efferocytosis are observed in CGD (15, 16). Given that G. bethesdensis persists for 24 hours in CGD PMN, in contrast to ~50% clearance of the organism by normal PMN in the same time frame (13), we hypothesized that long-term intracellular bacterial survival in the host might rely on longer-lived macrophages and precursor monocytes. Here we show for the first time that normal monocytes and monocyte-derived macrophages (MDM) internalize G. bethesdensis and kill it in an NADPH oxidase-dependent manner while this organism persists and proliferates in CGD monocytes and MDM. Resistance of G. bethesdensis to NADPH oxidase-independent myeloid host defenses likely plays an important role in long-term infections observed in CGD patients.
Materials and Methods
Ethics statement and cell isolation
Blood samples were obtained after informed consent from healthy and CGD donors enrolled at the NIH Clinical Center as defined in NIH institutional review board-approved protocols. Human polymorphonuclear leukocytes (PMN) were isolated from citrated peripheral blood as previously described (17). For the monocyte isolation, every 10 ml of citrated peripheral blood was diluted 1:3 in HBSS(−) and layered on 15ml of 53% percoll (diluted in PBS without Ca2+, Mg2+). PBMC layers were collected and cells washed twice in HBSS(−). Monocytes were isolated with CD14 antibody-coupled magnetic beads according to the manufacturer’s instructions (Miltenyi Biotec, Auburn, CA). Monocyte purity (~85–95%) was confirmed using CD14 surface staining and flow cytometry as well as differential staining and light microscopy. In some cases, CD14+ cells were further differentiated into monocyte-derived macrophages (MDM) by culturing in RPMI 1640 containing 2mM L-glutamine, 10mM HEPES, pH 7.2, 10% autologous serum, and 50 ng/ml M-CSF (Invitrogen, Carlsbad, CA) for 7 days. Media was replaced every 3–4 days. Where indicated, monocytes and MDM were treated with 65 units IFN-γ/ml (eBioscience, San Diego, CA) or PBS control for 2 days. Serum was collected from BD serum separator tubes according to the manufacturer’s instructions and used fresh or snap frozen and stored at −80°C.
Bacterial strains and cultures
G. bethesdensis NIH1.1 was cultured for 2 days and subcultured for 1 day to mid-log phase in YPG medium (13). Escherichia coli TOP10 was grown for 1 day and subcultured for 3 hours to mid-log phase in Lysogeny broth. Washed bacteria were enumerated by OD 600nm using a NanoDrop® ND-1000 (Grace Scientific, LLC, Clarksburg, MD).
Bacterial internalization assays
Cytospins
Freshly isolated monocytes (2.5 × 105/condition) were infected with G. bethesdensis for 1 hr at the indicated multiplicity of infections (MOIs) in 96-well plates. Plates had been precoated with 10% autologous serum for at least 1 hr at 37ºC and washed three times with HBSS(−). Cells were incubated with no serum, autologous serum, or autologous serum incubated for 30 min at 56°C, which inactivates complement. Heat-inactivated serum was centrifuged at 10,000 × g for 10 min at 4°C to pellet protein aggregates. Plates were centrifuged at 362 × g for 8 min at 4ºC to synchronize phagocytosis. Cells were removed with Cellstripper™ (Mediatech, Inc., Manassas, VA), subjected to cytospin, and differentially stained with HARLECO Hemacolor stain set (EMD Chemicals, Billerica, MA). Each sample was imaged and intracellular bacteria scored in a blinded manner.
Confocal and epifluorescence microscopy
3 × 105 or 5 × 105 freshly isolated monocytes were adhered to 12 mm or 18 mm ethanol-washed cover glasses in 24-well or 12-well plates, respectively. G. bethesdensis was labeled with pHrodo™ Red succinimidyl ester (Invitrogen) according to the manufacturer’s instructions. pHrodo-labeled bacteria were incubated with cells at the indicated MOIs and time points in the presence of serum or heat-treated serum (as described above) in RPMI 1640 containing 25 mM HEPES, pH 7.2. Plates were centrifuged at 362 × g for 8 min at 4ºC to synchronize phagocytosis. Cells were washed with cold PBS twice, fixed with 4% PFA for 5 min at RT in the dark, and washed with cold PBS or HBSS twice. Cover glasses were mounted on glass slides using VECTASHIELD® Mounting Medium with DAPI (Vector Laboratories, Inc., Burlingame, CA), or 1 μg/ml DAPI was added to the last wash prior to mounting, and imaged using a Leica SP5 X-WLL confocal microscope or a Leica Episcope with a DFC360FX camera (Leica Microsystems). For kinetic analyses, at least 100 cells were scored for the presence of internalized pHrodo bacteria using Imaris software (Bitplane, South Windsor, CT).
Luminol-enhanced chemiluminescence
Bacterial activation of PMN or monocyte luminol-enhanced chemiluminescence was measured at the indicated MOIs in an assay composed of PMN or monocytes at 1 × 106/ml in RPMI 1640 medium with 10% autologous serum, 25mM HEPES (pH 7.4), and 50 μM luminol essentially as previously described (13).
Bacterial killing assays
Freshly isolated PMN, freshly isolated monocytes, or 2-day old monocytes or MDM treated with 65U IFN-γ/ml or PBS control were infected with G. bethesdensis (pre-opsonized with serum) in 96-well plates for the indicated times and at the indicated MOIs. Plates were pre-coated with 10% autologous serum as described above. At each time point, a final concentration of 0.5% saponin was added per well. Plates were incubated on ice for 10 min before shearing the samples 10 times through a 28½-gauge needle to lyse phagocytes. Samples were diluted in saline, spread on YPG agar plates, and incubated 3–4 days at 37°C. In some cases, MDM were grown in 35 mm petri dishes, treated with IFN-γ for 2 days, and infected with pre-opsonized G. bethesdensis for 1 week before harvesting with Cellstripper™. Cells were then subjected to cytospin, differentially stained, and imaged via light microscope.
Cytokine analysis
Freshly isolated monocytes (1 × 105/200 μl/well) or MDM (5 × 104/200 μl/well) were not infected or infected with G. bethesdensis or E. coli at an MOI of 1 or 0.25, respectively, in 96-well plates (pre-coated with 10% autologous serum as described above) in the presence of 10 % autologous serum for 24 hours. Supernatants were collected and frozen until analysis. Thawed supernatants were diluted 1:3 and cytokines measured using a Bio-plex Pro 17-plex Human Cytokine Assay Kit analyzed on a Bio-plex system 200 running Bio-plex Manager (Version 5.0) according to the manufacturer’s instructions (Bio-rad, Hercules, CA).
XTT viability assay
Freshly isolated monocytes (2 × 105/200 μl/well) were infected with G. bethesdensis at the indicated MOIs in 96-well plates (pre-coated with 10% autologous serum as described above) in the presence of autologous serum for 24 hrs before addition of 50 μl XTT reagent (1 mg/ml XTT, 25 μM phenazine methosulfate). After 4 hrs, the absorbance was measured at 450 nm with a Beckman Coulter DTX 880 Multimode Detector and the background OD of media alone was subtracted to give corrected OD.
Data analyses
Flow cytometry was performed on a BD FACS Canto™ and the data analyzed using FlowJo (Tree Star, Inc., Ashland, OR). All other data was graphed and analyzed statistically using GraphPad Prism (version 5, GraphPad Software, La Jolla, CA). For statistical tests, * = p≤0.05, ** = p≤0.01, *** = p≤0.001, **** = p ≤ 0.0001.
Results
Internalization of opsonized G. bethesdensis by CD14+ monocytes
Serum contains opsonins, including heat-stable antibodies and heat-labile complement proteins, which coat microbes and can trigger their uptake by phagocytes. We tested whether normal CD14+ monocytes internalize G. bethesdensis and whether this process was serum-dependent. Most bacteria were internalized in the presence of serum after 1 hour at multiplicity of infections (MOIs) of 10:1 (Fig. 1A) and 20:1 (data not shown) as assessed by light microscopy. Little internalization occurred in the absence of serum and there was about a 50% reduction in internalization by normal monocytes incubated with heat-treated serum. Confocal microscopy of normal monocytes incubated with G. bethesdensis labeled with pHrodo, a dye that fluoresces more intensely at an acidic pH, also demonstrated greater internalization of bacteria in the presence of serum but not heat-treated serum (Fig. 1B). CGD monocytes internalized a similar amount of bacteria as normal monocytes in the presence of fresh serum (p=0.114, Mann Whitney test) and less so in the presence of heat-treated serum (p=0.081, Mann Whitney test) (Fig. 1A). CGD monocytes followed a similar time course of pHrodo-bacteria internalization as normal monocytes, with maximal uptake by 30 minutes post-infection (Fig. 1C). Maximal internalization at 30 minutes was also confirmed by scoring the number of pHrodo-labeled bacteria per cell (data not shown). Little to no internalization occurred in the absence of serum for either cell type (data not shown). These results show that monocytes internalize G. bethesdensis and this is partially dependent on a temperature-sensitive serum component for normal monocytes.
FIGURE 1.

Serum-dependent internalization of G. bethesdensis by CD14+ monocytes. (A) CD14+ monocytes from normal (n=5–12) and CGD (n=5) donors were incubated with G. bethesdensis at an MOI of 10 bacteria per host cell for 1 hour in the absence of serum, presence of autologous serum or heat-treated (HT, 56°C, 30 min) serum as described in methods (mean ± SD, non-parametric t-tests where ** = p ≤ 0.01). Internalization of bacteria by CGD monocytes in the absence of serum was not determined. (B) Confocal microscopy imaging of normal CD14+ monocytes incubated with pHrodo-labeled G. bethesdensis (red) at an MOI of 10 bacteria per host cell for 1 hour in the presence of autologous serum or heat-treated serum. Nuclei are labeled with DAPI (blue). Bar length = 50 μm. (C) CD14+ monocytes from normal (n=3) and CGD (n=3) donors were incubated with pHrodo-labeled G. bethesdensis at an MOI of 1 bacterium per host cell for the indicated time points in the presence of autologous serum. Epifluorescence images were scored and data represented as mean ± SD.
G. bethesdensis activation of the NADPH oxidase in CD14+ monocytes
The phagocyte NADPH oxidase (NOX2), the enzyme complex defective in CGD, is activated by microbes and their products, and normally generates superoxide that is transformed into a variety of microbicidal reactive oxygen species (ROS) like hydrogen peroxide and hypohalous acids (18). Previous studies showed that G. bethesdensis was less effective at activating normal PMN NOX2 than E. coli (13). We measured normal monocyte superoxide production in response to G. bethesdensis and E. coli using luminol-enhanced chemiluminescence. A dose-dependent luminol-enhanced chemiluminescence response occurred when normal monocytes were incubated with G. bethesdensis (Fig. 2A and 2B). This response was dampened in the absence of serum (data not shown) suggesting that phagocytosis of G. bethesdensis or phagocyte interaction with serum components such as complement is at least partially required for the generation of superoxide in monocytes. E. coli was equivalently stimulatory compared to G. bethesdensis (Fig. 2B), but the kinetics of the response differed (Fig. 2A). The relative lag in response to G. bethesdensis (Fig. 2A) mirrored the kinetics of internalization (Fig. 1C).
FIGURE 2.

G. bethesdensis induces oxidative burst activity in normal CD14+ monocytes. (A) Luminol-enhanced chemiluminescence was measured in monocytes exposed to control buffer, G. bethesdensis or E. coli at MOIs of 100 and 10 bacteria per host cell in the presence of autologous serum. Data are mean + SD for 7 donors. (B) Data from (A) are represented as area under the curve (AUC). (C) Luminol-enhanced chemiluminescence was measured in PMN and monocytes, isolated from the same donors (n=6), exposed to control buffer, G. bethesdensis or E. coli at an MOI of 100 bacteria per host cell in the presence of autologous serum. Data are mean + SD. (D) AUC data for PMN and monocytes, from the same donors (n=6), exposed to bacteria at MOIs of 100 (shown in (C)) and 10 bacteria per host cell in the presence of autologous serum.
In comparison to PMN, monocytes generated a weaker response to G. bethesdensis and E. coli (Fig. 2C and 2D). This difference in respiratory burst has been previously shown for PMN and monocytes stimulated with E. coli, PMA, or zymosan (19–22). It should also be noted that E. coli induced a biphasic response in monocytes and PMN (Fig. 2A and 2C). Based on experiments where the stimulus was mixed in with the cells versus gently added, it was determined that the first peak was due in part to mechanical stimulation during initial stimulus addition while the second peak occurred as a direct result of the stimulus interacting with the phagocytes (data not shown). Taken together, G. bethesdensis induces a respiratory burst in normal monocytes and to a greater extent in normal PMN.
NADPH oxidase-dependent killing of G. bethesdensis by CD14+ monocytes
Stimulation of a respiratory burst in monocytes has been linked to killing of internalized organisms such as Staphylococcus aureus (23). Given that G. bethesdensis induces superoxide in monocytes, we hypothesized that monocytes would also be able to kill G. bethesdensis. Normal monocytes killed G. bethesdensis in a dose-dependent manner after 24 hours of infection (Fig. 3A). In contrast, CGD monocytes were incapable of killing G. bethesdensis and, moreover, provided a suitable environment for growth of the organism (Fig. 3A). To exclude outgrowth of extracellular bacteria, gentamicin protection assays were performed. Although qualitatively similar results were obtained 1 hour post-infection, with or without gentamicin, almost complete killing occurred in both normal and CGD monocytes by 24 hours after gentamicin washout, indicating that this organism is sensitive to gentamicin internalized by monocytes prior to washing (data not shown). A comparison of PMN and monocytes isolated from the same normal donors showed that both cell types killed similar amounts of G. bethesdensis (Fig. 3B). Therefore, monocytes require the NADPH oxidase for the killing of G. bethesdensis and exhibit bactericidal activity on par with PMN.
FIGURE 3.
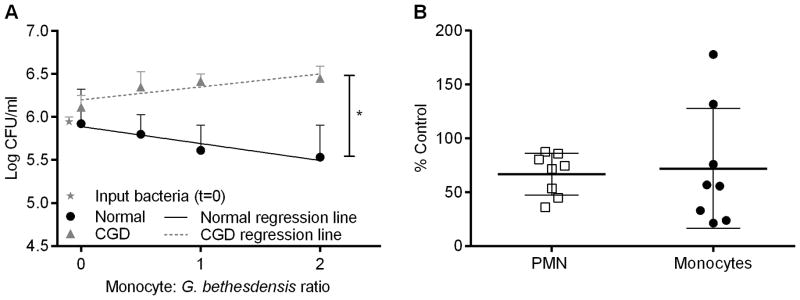
Freshly isolated normal but not CGD CD14+ monocytes kill G. bethesdensis. (A) CD14+ monocytes from normal (n=9) and CGD (n=3) donors were incubated with G. bethesdensis at MOIs of 2, 1, and 0.5 bacteria per host cell in the presence of autologous serum for 24 hours. Data are represented as mean + SD. Linear regression analysis was conducted to assess differences in slope of lines (* = p ≤ 0.05) for normal versus CGD. (B) PMN and monocytes isolated from the same normal donors (n=8) were incubated with G. bethesdensis at an MOI of 1 bacteria per host cell in the presence of autologous serum for 24 hours. Data are represented as % control input (mean ± SD).
Treatment of monocytes with interferon-gamma (IFN-γ) significantly enhances their microbicidal activities (24–26), possibly through the upregulation of NOX2 expression (27, 28). We tested whether treatment of normal monocytes with IFN-γ for two days altered their ability to kill G. bethesdensis. While normal monocytes cultured for two days without IFN-γ lost their ability to kill, IFN-γ treatment maintained the killing abilities of normal monocytes (Fig. 4A). Similar amounts of G. bethesdensis were recovered when the bacteria were incubated with PBS control or IFN-γ in the absence of monocytes (data not shown). G. bethesdensis-stimulated normal monocytes pre-treated with IFN-γ produced significantly more superoxide, as measured by chemiluminescence, compared to monocytes not receiving IFN-γ (data not shown), suggesting a link between induced superoxide and enhanced antibacterial activity after IFN-γ treatment. These data confirm that IFN-γ treatment is important for the maintenance of in vitro monocyte bactericidal activity and may augment phagocytic killing in vivo (29, 30).
FIGURE 4.
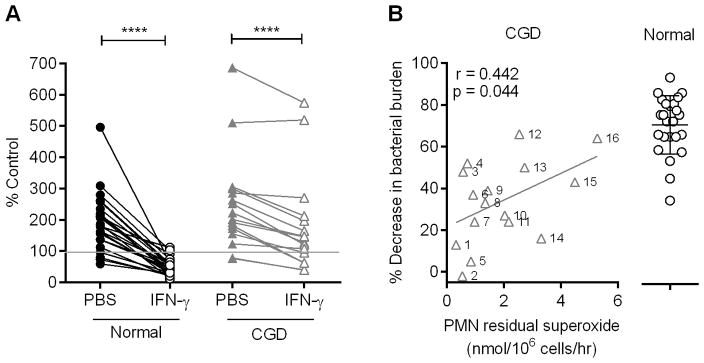
IFN-γ maintains microbicidal activity of normal monocytes and stimulates monocytes from some CGD subjects to control G. bethesdensis growth. (A) Monocytes from normal (n=24) and CGD (n=16) donors were treated with PBS control or 65U IFN-γ/ml for 2 days before infection with G. bethesdensis for 24 hours at an MOI of 1 bacterium per host cell in the presence of autologous serum. Data are represented as % control input. Significance testing of PBS control versus IFN-γ treatment was by Wilcoxon paired t-test (**** = p ≤ 0.0001). (B) Percentage decrease in bacterial burden after IFN-γ treatment relative to PBS control for all normal and CGD monocytes in panel (A). Percentages for CGD monocytes are plotted against residual superoxide in PMN from the same CGD patients. The relationship of the two variables was assessed using a Spearman correlation and the best fit (Y = 6.409X + 21.59) is depicted.
IFN-γ stimulates some CGD monocytes to control G. bethesdensis growth
Since IFN-γ maintains normal monocyte killing of G. bethesdensis, we next tested the capacity of IFN-γ-treated CGD monocytes to kill G. bethesdensis. When considered as a group, monocytes from CGD patients were responsive to IFN-γ treatment as indicated by significantly reduced bacterial burdens compared to CGD monocytes that had not received the cytokine (Fig. 4A). Interestingly, the magnitude of the monocyte response to IFN-γ varied considerably from patient to patient. Previous studies indicated that IFN-γ treatment can enhance superoxide production in CGD monocytes if there are detectable levels of residual superoxide at baseline (29, 31). Moreover, enhanced monocyte superoxide production accompanied bactericidal activity against S. aureus for a CGD patient receiving in vivo IFN-γ treatment (29). Given these previous studies, we predicted a priori a positive relationship between residual PMN ROS production (32) and monocyte killing of G. bethesdensis. Thus, we employed a one-tailed pvalue to test this prediction. A Spearman correlation demonstrated a positive relationship (r = 0.442) between reduction in bacterial burden and CGD neutrophil residual superoxide that was statistically significant (one-tailed p-value = 0.044) (Fig. 4B). The genetic background of these CGD patients is described in Table I. Understanding the mechanism of IFN-γ-induced bactericidal activity of CGD monocytes could prove a powerful diagnostic tool to determine the likelihood that IFN-γ treatment will benefit specific CGD patients.
Table I.
CGD patient subtype and specific mutation
| CGD Patient | Gene affected | DNA mutation | Protein mutation | Mutation type |
|---|---|---|---|---|
| 1 | gp91 | c.676 C>T | p.Arg226X | Nonsense |
| 2 | gp91 | c.483+1 G>C | Del exon 5 | Splice |
| 3 | gp91 | c.80-83 del TCTG | p.Val27GlyfsX33 | Frameshift |
| 4 | gp91 | c.742 dup A | p.Ile248fsX37 | Frameshift |
| 5 | gp91 | c.868 C>T | p.Arg290X | Nonsense |
| 6 | gp91 | c.676 C>T | p.Arg226X | Nonsense |
| 7 | gp91 | c.676 C>T | p.Arg226X | Nonsense |
| 8 | gp91 | c.675-1 G>A | Del exons 6 & 7 | Splice |
| 9 | p47 | Not determined | Not determined | Not determined |
| 10 | gp91 | c.909 C>A | p.His303Gln | Missense |
| 11 | gp91 | c.388 C>T | p.Arg130X | Nonsense |
| 12 | p47 | c.75_76 del GT | p.Tyr26fsX26 | Frameshift |
| 13 | gp91 | c.868 C>T | p.Arg290X | Nonsense |
| 14 | p47 | c.75_76 del GT | p.Tyr26HisfsX26 | Frameshift |
| 15 | p47 | Not determined | Not determined | Not determined |
| 16 | p47 | c.75_76 del GT | p.Tyr26HisfsX26 | Frameshift |
G. bethesdensis infection does not affect monocyte survival
We next assessed the impact of G. bethesdensis infection on monocyte viability using an XTT assay, which measures the reduction of XTT to an orange formazan product by metabolically active cells. No significant difference was detected between normal and CGD monocytes either uninfected or incubated for 24 hrs with G. bethesdensis at MOIs of 0.5:1, 1:1, and 10:1 in the presence of serum (Fig. 5), indicating that monocytes remain viable in the presence of G. bethesdensis.
FIGURE 5.

G. bethesdensis infection does not alter monocyte survival. Monocytes from normal (n=6) or CGD (n=3) donors were incubated alone or with G. bethesdensis at MOIs of 0.5, 1, or 10 bacteria per host cell in the presence of autologous serum for 24 hours. Cell viability was assessed using an assay that measures conversion of XTT to an orange formazan product by metabolically active cells (mean optical density + SD).
G. bethesdensis persistence in CGD monocyte-derived macrophages
Peripheral blood monocytes are precursors to macrophages, supplying tissues during steady-state conditions or in the setting of infection (34). Culturing CD14+ monocytes in the presence of macrophage colony-stimulating factor (M-CSF) for 1 week results in a macrophage-like phenotype (35). While normal monocyte-derived macrophages (MDM) enabled growth of G. bethesdensis, treatment of these cells for 2 days with IFN-γ generated MDM capable of controlling G. bethesdensis growth in a dose-dependent manner (Fig. 6A). CGD MDM were significantly less effective in controlling G. bethesdensis growth either in the absence or presence of IFN-γ (Fig. 6B). We confirmed via confocal microscopy that normal and CGD MDM internalized similar amounts of bacteria at various MOIs in the presence of serum after 1 hour (data not shown).
FIGURE 6.
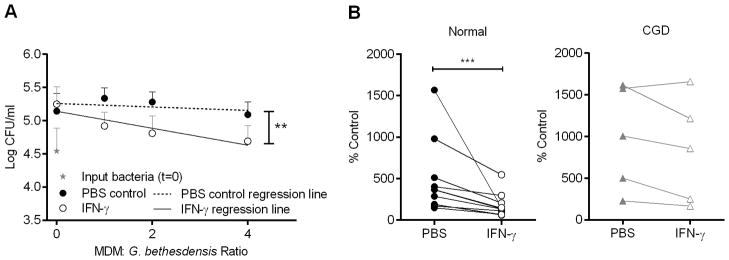
Normal but not CGD MDM control G. bethesdensis growth after 24 hours of infection. (A) Normal MDM were incubated ± 65 U IFN-γ/ml for 2 days before co-culture with G. bethesdensis for 24 hours at the indicated MOI in the presence of autologous serum. Data are presented as mean + SD (n=11 donors). Linear regression analysis was conducted to assess differences in slope of lines (** = p ≤ 0.01) for PBS control versus IFN-γ treatment. (B) MDM were treated as in (A) at an MOI of 0.25 bacteria per host cell. Data are % control input for 11 normal donors and 5 CGD donors. Wilcoxon paired t-test was used to compare PBS control versus IFN-γ treatment (*** = p ≤ 0.001).
Macrophages are highly phagocytic cells that support the persistence of several bacterial pathogens such as Salmonella typhimurium, Mycobacterium tuberculosis, Legionella pneumophila, and Brucella species (36–38). Given that normal MDM were incapable of controlling G. bethesdensis growth at the MOIs tested after 24 hours, unless supplemented with IFN-γ, we examined the survival of these bacteria after 1 week of co-culture with MDM. IFN-γ-treated normal MDM infected for 1 week killed G. bethesdensis at a MOI of 0.25 bacteria per host cell (Fig. 7A and 7B). However, IFN-γ-treated CGD MDM maintained a favorable environment for G. bethesdensis survival (Fig. 7B and 7C). No decrease in viability, as assessed by LDH assay and XTT assay, was observed for normal or CGD MDM infected with G. bethesdensis for 1 week compared to non-infected MDM incubated for the same time period (data not shown). Based on these findings, the CGD macrophage may contribute to the persistence of this bacterium.
FIGURE 7.

G. bethesdensis persists inside CGD MDM for 1 week. (A) Normal MDM were incubated ± 65 U IFN-γ/ml for 2 days before co-culture with G. bethesdensis for 1 week at the indicated MOI in the presence of autologous serum. Data are presented as mean + SD (n=6 donors). Linear regression analysis was conducted to assess differences in slope of lines (* = p ≤ 0.05) for PBS control versus IFN-γ treatment. (B) MDM were treated as in (A) at an MOI of 0.25 bacteria per host cell. Data are % control input for 6 normal donors and 6 CGD donors. Wilcoxon paired t-test was used to compare PBS control versus IFN-γ treatment (* = p ≤ 0.05). (C) IFN-γ-treated normal and CGD MDM infected for 1 week with G. bethesdensis at an MOI of 1 bacterium per host cell. Black arrows indicate the presence of bacteria.
G. bethesdensis induces cytokine release from normal and CGD monocytes and MDM
Pro-inflammatory cytokines initiate host responses during infection, whereas anti-inflammatory cytokines play a significant role during the resolution phase once infection has been contained. Increased pro-inflammatory and decreased anti-inflammatory cytokine production have been reported for activated CGD cells compared to activated normal cells in some settings (16, 39) whereas in others, the opposite trend was observed (40). We measured cytokine concentrations in supernatants from normal and CGD monocytes and MDM cultured with G. bethesdensis or E. coli for 24 hours. Both bacteria induced pro-inflammatory and anti-inflammatory cytokine release from all cell types tested with E. coli eliciting significantly greater responses than G. bethesdensis (Table II). No significant differences could be detected between non-infected normal and CGD cell cultures. However, CGD monocytes produced significantly more cytokine than normal monocytes when infected with either bacterium while MDM responses were similar in magnitude for normal versus CGD. There was little to no production of IL-1β (for MDM), IL2, IL-4, IL-5, IL-7, IL-12p70, IL-13, and GM-CSF (data not shown). Taken together, the lower cytokine responses of monocytes and MDM to G. bethesdensis, compared to E. coli, may help G. bethesdensis persist in these cells.
Table II.
Cytokine profiles of normal and CGD monocytes and MDM infected with G. bethesdensis or E. coli for 24 hours
| Monocytes | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Normal (n = 7) | CGD (n = 4) | P-values | ||||||||
|
| ||||||||||
| Cytokinea | (I) Control | (II) G. bethesdensis | (III) E. coli | (IV) Control | (V) G. bethesdensis | (VI) E. coli | II vs. IIIc | V vs. VIc | II vs. Vd | III vs. VId |
| IL-1β | 103 (52) | 608 (126) | 5129 (2502) | 74 (84) | 1088 (462) | 14244 (3620) | * | 0.13 | ** | * |
| IL-6 | 278 (180) | 5404 (2619) | 13462 (4781) | 2605 (3102) | 26699 (8989) | 31512 (18983) | * | ns | ** | * |
| IL-8 | 15089 (7437) | 22252 (3276) | 27901 (6308) | 9794 (8272) | 20904 (1738) | 27254 (5620) | 0.08 | 0.13 | ns | ns |
| IL-10 | 1 (1) | 38(47) | 127 (94) | 9(13) | 539 (147) | 528 (469) | * | ns | ** | ** |
| IL-17 | 29 (14) | 76 (13) | 111 (12) | 37 (33) | 93 (11) | 134 (13) | * | 0.13 | 0.11 | * |
| G-CSF | 10 (4) | 217 (160) | 662 (360) | 73 (108) | 2310 (895) | 1938 (1495) | * | ns | ** | * |
| IFN-γ | 82 (45) | 437 (119) | 815 (208) | 177 (187) | 714 (145) | 1154 (130) | * | 0.13 | * | ** |
| CCL-2 | 1587 (1390) | 3004 (950) | 1435 (1047) | 2334 (2218) | 2990 (794) | 873 (569) | * | 0.13 | ns | ns |
| CCL-4 | 1016 (601) | 3974 (1582) | 7047 (2495) | 2275 (2216) | 5155 (485) | 7634 (3947) | * | ns | ns | ns |
| TNF-α | 101 (87) | 2613 (2178) | 11868 (5249) | 223 (241) | 6814 (6075) | 39407 (12606) | * | 0.13 | * | ** |
|
| ||||||||||
| MDM | ||||||||||
| Normal (n=5-7) | CGD (n=4-5) | P-values | ||||||||
|
| ||||||||||
| Cytokineb | (I) Control (n=7) | (II) G. bethesdensis (n=7) | (III) E. coli (n=5) | (IV) Control (n=5) | (V) G. bethesdensis (n=5) | (VI) E. coli (n=4) | II vs. IIIc | V vs. VIc | II vs. Vd | III vs. VId |
|
| ||||||||||
| IL-6 | 2 (2) | 84 (95) | 1890 (1904) | 4 (2) | 60 (78) | 2834 (1912) | ** | * | ns | ns |
| IL-8 | 185 (117) | 1885 (1604) | 6667 (4561) | 436 (418) | 3264 (3083) | 11548 (6601) | * | 0.06 | ns | ns |
| IL-10 | 2 (2) | 10 (11) | 214 (182) | 5 (3) | 13 (10) | 940 (747) | * | * | ns | 0.11 |
| G-CSF | 2 (1) | 6 (4) | 81 (48) | 3 (1) | 8 (4) | 184 (137) | ** | * | ns | ns |
| IFN-γ | 4 (5) | 35 (32) | 151 (85) | 20 (15) | 47 (35) | 198 (91) | * | * | ns | ns |
| CCL-2 | 547 (398) | 972 (880) | 1318 (862) | 1979 (1104) | 2865 (1931) | 2602 (1862) | ns | ns | * | ns |
| CCL-4 | 139 (210) | 902 (1077) | 6330 (10096) | 136 (71) | 1111 (1476) | 8350 (6852) | ns | 0.11 | ns | ns |
| TNF-α | 3 (2) | 118 (114) | 5502 (6627) | 5 (4) | 115 (132) | 8274 (6333) | ** | * | ns | ns |
Cytokines from 5×105 monocytes/ml infected at an MOI of 1 are represented as mean (SD) (pg/ml).
Cytokines from 2.5×105 MDM/ml infected at an MOI of 0.25 are represented as mean (SD) (pg/ml).
Statistical comparisons by exact paired nonparametric Wilcoxon signed rank test,* =p ≤ 0.05, ns = not significant.
Statistical comparisons by exact unpaired nonparametric Mann-Whitney test, *=p ≤ 0.05, **=p ≤ 0.01, ns = not significant.
Discussion
Granulibacter bethesdensis is a bacterial pathogen that can infect patients with CGD. Isolation of genetically indistinguishable strains from individual subjects over 2–3 years, despite periods without clinically evident infections, may reflect either reinfection or reactivation of a latent infection. Persistence of bacteria has been suggested for a number of human diseases including Legionnaires’ disease (Legionella pneumophila) (41), tuberculosis (Mycobacterium tuberculosis) (42), and brucellosis (Brucella species) (43). G. bethesdensis shares several attributes with these microbes that may explain its ability to survive in the host for a long period of time: (1) resistance to killing by non-oxidative host defenses, (2) replication and survival inside host cells and (3) promotion of host cell survival.
The natural reservoir for G. bethesdensis and the mode of entry into the human host are unknown. Regardless of the site of entry, G. bethesdensis must resist a variety of innate immune host defenses to establish infection. G. bethesdensis is resistant to killing by human serum and the cathelicidin cationic antimicrobial peptide (13). Serum resistance has been linked to increased virulence in other Gram-negative bacilli such as L. pneumophila strain UH1 (44) and O-antigen-positive B. abortus strains (45), and many others. Virulence factors also enable L. pneumophila (46, 47), M. tuberculosis (48), and Brucella species (49) to resist killing by a variety of cationic antimicrobial peptides that compose a major first line of host defenses.
G. bethesdensis may also come into contact with innate immune phagocytes. One of the first cellular responders recruited to the site of bacterial infection is the neutrophil. In vitro studies show that healthy neutrophils internalize G. bethesdensis in a complement-dependent manner and kill ~50% of input bacteria after 24 hours of infection at an MOI of 1 (13). CGD neutrophils internalize G. bethesdensis normally but fail to kill the organism and instead control its growth, highlighting the importance of the NADPH oxidase in G. bethesdensis killing by PMN. Survival of this bacterium in neutrophils may be further enhanced by the ability of G. bethesdensis to inhibit neutrophil apoptosis (13), which would normally aid in the containment and clearance of the microbe by macrophages. Although the neutrophil may play an important role in survival of G. bethesdensis, it is relatively short-lived (~5–8-hour half-life in circulation (50)) compared to other innate immune phagocytes like monocytes (~3-day half-life in circulation (51)) and macrophages (~30-day half-life (52)).
Monocytes are recruited to the site of infection and undergo differentiation to macrophages within the infected tissue, which also houses resident macrophages. We demonstrate in this study that monocytes from healthy and CGD donors internalize G. bethesdensis. Although normal PMN internalization of G. bethesdensis relies almost entirely on a temperature-sensitive serum component (13), normal monocyte internalization only partially depends on a heat-labile serum component. This difference in phagocytic mechanism may be explained by the differential expression of Fc receptors on the two cell types (53) or the wider repertoire of receptors (e.g., scavenger receptors (54)) on monocytes. Differential expression or activation of phagocytic receptors, as well as pattern recognition receptors (PRRs) (55, 56), on PMN and monocytes may also explain the difference in relative magnitude of the respiratory burst in these two cell types, and in response to E. coli versus G. bethesdensis. For instance, PRRs dectin-1 and dectin-2 on inflammatory PMN and monocytes/macrophages have been shown to differentially contribute to fungal particle association and subsequent ROS production (57). The same study also showed that serum opsonization of zymosan or Candida albicans was important for interaction with PMN, but less so for monocytes/macrophages.
One downstream effect of phagocytosis and PRR activation is the production of cytokines, which contribute to the inflammation and resolution phases of infection. In this study, we show that G. bethesdensis induces cytokine release from normal and CGD monocytes and macrophages, but to a significantly lesser extent than E. coli. Suppressing cytokine responses can be a microbial evasion mechanism as in the case of L. pneumophila, which requires a type II secretion system to dampen cytokine release from and permit growth within infected monocytes (58), although such mechanisms have yet to be described for Granulibacter. Our laboratory is currently characterizing structural differences between G. bethesdensis and E. coli endotoxin that may also contribute to the observed differences in cytokine secretion.
Similar to neutrophils from normal subjects, freshly isolated and IFN-γ-treated normal monocytes kill similar amounts of input bacteria after 24 hours of infection at an MOI of 1. However, unlike CGD neutrophils (13), freshly isolated CGD monocytes allow a two- to threefold expansion of G. bethesdensis over 24 hours, demonstrating that these cells provide a favorable environment for survival of the bacterium. Though it does not induce killing, IFN-γ treatment of CGD monocytes significantly limits G. bethesdensis outgrowth, and this effect correlates with the amount of residual ROS in CGD neutrophils. In comparison to neutrophils and monocytes, MDM are less successful in exerting bactericidal activity against G. bethesdensis even with the help of IFN-γ. Our studies demonstrate that 4 normal IFN-γ-treated MDM are required to control the growth of a single bacterium after 24 hours of infection and only modest killing occurs by 1 week post-infection. This inability to kill G. bethesdensis is more pronounced in CGD MDM where IFN-γ could not significantly control intracellular replication of the organism up to 1 week after infection.
These data suggest that the CGD macrophage is a potential niche for G. bethesdensis growth and survival in vivo. The macrophage as a niche for bacterial replication and persistence is not unprecedented. Alveolar macrophages internalize L. pneumophila via conventional complement-mediated phagocytosis (59) or coiling phagocytosis (60) into phagosomes that permit bacterial replication and evade lysosomal fusion (61). This suitable environment for L. pneumophila replication may allow the organism to survive for a long period of time. Moreover, L. pneumophila inhibits macrophage apoptosis by blocking the effects of pro-apoptotic Bcl2 family members (62). Latency of the organism in patients from the 1976 Philadelphia epidemic of Legionnaires’ disease has been suggested, where specific IgM antibodies persisted for 2 years after initial infection (41).
Latent infection by M. tuberculosis in one third of the human population has been reported (42). Infected macrophages play a central role in latency by forming granulomas with antigen-specific T cells that inhibit the growth of M. tuberculosis, while the bacterium resists macrophage bactericidal killing (63). Like L. pneumophila, M. tuberculosis also inhibits phagosomal maturation and replicates within macrophages (64). Additionally, its virulence factors are critical for the inhibition of macrophage apoptosis (65) and pyroptosis (66). Lastly, O-antigen-positive Brucella species also prevent phagosome-lysosome fusion inside macrophages (67) and inhibit macrophage apoptosis (68), which likely prolongs the bacterium’s intracellular survival. Cases of chronic brucellosis have been described as lasting from several months to several years after acute infection (43, 69). Taken together, the macrophage centrally contributes to the persistence of bacteria that cause chronic disease in humans.
G. bethesdensis causes recurrent infections in CGD patients for years and can be recovered from normal mouse spleens up to 76 days post-inoculation (3, 4). Like the other persistent bacterial pathogens mentioned above, G. bethesdensis resists killing by serum and cationic antibacterial peptides, replicates within CGD macrophages, and does not compromise the viability of infected macrophages. G. bethesdensis resists some oxygen-independent microbicides but it may not be impervious to all non-oxidative killing mechanisms. Though the NADPH oxidase is required for G. bethesdensis clearance, it is possible that the products of the NADPH oxidase interact with and activate oxygen-independent bactericidal systems.
Interestingly, all of the aforementioned bacteria have slow rates of division with G. bethesdensis doubling every 5–6 hours, L. pneumophila every 3 hours (70), M. tuberculosis every 24 hours (71), and Brucella species every 2.5–3.5 hours (72), which contrasts to E. coli’s faster doubling time of 0.5–1.5 hours (73). This trait could be important for the maintenance of an intact host phagocyte and bacterial replication niche, which might otherwise lyse if overwhelmed with too many intracellular bacteria. It remains to be determined if in vitro survival of G. bethesdensis in macrophages correlates to persistence of G. bethesdensis in vivo.
The intracellular nature of G. bethesdensis infection may pose a challenge for clinical treatment. G. bethesdensis is innately multidrug resistant and therapy of infected individuals requires aggressive antimicrobial treatments and sometimes surgical intervention (4). Ensuring the efficacy of antimicrobial therapies may require that they can penetrate infected phagocytes as has been shown for the treatment of CGD, tuberculosis, and legionellosis (74). IFN-γ is critical for activating bactericidal mechanisms in macrophages infected with L. pneumophila (75, 76) and M. tuberculosis (77), and may prove an effective therapy for in vivo treatment of infections with these organisms (78, 79). IFN-γ may also be an effective treatment for some CGD patients infected with G. bethesdensis, as was shown for CGD patients infected with S. aureus (29). We show for the first time that in vitro IFN-γ-enhanced control of G. bethesdensis outgrowth in CGD monocytes correlates with higher residual superoxide levels in PMN from the same CGD patients. Since higher residual PMN superoxide correlated with increased survival in a large CGD cohort (32), this in vitro finding may be a useful test to identify patients that are likely to respond to IFN-γ therapy and those who might not.
Given the high morbidity associated with IFN-γ therapy as well as the high cost, this finding has important clinical implications. It is likely that IFN-γ boosts superoxide levels in CGD phagocytes with a certain threshold of residual superoxide leading to enhanced control of bacterial burden. However, we cannot rule out that IFN-γ stimulates ROS-independent bactericidal or bacteriostatic pathways, such as the downregulation of transferrin receptors that limits iron availability required for the growth of some organisms (24). IFN-γ also plays a role in vitamin D-dependent upregulation of antimicrobial peptides and induction of autophagy (33); however, we found no contribution of vitamin D in the control of G. bethesdensis burden (data not shown). It will be important to establish further correlations between in vitro and in vivo IFN-γ efficacy to help identify specific CGD patients who will respond to treatment. Finding novel therapies to treat chronic intracellular infections, especially in immunocompromised populations, is crucial given that many persistent bacterial pathogens are resistant to antibiotics.
Acknowledgments
We acknowledge Drs. Harry L. Malech, Suk See DeRavin, and Stephen M. Holland for generously providing us with CGD patient samples and thank the CGD patients who have contributed to this study. We also thank Drs. Margery Smelkinson and Stephen Becker for assistance with epifluorescence and confocal microscopy, Anthony Pettinato for blindly scoring monocyte internalization images, Drs. Mark VanRaden and Chiung-Yu Huang for statistical help, Neysha Martinez for preliminary studies provoking us to pursue this study, and Dr. David Greenberg for helpful suggestions during the beginning of this study and critical review of this manuscript.
The views expressed here are those of the authors and not necessarily those of the U.S. Government.
Footnotes
This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases at the National Institutes of Health.
References
- 1.Winkelstein JA, Marino MC, Johnston RB, Jr, Boyle J, Curnutte J, Gallin JI, Malech HL, Holland SM, Ochs H, Quie P, Buckley RH, Foster CB, Chanock SJ, Dickler H. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore) 2000;79:155–169. doi: 10.1097/00005792-200005000-00003. [DOI] [PubMed] [Google Scholar]
- 2.Song E, Jaishankar GB, Saleh H, Jithpratuck W, Sahni R, Krishnaswamy G. Chronic granulomatous disease: a review of the infectious and inflammatory complications. Clin Mol Allergy. 2011;9:10. doi: 10.1186/1476-7961-9-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Greenberg DE, Ding L, Zelazny AM, Stock F, Wong A, Anderson VL, Miller G, Kleiner DE, Tenorio AR, Brinster L, Dorward DW, Murray PR, Holland SM. A novel bacterium associated with lymphadenitis in a patient with chronic granulomatous disease. PLoS Pathog. 2006;2:e28. doi: 10.1371/journal.ppat.0020028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greenberg DE, Shoffner AR, Zelazny AM, Fenster ME, Zarember KA, Stock F, Ding L, Marshall-Batty KR, Wasserman RL, Welch DF, Kanakabandi K, Sturdevant DE, Virtaneva K, Porcella SF, Murray PR, Malech HL, Holland SM. Recurrent Granulibacter bethesdensis infections and chronic granulomatous disease. Emerg Infect Dis. 2010;16:1341–1348. doi: 10.3201/eid1609.091800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lopez FC, de Luna FF, Delgado MC, Ibarra de la Rosa I, Valdezate S, Nieto JA, Casal M. Granulibacter bethesdensis isolated in a child patient with chronic granulomatous disease. J Infect. 2008;57:275–277. doi: 10.1016/j.jinf.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 6.Abdel-Haq N, Savasan S, Davis M, Asmar BI, Painter T, Salimnia H. Asaia lannaensis bloodstream infection in a child with cancer and bone marrow transplantation. J Med Microbiol. 2009;58:974–976. doi: 10.1099/jmm.0.008722-0. [DOI] [PubMed] [Google Scholar]
- 7.Alauzet C, Teyssier C, Jumas-Bilak E, Gouby A, Chiron R, Rabaud C, Counil F, Lozniewski A, Marchandin H. Gluconobacter as well as Asaia species, newly emerging opportunistic human pathogens among acetic acid bacteria. J Clin Microbiol. 2010;48:3935–3942. doi: 10.1128/JCM.00767-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bittar F, Reynaud-Gaubert M, Thomas P, Boniface S, Raoult D, Rolain JM. Acetobacter indonesiensis pneumonia after lung transplant. Emerg Infect Dis. 2008;14:997–998. doi: 10.3201/eid1406.071236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gouby A, Teyssier C, Vecina F, Marchandin H, Granolleras C, Zorgniotti I, Jumas-Bilak E. Acetobacter cibinongensis bacteremia in human. Emerg Infect Dis. 2007;13:784–785. doi: 10.3201/eid1305.060532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Juretschko S, Beavers-May TK, Stovall SH. Nosocomial infection with Asaia lannensis in two paediatric patients with idiopathic dilated cardiomyopathy. J Med Microbiol. 2010;59:848–852. doi: 10.1099/jmm.0.019067-0. [DOI] [PubMed] [Google Scholar]
- 11.Snyder RW, Ruhe J, Kobrin S, Wasserstein A, Doline C, Nachamkin I, Lipschutz JH. Asaia bogorensis peritonitis identified by 16S ribosomal RNA sequence analysis in a patient receiving peritoneal dialysis. Am J Kidney Dis. 2004;44:e15–17. doi: 10.1053/j.ajkd.2004.04.042. [DOI] [PubMed] [Google Scholar]
- 12.Tuuminen T, Heinasmaki T, Kerttula T. First report of bacteremia by Asaia bogorensis, in a patient with a history of intravenous-drug abuse. J Clin Microbiol. 2006;44:3048–3050. doi: 10.1128/JCM.00521-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zarember KA, Marshall-Batty KR, Cruz AR, Chu J, Fenster ME, Shoffner AR, Rogge LS, Whitney AR, Czapiga M, Song HH, Shaw PA, Nagashima K, Malech HL, DeLeo FR, Holland SM, Gallin JI, Greenberg DE. Innate immunity against Granulibacter bethesdensis, an emerging gram-negative bacterial pathogen. Infect Immun. 2012;80:975–981. doi: 10.1128/IAI.05557-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Savill JS, Wyllie AH, Henson JE, Walport MJ, Henson PM, Haslett C. Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. J Clin Invest. 1989;83:865–875. doi: 10.1172/JCI113970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fernandez-Boyanapalli R, McPhillips KA, Frasch SC, Janssen WJ, Dinauer MC, Riches DW, Henson PM, Byrne A, Bratton DL. Impaired phagocytosis of apoptotic cells by macrophages in chronic granulomatous disease is reversed by IFNgamma in a nitric oxide-dependent manner. J Immunol. 2010;185:4030–4041. doi: 10.4049/jimmunol.1001778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown JR, Goldblatt D, Buddle J, Morton L, Thrasher AJ. Diminished production of anti-inflammatory mediators during neutrophil apoptosis and macrophage phagocytosis in chronic granulomatous disease (CGD) J Leukoc Biol. 2003;73:591–599. doi: 10.1189/jlb.1202599. [DOI] [PubMed] [Google Scholar]
- 17.Zarember KA, Sugui JA, Chang YC, Kwon-Chung KJ, Gallin JI. Human Polymorphonuclear Leukocytes Inhibit Aspergillus fumigatus Conidial Growth by Lactoferrin-Mediated Iron Depletion. The Journal of Immunology. 2007;178:6367–6373. doi: 10.4049/jimmunol.178.10.6367. [DOI] [PubMed] [Google Scholar]
- 18.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 19.Ballabh P, Simm M, Kumari J, Califano C, Aghai Z, Laborada G, Sison C, Cunningham-Rundles S. Respiratory burst activity in bronchopulmonary dysplasia and changes with dexamethasone. Pediatr Pulmonol. 2003;35:392–399. doi: 10.1002/ppul.10279. [DOI] [PubMed] [Google Scholar]
- 20.Bruns T, Peter J, Hagel S, Herrmann A, Stallmach A. The augmented neutrophil respiratory burst in response to Escherichia coli is reduced in liver cirrhosis during infection. Clin Exp Immunol. 164:346–356. doi: 10.1111/j.1365-2249.2011.04373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Rossi L, Gott K, Horn N, Hecker K, Hutschenreuter G, Rossaint R. Xenon preserves neutrophil and monocyte function in human whole blood. Can J Anaesth. 2002;49:942–945. doi: 10.1007/BF03016879. [DOI] [PubMed] [Google Scholar]
- 22.Reiss M, Roos D. Differences in oxygen metabolism of phagocytosing monocytes and neutrophils. J Clin Invest. 1978;61:480–488. doi: 10.1172/JCI108959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leijh PC, Nathan CF, van den Barselaar MT, van Furth R. Relationship between extracellular stimulation of intracellular killing and oxygen-dependent microbicidal systems of monocytes. Infect Immun. 1985;47:502–507. doi: 10.1128/iai.47.2.502-507.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Byrd TF, Horwitz MA. Interferon gamma-activated human monocytes downregulate transferrin receptors and inhibit the intracellular multiplication of Legionella pneumophila by limiting the availability of iron. J Clin Invest. 1989;83:1457–1465. doi: 10.1172/JCI114038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gyan B, Troye-Blomberg M, Perlmann P, Bjorkman A. Human monocytes cultured with and without interferon-gamma inhibit Plasmodium falciparum parasite growth in vitro via secretion of reactive nitrogen intermediates. Parasite Immunol. 1994;16:371–375. doi: 10.1111/j.1365-3024.1994.tb00362.x. [DOI] [PubMed] [Google Scholar]
- 26.Peck R. Gamma interferon induces monocyte killing of Listeria monocytogenes by an oxygen-dependent pathway; alpha- or beta-interferons by oxygen-independent pathways. J Leukoc Biol. 1989;46:434–440. doi: 10.1002/jlb.46.5.434. [DOI] [PubMed] [Google Scholar]
- 27.Mazzi P, Donini M, Margotto D, Wientjes F, Dusi S. IFN-gamma induces gp91phox expression in human monocytes via protein kinase C-dependent phosphorylation of PU.1. J Immunol. 2004;172:4941–4947. doi: 10.4049/jimmunol.172.8.4941. [DOI] [PubMed] [Google Scholar]
- 28.Dusi S, Donini M, Lissandrini D, Mazzi P, Bianca VD, Rossi F. Mechanisms of expression of NADPH oxidase components in human cultured monocytes: role of cytokines and transcriptional regulators involved. Eur J Immunol. 2001;31:929–938. doi: 10.1002/1521-4141(200103)31:3<929::aid-immu929>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 29.Sechler JM, Malech HL, White CJ, Gallin JI. Recombinant human interferon-gamma reconstitutes defective phagocyte function in patients with chronic granulomatous disease of childhood. Proc Natl Acad Sci U S A. 1988;85:4874–4878. doi: 10.1073/pnas.85.13.4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Czarniecki CW, Sonnenfeld G. Interferon-gamma and resistance to bacterial infections. APMIS. 1993;101:1–17. doi: 10.1111/j.1699-0463.1993.tb00073.x. [DOI] [PubMed] [Google Scholar]
- 31.Ezekowitz RA, Orkin SH, Newburger PE. Recombinant interferon gamma augments phagocyte superoxide production and X-chronic granulomatous disease gene expression in X-linked variant chronic granulomatous disease. J Clin Invest. 1987;80:1009–1016. doi: 10.1172/JCI113153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuhns DB, Alvord WG, Heller T, Feld JJ, Pike KM, Marciano BE, Uzel G, DeRavin SS, Priel DA, Soule BP, Zarember KA, Malech HL, Holland SM, Gallin JI. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. 2010;363:2600–2610. doi: 10.1056/NEJMoa1007097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fabri M, Stenger S, Shin DM, Yuk JM, Liu PT, Realegeno S, Lee HM, Krutzik SR, Schenk M, Sieling PA, Teles R, Montoya D, Iyer SS, Bruns H, Lewinsohn DM, Hollis BW, Hewison M, Adams JS, Steinmeyer A, Zugel U, Cheng G, Jo EK, Bloom BR, Modlin RL. Vitamin D is required for IFN-gamma-mediated antimicrobial activity of human macrophages. Sci Transl Med. 3:104ra102. doi: 10.1126/scitranslmed.3003045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Serbina NV, Jia T, Hohl TM, Pamer EG. Monocyte-mediated defense against microbial pathogens. Annu Rev Immunol. 2008;26:421–452. doi: 10.1146/annurev.immunol.26.021607.090326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lacey DC, Achuthan A, Fleetwood AJ, Dinh H, Roiniotis J, Scholz GM, Chang MW, Beckman SK, Cook AD, Hamilton JA. Defining GM-CSF- and Macrophage-CSF-Dependent Macrophage Responses by In Vitro Models. J Immunol. doi: 10.4049/jimmunol.1103426. [DOI] [PubMed] [Google Scholar]
- 36.Cianciotto NP. Pathogenicity of Legionella pneumophila. Int J Med Microbiol. 2001;291:331–343. doi: 10.1078/1438-4221-00139. [DOI] [PubMed] [Google Scholar]
- 37.Raupach B, Kaufmann SH. Immune responses to intracellular bacteria. Curr Opin Immunol. 2001;13:417–428. doi: 10.1016/s0952-7915(00)00236-3. [DOI] [PubMed] [Google Scholar]
- 38.Dornand J, Gross A, Lafont V, Liautard J, Oliaro J, Liautard JP. The innate immune response against Brucella in humans. Vet Microbiol. 2002;90:383–394. doi: 10.1016/s0378-1135(02)00223-7. [DOI] [PubMed] [Google Scholar]
- 39.Bylund J, MacDonald KL, Brown KL, Mydel P, Collins LV, Hancock RE, Speert DP. Enhanced inflammatory responses of chronic granulomatous disease leukocytes involve ROS-independent activation of NF-kappa B. Eur J Immunol. 2007;37:1087–1096. doi: 10.1002/eji.200636651. [DOI] [PubMed] [Google Scholar]
- 40.Rahman FZ, Hayee B, Chee R, Segal AW, Smith AM. Impaired macrophage function following bacterial stimulation in chronic granulomatous disease. Immunology. 2009;128:253–259. doi: 10.1111/j.1365-2567.2009.03112.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lattimer GL, I, Rhodes LV, Salventi JS, Galgon JP, Stonebraker V, Boley S, Haas G. The Philadelphia Epidemic of Legionnaire’s Disease: Clinical, Pulmonary, and Serologic Findings Two Years Later. Annals of Internal Medicine. 1979;90:522–526. doi: 10.7326/0003-4819-90-4-522. [DOI] [PubMed] [Google Scholar]
- 42.Riska PF, Carleton S. Latent tuberculosis: Models, mechanisms, and novel prospects for eradication. Seminars in Pediatric Infectious Diseases. 2002;13:263–272. doi: 10.1053/spid.2002.127198. [DOI] [PubMed] [Google Scholar]
- 43.Castaño MJ, Solera J. Chronic Brucellosis and Persistence of Brucella melitensis DNA. Journal of Clinical Microbiology. 2009;47:2084–2089. doi: 10.1128/JCM.02159-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Plouffe JF, Para MF, Fuller KA. Serum bactericidal activity against Legionella pneumophila. Journal of Clinical Microbiology. 1985;22:863–864. doi: 10.1128/jcm.22.5.863-864.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eisenschenk FC, Houle JJ, Hoffmann EM. Mechanism of serum resistance among Brucella abortus isolates. Veterinary Microbiology. 1999;68:235–244. doi: 10.1016/s0378-1135(99)00075-9. [DOI] [PubMed] [Google Scholar]
- 46.Edelstein PH, Hu B, Higa F, Edelstein MAC. lvgA, a Novel Legionella pneumophila Virulence Factor. Infection and Immunity. 2003;71:2394–2403. doi: 10.1128/IAI.71.5.2394-2403.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robey M, O’Connell W, Cianciotto NP. Identification of Legionella pneumophila rcp, a pagP-Like Gene That Confers Resistance to Cationic Antimicrobial Peptides and Promotes Intracellular Infection. Infection and Immunity. 2001;69:4276–4286. doi: 10.1128/IAI.69.7.4276-4286.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maloney E, Stankowska D, Zhang J, Fol M, Cheng QJ, Lun S, Bishai WR, Rajagopalan M, Chatterjee D, Madiraju MV. The Two-Domain LysX Protein of Mycobacterium tuberculosis Is Required for Production of Lysinylated Phosphatidylglycerol and Resistance to Cationic Antimicrobial Peptides. PLoS Pathog. 2009;5:e1000534. doi: 10.1371/journal.ppat.1000534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martinez de Tejada G, Pizarro-Cerda J, Moreno E, Moriyon I. The outer membranes of Brucella spp. are resistant to bactericidal cationic peptides. Infection and Immunity. 1995;63:3054–3061. doi: 10.1128/iai.63.8.3054-3061.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dancey JT, Deubelbeiss KA, Harker LA, Finch CA. Neutrophil kinetics in man. The Journal of Clinical Investigation. 1976;58:705–715. doi: 10.1172/JCI108517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Whitelaw DM, Bell M. The Intravascular Lifespan of Monocytes. Blood. 1966;28:455–464. [PubMed] [Google Scholar]
- 52.Murphy J, Summer R, Wilson AA, Kotton DN, Fine A. The Prolonged Life-Span of Alveolar Macrophages. American Journal of Respiratory Cell and Molecular Biology. 2008;38:380–385. doi: 10.1165/rcmb.2007-0224RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Buckle AM, Jayaram Y, Hogg N. Colony-stimulating factors and interferon-gamma differentially affect cell surface molecules shared by monocytes and neutrophils. Clin Exp Immunol. 1990;81:339–345. doi: 10.1111/j.1365-2249.1990.tb03342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stephen SL, Freestone K, Dunn S, Twigg MW, Homer-Vanniasinkam S, Walker JH, Wheatcroft SB, Ponnambalam S. Scavenger receptors and their potential as therapeutic targets in the treatment of cardiovascular disease. Int J Hypertens. 2010;2010:646929. doi: 10.4061/2010/646929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Muzio M, Bosisio D, Polentarutti N, D’Amico G, Stoppacciaro A, Mancinelli R, van’t Veer C, Penton-Rol G, Ruco LP, Allavena P, Mantovani A. Differential expression and regulation of toll-like receptors (TLR) in human leukocytes: selective expression of TLR3 in dendritic cells. J Immunol. 2000;164:5998–6004. doi: 10.4049/jimmunol.164.11.5998. [DOI] [PubMed] [Google Scholar]
- 56.Sabroe I, Jones EC, Usher LR, Whyte MK, Dower SK. Toll-like receptor (TLR)2 and TLR4 in human peripheral blood granulocytes: a critical role for monocytes in leukocyte lipopolysaccharide responses. J Immunol. 2002;168:4701–4710. doi: 10.4049/jimmunol.168.9.4701. [DOI] [PubMed] [Google Scholar]
- 57.McDonald JU, Rosas M, Brown GD, Jones SA, Taylor PR. Differential dependencies of monocytes and neutrophils on dectin-1, dectin-2 and complement for the recognition of fungal particles in inflammation. PLoS One. 2012;7:e45781. doi: 10.1371/journal.pone.0045781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McCoy-Simandle K, Stewart CR, Dao J, DebRoy S, Rossier O, Bryce PJ, Cianciotto NP. Legionella pneumophila type II secretion dampens the cytokine response of infected macrophages and epithelia. Infect Immun. 2011;79:1984–1997. doi: 10.1128/IAI.01077-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Payne NR, Horwitz MA. Phagocytosis of Legionella pneumophila is mediated by human monocyte complement receptors. The Journal of Experimental Medicine. 1987;166:1377–1389. doi: 10.1084/jem.166.5.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Horwitz MA. Phagocytosis of the legionnaires’ disease bacterium (legionella pneumophila) occurs by a novel mechanism: Engulfment within a Pseudopod coil. Cell. 1984;36:27–33. doi: 10.1016/0092-8674(84)90070-9. [DOI] [PubMed] [Google Scholar]
- 61.Kagan JC, Roy CR. Legionella phagosomes intercept vesicular traffic from endoplasmic reticulum exit sites. Nat Cell Biol. 2002;4:945–954. doi: 10.1038/ncb883. [DOI] [PubMed] [Google Scholar]
- 62.Banga S, Gao P, Shen X, Fiscus V, Zong WX, Chen L, Luo ZQ. Legionella pneumophila inhibits macrophage apoptosis by targeting pro-death members of the Bcl2 protein family. Proceedings of the National Academy of Sciences. 2007;104:5121–5126. doi: 10.1073/pnas.0611030104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Saunders BM, Britton WJ. Life and death in the granuloma: immunopathology of tuberculosis. Immunol Cell Biol. 2007;85:103–111. doi: 10.1038/sj.icb.7100027. [DOI] [PubMed] [Google Scholar]
- 64.Sullivan JT, Young EF, McCann JR, Braunstein M. The Mycobacterium tuberculosis SecA2 System Subverts Phagosome Maturation To Promote Growth in Macrophages. Infection and Immunity. 2012;80:996–1006. doi: 10.1128/IAI.05987-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Velmurugan K, Chen B, Miller JL, Azogue S, Gurses S, Hsu T, Glickman M, Jacobs WR, Jr, Porcelli SA, Briken V. Mycobacterium tuberculosis nuoG Is a Virulence Gene That Inhibits Apoptosis of Infected Host Cells. PLoS Pathog. 2007;3:e110. doi: 10.1371/journal.ppat.0030110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Danelishvili L, Everman J, McNamara M, Bermudez L. Inhibition of the Plasma-Membrane Associated Serine Protease Cathepsin G by Mycobacterium tuberculosis Rv3364c Suppresses Caspase-1 and Pyroptosis in Macrophages. Frontiers in Microbiology. 2012;2:281. doi: 10.3389/fmicb.2011.00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Porte F, Naroeni A, Ouahrani-Bettache S, Liautard JP. Role of the Brucella suis Lipopolysaccharide O Antigen in Phagosomal Genesis and in Inhibition of Phagosome-Lysosome Fusion in Murine Macrophages. Infection and Immunity. 2003;71:1481–1490. doi: 10.1128/IAI.71.3.1481-1490.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fernandez-Prada CM, Zelazowska EB, Nikolich M, Hadfield TL, Roop RM, II, Robertson GL, Hoover DL. Interactions between Brucella melitensis and Human Phagocytes: Bacterial Surface O-Polysaccharide Inhibits Phagocytosis, Bacterial Killing, and Subsequent Host Cell Apoptosis. Infection and Immunity. 2003;71:2110–2119. doi: 10.1128/IAI.71.4.2110-2119.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Davies JE. Chronic brucellosis in general practice. Br Med J. 1957;2:1082–1087. doi: 10.1136/bmj.2.5053.1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bandyopadhyay P, Steinman HM. Legionella pneumophila Catalase-Peroxidases: Cloning of the katB Gene and Studies of KatB Function. Journal of Bacteriology. 1998;180:5369–5374. doi: 10.1128/jb.180.20.5369-5374.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ginsberg AM, Spigelman M. Challenges in tuberculosis drug research and development. Nat Med. 2007;13:290–294. doi: 10.1038/nm0307-290. [DOI] [PubMed] [Google Scholar]
- 72.Rest RF, Robertson DC. Characterization of the electron transport system in Brucella abortus. Journal of Bacteriology. 1975;122:139–144. doi: 10.1128/jb.122.1.139-144.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Plank LD, Harvey JD. Generation time statistics of Escherichia coli B measured by synchronous culture techniques. J Gen Microbiol. 1979;115:69–77. doi: 10.1099/00221287-115-1-69. [DOI] [PubMed] [Google Scholar]
- 74.Schwab JC, Mandell GL. The importance of penetration of antimicrobial agents into cells. Infect Dis Clin North Am. 1989;3:461–467. [PubMed] [Google Scholar]
- 75.Gebran SJ, Yamamoto Y, Newton C, Klein TW, Friedman H. Inhibition of Legionella pneumophila growth by gamma interferon in permissive A/J mouse macrophages: role of reactive oxygen species, nitric oxide, tryptophan, and iron(III) Infection and Immunity. 1994;62:3197–3205. doi: 10.1128/iai.62.8.3197-3205.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nash TW, Libby DM, Horwitz MA. IFN-gamma-activated human alveolar macrophages inhibit the intracellular multiplication of Legionella pneumophila. The Journal of Immunology. 1988;140:3978–3981. [PubMed] [Google Scholar]
- 77.Herbst S, Schaible UE, Schneider BE. Interferon Gamma Activated Macrophages Kill Mycobacteria by Nitric Oxide Induced Apoptosis. PLoS ONE. 2011;6:e19105. doi: 10.1371/journal.pone.0019105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Deng JC, Tateda K, Zeng X, Standiford TJ. Transient Transgenic Expression of Gamma Interferon Promotes Legionella pneumophila Clearance in Immunocompetent Hosts. Infection and Immunity. 2001;69:6382–6390. doi: 10.1128/IAI.69.10.6382-6390.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Reljic R. IFN-gamma therapy of tuberculosis and related infections. J Interferon Cytokine Res. 2007;27:353–364. doi: 10.1089/jir.2006.0103. [DOI] [PubMed] [Google Scholar]