

Abstract
IL-21 is a pluripotent cytokine that regulates B cell and plasma cell differentiation, and is thought be an autocrine factor for TFH and TH17 differentiation. Although IL-21 has been implicated in autoimmune diseases, its relevant cellular source and target cells have not been well characterized. We investigated this issue in the K/BxN mouse model of autoimmune arthritis. Adoptive transfer of KRN transgenic CD4+ T cells into appropriate hosts drives germinal center formation and autoantibody production against glucose-6-phosphate isomerase, leading to joint inflammation and destruction. By comparing transfer of T or B cells deficient in IL-21 or IL-21R we were able to dissect the contribution of each cell type. T cells deficient in IL-21 did not induce germinal center (GC) formation or autoantibody production, but went through normal TFH differentiation. However, T cells lacking IL-21R induced similar antibody titer, GC B cell frequency, and arthritis development as wild type T cells, suggesting that IL-21 is not required for TFH differentiation and function. IL-21 acts on B cells, as IL-21R expression on B cells was required to induce disease. In contrast, TH17 cells, a T cell subset that also produces IL-21 and can provide help to B cells, are not required for the GC response and arthritis. These data have implications in developing effective therapies for rheumatoid arthritis and other antibody-mediated autoimmune diseases.
Introduction
IL-21, a member of the common γ chain–signaling family of cytokines, plays an important role in lymphocyte activation, survival and differentiation (1). IL-21 production is restricted to activated T cells, such as follicular helper T cells (TFH), TH17, and NKT cells. The receptor for IL-21 is widely expressed on a variety of cell types, including B cells, activated T cells, NK cells and dendritic cells. IL-21 promotes B cell proliferation, immunoglobulin class switching and production, and plasma cell differentiation (2). IL-21 also enhances the proliferation of T cells stimulated through their T cell receptors (3) and has been shown to be an autocrine growth factor for TFH and TH17 cell differentiation (4-8).
The TFH cell subset, a canonical producer of IL-21, is controlled by the transcription factor Bcl6. Changes in chemokine receptor expression allow TFH to migrate from the T cell zone into B cell follicles. Expression of cell surface molecules promote cell-cell contacts with B cells presenting cognate antigen. It is in these intimate interactions that IL-21 from TFH is thought to act on B cells to promote germinal center and plasma cell differentiation (reviewed in (9, 10)). In addition to TFH, the TH17 cell subset also produces IL-21. TH17 is a dominant proinflammatory T cell subset, controlled by the transcription factor RORγt, and is involved in a number of autoimmune diseases (reviewed in (11)). TH17 cells have been shown to directly interact with and help B cells (12) and promote spontaneous GC formation in autoimmune BXD2 mice (13).
IL-21 is important in a number of animal models of SLE and rheumatoid arthritis (14-18). Accordingly, an association of certain IL-21 and IL-21R alleles with risk for SLE in humans was reported (19, 20). In the NOD mouse model IL-21 is required for the development of type I diabetes (21, 22). Given the complex biological functions of IL-21, it is important to understand the relevant cells producing and responding to the cytokine in the context of B cell-mediated autoimmunity.
We investigated this question of the relevant targets of IL-21 using the K/BxN model of rheumatoid arthritis. K/BxN mice develop arthritis by 4 weeks of age (23). The disease is initiated by KRN TCR transgenic CD4+ T cells that recognize a peptide from the ubiquitously expressed self-protein glucose-6-phosphate isomerase (GPI) presented by the NOD-derived MHC class II molecule I-Ag7 (24). Activated KRN T cells drive B cells to form germinal centers and to produce anti-GPI IgG autoantibodies, which induce joint pathology (25). Unlike other models of arthritis, K/BxN mice develop spontaneous disease and do not involve antigen vaccination with adjuvants, which can affect TFH development (26). The advantage of the model for these studies is that the disease can be induced by transferring naïve KRN T cells into T cell deficient hosts expressing the MHC class II molecule I-Ag7 (25, 27).
Here, we have utilized this cell transfer approach of the K/BxN model to determine the source and action of IL-21 in arthritis. We showed that T cells deficient in IL-21 did not induce GC formation or autoantibody production, but went through normal TFH differentiation. However, T cells lacking IL-21R induced similar antibody titer, GC B cell frequency, and arthritis development as wild type T cells, suggesting that IL-21 is not required for TFH differentiation and function. IL-21 must act on B cells, as IL-21R expression on B cells was required to induce disease. Surprisingly, TH17 cells are not required for arthritis development, stressing the importance of IL-21 production specifically from the TFH subset. These results have implications for developing effective therapies for rheumatoid arthritis and other antibody-mediated autoimmune diseases.
Materials and Methods
Mice
KRN TCR transgenic mice (23), IL-21R−/− mice (28), and RORγtGFP/GFP mice (29) were maintained on C57/BL6 background. IL-21−/− mice (B6;129S5-Il21tm1Lex, obtained from the Mutant Mouse Regional Resource Center) were maintained on a B6;129S5 mixed background. KRN was crossed to IL-21R−/− or IL-21−/− mice to generate K/IL-21R−/− or K/IL-21−/− mice respectively. TCR Cα−/− BxN mice used as hosts were F1 of Cα−/− B6 and Cα−/− NOD (25). IL21R knockout mice crossed to B6.H2g7 congenic mice (30) were used as donors for purified B cells. All experiments were approved by IACUC of the University of Chicago.
Cell transfer induction of arthritis
CD4+ cells were isolated from splenocytes using AutoMACS positive selection program. 1×106 cells in sterile DMEM were injected into the tail vein of Cα−/− BxN mice. B cells were enriched from splenocytes by depleting T cells with anti-CD90.2 antibody (clone 53-2.1, BioLegend) followed by rabbit complement (Cedar Lane).
Abs and flow cytometry
Anti-KRN TCRα specific antibody 3-4G-B7 (manuscript in preparation) was labeled with AlexFluor 647 or biotin. Antibodies used were against CXCR5, Bcl6, and Fas (BD Biosciences); CD45.1, TCRβ, GL-7, CD19, IL-17A, and B220 (eBioscience); and CD4 and PD-1 (BioLegend). Intracellular staining for Bcl-6 was performed using the Foxp3 intracellular staining kit from eBioscience, according to the manufacturer’s protocol. Intracellular staining for IL-17 was performed using the intracellular staining kit from BD Biosciences after cells were stimulated with PMA and ionomycin as described (12). Multicolor flow cytometric analysis was performed using a FACSCanto (BD Biosciences). Data analysis was conducted using FlowJo (Tree Star, Inc.).
Immunohistochemistry staining of splenic sections
Sections of frozen spleen (5um) were thawed, rehydrated, and then stained. Peanut agglutinin (PNA, Alexa Fluor 488), anti-mouse IgD (PE), and anti-mouse Vβ6 (Alexa Fluor 647) or anti-KRN TCRa (Alexa Fluor 647) were used. Images were taken on an Axiovert200m (Zeiss) and visualized with ImageJ.
ELISA for anti-GPI total IgG
96-well plates were coated with 5ug/mL of recombinant GPI in PBS overnight at 4°C and blocked with 1% BSA 0.05% Tween-20 in PBS at room temperature. A serial dilution of the samples added to the plate was detected with a biotinylated goat anti-mIgG (subclasses 1+2a+2b+3) Fcγ or goat anti-mIgM Fcμ fragment specific antibody followed by alkaline phosphatase-conjugated streptavidin (all from Jackson ImmunoResearch Laboratories, Inc.). Samples were developed with phosphatase substrate (Sigma) and were read at 405 nm. A four-parameter variable slope was fitted to the data points and the EC50 (inflection point) for a standard sample was calculated from this non-linear regression. Serum titers were calculated as the serum dilution (x value) that gave the calculated EC50 (y value) based on the fitted non-linear regression for each sample. Samples where the curve could not be fitted because of low signal (low antibody binding) are indicated as ND (not detectable) and a titer of 1 was assigned for statistical comparisons. All analyses were conducted using Prism 5.0b software (GraphPad).
Statistical analysis
Normally distributed data were analyzed by the unpaired t test using Prism 5.0b software (GraphPad).
Results
TFH differentiation in the cell transfer model of autoimmune arthritis
To investigate the role of IL-21 in a cell specific manner, we took advantage of the cell transfer model of the K/BxN mouse. Naïve CD4+ KRN T cells are isolated from healthy KRN/B6 (KRN maintained on C57/BL6 background) and are transferred into Cα−/− BxN hosts (TCR Cα−/− on B6xNOD F1 background) (25, 27). These hosts lack αβ T cells and express the MHC class II allele I-Ag7, which is required for the KRN TCR to recognize a peptide from the self-antigen GPI. Transferred KRN T cells are activated and induce high titers of anti-GPI IgG antibodies resulting in ankle swelling and joint remodeling. To follow autoreactive TFH differentiation and germinal center response, TFH cells and germinal center B cells were characterized after T cell transfer. For comparison, naïve KRN/B6 T cells were transferred into Cα−/− B6 hosts. Because Cα−/− B6 mice do not carry the MHC class II allele I-Ag7, KRN T cells do not precipitate disease upon transfer. This allowed us to verify that TFH differentiation is dependent on antigen recognition rather than lymphopenia-induced homeostatic proliferation. PD-1+ CXCR5+ or Bcl6+ CXCR5+ TFH cells were identified in Cα−/− BxN hosts, but not in Cα−/− B6 hosts or KRN/B6 mice (Fig. 1A). In the following experiments we used CXCR5 and intracellular Bcl6 to mark TFH cells since these markers showed a more distinct pattern. Consistent with the TFH staining, germinal center B cells (identified as GL-7+ Fas+) were induced in Cα−/− BxN hosts while Cα−/− B6 hosts had a small germinal center population, similar to what was observed in naïve KRN/B6 mice (Fig. 1B).
FIGURE 1.
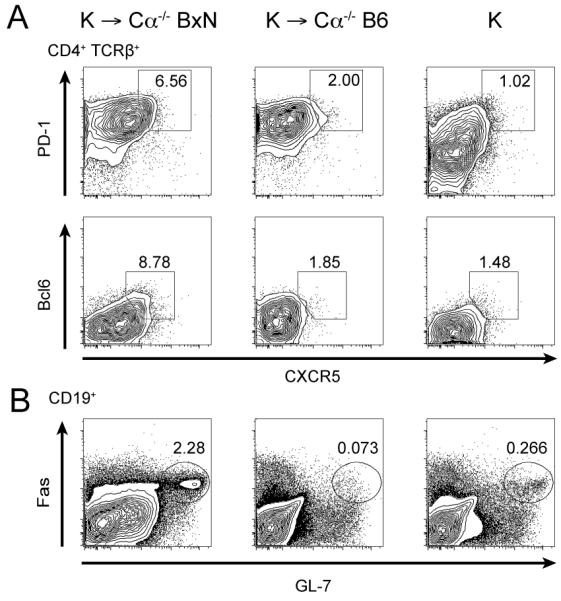
KRN T cell transfer leads to TFH and GC B cell differentiation upon antigen recognition. (A) CD4+ cells from KRN/B6 (K) mice were transferred into Cα−/− BxN or Cα−/− B6 hosts and splenocytes were analyzed 8 days later. Transferred cells, identified as CD4+ TCRβ+, were assessed for TFH differentiation by CXCR5 and PD-1 expression as well as CXCR5 and intracellular Bcl6 expression. Naïve CD4+TCRβ+ cells in KRN/B6 splenocytes were used as controls. (B) GC B cells were assessed by Fas and GL-7 expression in the CD19+ population. Results are representative of three independent experiments.
IL-21 production by T cells is required to induce arthritis
To determine whether IL-21 production by T cells acts on T cells in an autocrine manner to induce arthritis, we compared naïve CD4+ KRN T cells purified from wild-type K/B6 mice (in figures denoted as K) to those purified from KRN mice deficient in IL-21 (referred to as K/IL-21−/−) or IL-21R (referred to as K/IL-21R−/−) after transfer into Cα−/− BxN hosts. In the K/IL-21−/− transfer, KRN T cells could not produce IL-21, and in the K/IL-21R−/− transfer KRN T cells were able to produce IL-21 but unable to receive IL-21R signaling. These two groups allowed us to test whether IL-21 production by autoreactive T cells is required and whether autoreactive T cells require IL-21 as an autocrine factor for disease. As shown in Fig. 2A, K/IL-21−/− CD4+ T cells transferred into Cα−/− BxN hosts did not induce arthritis. In contrast, K/IL-21R−/− CD4+ T cells induced severe arthritis with the same kinetics as the wild-type KRN T cells. We determined the anti-GPI IgG titers both early (8 days) and late (29-31 days) in disease. Wild-type KRN T cells and K/IL21R−/− T cells induced high titers of anti-GPI IgG at both time points. In contrast, anti-GPI IgG titers were two to three orders of magnitude lower in K/IL21−/− T cell transfer (Fig. 2B). These data demonstrate that IL-21 production by T cells is crucial for IgG antibody response and arthritis, but that IL-21R is not required on T cells.
FIGURE 2.

T cell production of IL-21 is required for arthritis development. (A) 1×106 CD4+ cells purified from KRN/B6 (K), KRN IL-21R−/− (K/IL-21R−/−), or KRN IL-21−/− (K/IL-21−/−) mice were transferred into Cα−/− BxN hosts and ankle thickening was measured over time. Results shown are from two experiments (n = 6 - 8 mice per group). Mean and standard deviation are shown. (B) Anti-GPI IgG titers were measured by ELISA from serum collected 8 and 29 - 31 days post-transfer. Mean is shown; each symbol represents an individual mouse. Day eight results are from four experiments (n = 5 - 10 mice per group). Results for days 29-31 are from the experiment described in A. (C) 8×106 splenocytes from KRN/B6 (K) or KRN IL-21−/− (K/IL-21−/−) mice were transferred into Cα−/− BxN hosts and serum was collected on the days indicated. Anti-GPI IgM and IgG titers were measured by ELISA. Mean is shown; each symbol represents an individual mouse (n = 4-5 mice per group). ND (not detectable). Student’s t-test: ns, p > 0.05; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To test the role of IL-21 in extrafollicular response in this model, we determined anti-GPI IgM and IgG at earlier timepoints after transferring wild-type KRN or K/IL-21−/− splenocytes. In wild-type KRN transfer, anti-GPI IgM titers were elevated by day four and continued to increase over time, occurring before IgG titers increased. In contrast, both anti-GPI IgM and IgG titers remained low in K/IL-21−/− transfers (Fig. 2C). These results suggest that IL-21 is required in even the early extrafollicular response in this model.
IL-21 is not required for TFH differentiation in vivo
We next compared the fate of transferred T cells and TFH differentiation in all three transfer settings. Congenic markers on transferred cells (CD45.2+) and Cα−/− BxN host cells (CD45.1+/CD45.2+) allowed us to identify the transferred T cells as the CD45.1− CD4+ population (Fig. 3A). Eight days after cell transfer, just after disease onset, there was a small but significant increase in the percentage and number of K/IL-21−/− T cells compared to WT KRN T cells in the spleen. The percentage and number of K/IL-21R−/− T cells were comparable to WT KRN T cells. At 29 to 31 days, when disease was fully established in WT KRN and K/IL-21R−/− T cell transfer, there was no significant difference in percentage and numbers of transferred T cells in all three groups of transfer. These data suggest that the survival of transferred CD4+ KRN T cells was not affected by their ability to produce or respond to IL-21.
FIGURE 3.

IL-21 production and IL-21 receptor signaling are not required for KRN T cell survival or TFH differentiation. (A) Transferred KRN T cells were identified in the spleen 8 and 29-31 days post-transfer by flow cytometry as CD45.1− CD4+. Percent and total number of transferred cells were shown. (B) TFH differentiation of transferred cells was determined by CXCR5 and intracellular Bcl6 expression. Percent and total number of TFH cells are shown. Day 8 results are from four experiments (n = 5 - 10 mice per group), Days 29-31 results are from two experiments (n = 6 - 8 mice per group). Mean and standard error are shown. Student’s t-test: ns, p ≥ 0.05; *, p < 0.05; **, p< 0.01; ***, p < 0.001.
To determine how TFH differentiation was affected, CXCR5 and intracellular Bcl6 staining were used to identify TFH cells. WT KRN, K/IL-21R−/−, and K/IL-21−/− T cells differentiated into TFH at similar frequencies in the spleen 8 days after transfer (Fig. 3B). The absolute number of TFH from K/IL-21−/− animals was transiently higher than that from WT donors due to a higher total number of CD4+ T cells, as shown in Fig. 3A. However, by 29-31 days K/IL-21−/− TFH cell percentage and numbers decreased to half of those in WT KRN transfer. This presumably reflects the defects in the maintenance phase of TFH differentiation (31) given that there were no germinal centers formed in these mice (see below). However, there were comparable numbers of WT and K/IL-21R−/− TFH cells 8 days after transfer, and a small but not statistically significant decrease in TFH numbers in K/IL-21R−/− cells compared to in WT at days 29-31 (Fig. 3B). These data suggest that IL-21 is not a requisite autocrine factor for KRN TFH cell differentiation.
IL-21 is required for GC formation
Although TFH differentiation was normal in all transfers, anti-GPI IgG antibody production was severely impaired following the transfer of K/IL-21−/− T cells (Fig. 2B). Therefore, we investigated the GC formation and T cell migration by immunofluorescence on spleen sections. As shown in Fig. 4A, there were abundant GCs in both KRN WT and K/IL-21R−/− T cell transfers. In contrast, GCs in K/IL-21−/− T cell transfer were rarely observed and the few germinal centers were amorphous and dimly labeled with PNA. The number of GCs was counted from multiple spleen sections from multiple mice (Fig. 4B). KRN WT and K/IL-21R−/− T cell transfer induced equivalent numbers of GCs per section. GC size was measured and KRN WT had slightly larger GCs on average compared to K/IL-21R−/− T cell transfer. We also assessed the GC B cells by flow cytometry (Fig. 4C). Consistent with the immunohistology, there was a significant increase in the GL-7+ Fas+ GC B cell population in KRN WT and K/IL21R−/− T cell transfer when compared to Cα−/− BxN hosts without transfers, whereas K/IL-21−/− T cells induced very few GL-7+ Fas+ GC B cells.
FIGURE 4.

GC B cell formation is impaired in K/IL-21−/− T cell transfer. (A) Representative immunohistology of spleens 29 days after transfer of CD4+ KRN cells of the indicated genotype, stained with anti-IgD (red) and PNA (green), 4× magnification. (B) Quantification of number of GC in each spleen section (normalized by section size), and size of GCs as measured by the number of pixels (n = 6 - 8 mice per group). (C) GC B cells as assessed by Fas and GL-7 expression in the CD19+ population. Percent and total number in the spleen were shown. (D) Immunohistology of spleens 29 days after transfer of CD4+ KRN cells of the indicated genotype, labeled with anti-KRN TCRα chain specific monoclonal Ab 3-4G-B7 (blue), anti-IgD (red), and PNA (green) (top), anti-KRN staining was shown again (bottom) separately, 20× magnification. For all bar graphs mean and standard deviation are shown. Results are from two experiments (n = 6 - 8 mice per group). Student’s t-test: ns, p > 0.05; *, p < 0.05; **, p< 0.01; ***, p < 0.001.
To determine if the lack of GCs in the K/IL21−/− transfer was due to defective TFH migration into the B cell follicles or defective T-B interactions, we examined spleen sections for the presence of transferred T cells in B cell follicles. As shown in Fig. 4D, staining with an anti-KRN Vα4 specific antibody 3-4G-B7 (or anti-TCR Vβ6, data not shown) revealed KRN T cells throughout the B cell follicle in all transfers. These data suggest that the lack of GCs in K/IL-21−/− is due to impaired T cell help and not T cell migration.
TH17 cells are not essential for GC responses and arthritis
IL-21 is not exclusively a TFH cytokine. The TH17 subset produces IL-21 in addition to production of Il-17A, IL-17F, and IL-22 (11). TH17 cells were shown to drive the formation of spontaneous GCs in autoimmune responses of the BXD2 mice (13) and TH17 cells can provide effective help to B cells (12, 32). We therefore tested the contribution of TH17 cells in initiating GC responses and arthritis. K/B6 mice were crossed to RORγtGFP/GFP mice (33) to generate K/RORγtGFP/GFP mice. In these mice, GFP insertion inactivates RORγt expression and TH17 differentiation is defective. CD4+ T cells were purified from either K/RORγt+/+ or K/RORγtGFP/GFP mice and transferred into Cα−/− BxN hosts. As shown in Fig. 5A, K/RORγt+/+ and K/RORγtGFP/GFP CD4+ T cells induced arthritis with a similar kinetics and severity. There was also no difference in the serum anti-GPI IgG titers (Fig. 5B). The transferred K/RORγtGFP/GFP cells were indeed deficient in generating TH17 cells as determined by IL-17 intracellular staining after in vitro stimulation (Fig. 5C). However, there was no difference in the differentiation of TFH cells (Fig. 5D). These results demonstrate that TH17 cells and their production of IL-21 are not essential for GC responses and arthritis development, supporting the conclusion that TFH cells are the major source of IL-21 production.
FIGURE 5.

TH17 cells are dispensable for arthritis. (A) KRN splenocytes purified from WT KRN/B6 (K/RORγt+/+) or KRN RORγt deficient mice (K/RORγtGFP/GFP) were transferred into Cα−/− BxN hosts. Ankle thickening was monitored over time. (B) Anti-GPI IgG titers were detected by ELISA. (C) Splenocytes were cultured in PMA and ionomycin with BFA for five hours and IL-17A production was detected by intracellular staining. Percent and total number of IL-17A producing transferred cells (CD45.1− CD4+) in the spleen are shown. (D) Percent and total number of TFH (CXCR5+ Bcl6+) of the CD45.1− CD4+ transferred cell gate in the spleen are shown. Representative of two independent experiments (n = 4 - 5 mice per group). Mean and standard deviation are shown. Student’s t-test: ns, p > 0.05; *, p < 0.05; **, p< 0.01. (E) Cells from the spleen, draining lymph nodes (dLN, pooled popliteal and inguinal lymph nodes), and mesenteric lymph nodes (mLN) from K/BxN mice and Cα−/− BxN hosts 20 days after KRN/B6 cell transfer were stimulated and assessed for IL-17A production as in (C). Representative of one experiment with two mice per group.
It has been shown previously that TH17 cell development is correlated with disease induction in K/BxN mice (34). We investigated whether there were differences in TH17 cell development and distribution in K/BxN mice vs. the transfer model. Cells isolated from the spleen, draining lymph nodes (pooled popliteal and inguinal lymph nodes), and mesenteric lymph nodes stimulated in vitro revealed a similar profile of IL-17A production from K/BxN mice and Cα−/− BxN hosts after KRN T cell transfer (Fig. 5E). IL-17A production was detected from KRN T cells identified with the anti-KRN Vα4 specific antibody 3-4G-B7. However, non-KRN T cells were the major IL-17A producers in both K/BxN and KRN transferred mice.
B cells require IL-21R signaling to initiate GC
Because IL-21 production by T cells is necessary for GC formation, it suggests that the target of IL-21 is the B cell. To directly test the importance of IL-21R expression on B cells, we performed a B and T cell co-transfer experiment. B cells purified from IL-21R+/− or IL-21R−/− mice expressing I-Ag7/b MHC class II alleles were transferred along with naïve K/B6 CD4+ cells into Rag1−/− B6xNOD F1 hosts. Most mice that received IL-21R+/− B cells developed arthritis whereas mice that received IL-21R−/− B cells never showed signs of arthritis (Fig. 6A). The disease states were reflected in the dramatic difference in anti-GPI IgG titers between these two groups of mice (Fig. 6B). There was a moderate decrease in TFH cells in hosts receiving IL-21R−/− B cells compared to those receiving IL-21R+/− B cells (Fig. 6C). This result is consistent with what was observed in the K/IL-21−/− T cell transfer, where TFH numbers decreased after 29-30 days, likely due to a lack of interaction with GC B cells to promote TFH maintenance. There was a more dramatic decrease in GC B cells in hosts receiving IL-21R−/− B cells compared to those receiving IL-21R+/− B cells (Fig. 6D). Immunohistological analysis of GCs on spleen section confirmed the results obtained by flow cytometry, although there were few B cell follicles and smaller GCs in general compared to T cell transfer into Cα−/− BxN hosts (data not shown). This is not surprising considering that Rag1−/− mice have defective follicular structures and a much smaller population of transferred B cells. These data support the conclusion that IL-21R signaling in B cells is essential for GC formation, antibody production, and arthritis development and GC B cells are required for TFH cells maintenance.
FIGURE 6.
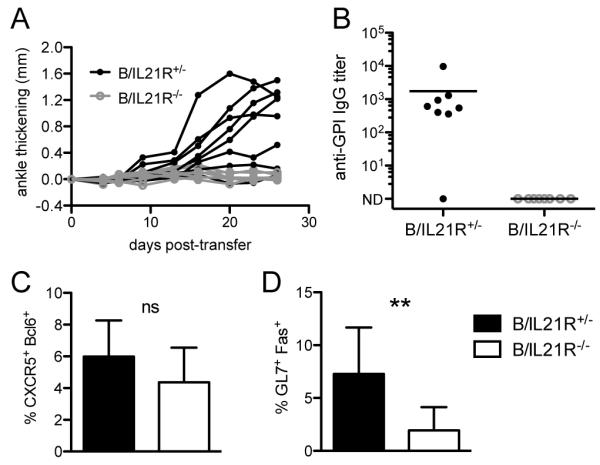
B cells require expression of IL-21R for GC formation and autoantibody production. (A) IL21R+/− or IL-21R−/− I-Ag7/b splenic B cells and CD4+ KRN/B6 T cells were transferred into Rag1−/− BxN hosts. (A) Ankle thickening was monitored over time. (B) Anti-GPI IgG titers were detected by ELISA, analyzed 37-40 days after transfer. ND (not detectable). (C) Percent of GC B cells (GL-7+Fas+) of the B220+ gate and (D) percent of TFH cells (CXCR5+Bcl6+) of the TCRβ+CD4+ gate were determined by flow cytometry. Mean and standard deviation are shown. Results shown are from two experiments (n = 8 per group). Student’s t-test: ns, p > 0.05; **, p< 0.01.
Discussion
IL-21 is a pleiotropic cytokine, affecting a diverse array of cell types (2). Without conditional deletion of IL-21 or IL-21R, it has been difficult to evaluate the specific roles of IL-21 in autoimmunity. We used a cell transfer system based on the K/BxN mouse model of autoimmune arthritis to address these roles in TFH differentiation and B cell activation. Our results demonstrate that IL-21 production by T cells is important in disease induction. However, IL-21 is not required for TFH differentiation, maintenance, or function. There was no defect in T cell survival when KRN T cells were deficient in either IL-21 or IL-21R, and in fact survival was increased one week after transfer in IL-21−/− T cells (Fig. 3A). A similar increase was observed in IL-21−/− mice after immunization with NP-KLH (35), although the mechanism for enhanced survival is unclear. Both IL-21−/− and IL-21R−/− KRN T cells proliferated and differentiated into TFH and were able to migrate into the B cell follicles in an antigen-specific manner, as this process did not take place when KRN T cells were transferred into Cα−/− B6 hosts that do not express the self-peptide-MHC complex.
In an earlier study using the IL-21R−/− K/BxN mice, it was shown that there were fewer CD4+ T cells in the spleen and joint draining lymph nodes compared to normal K/BxN mice (36). The IL-21R−/− K/BxN mice did not develop arthritis and it was attributed to a requirement of IL-21 by KRN T cells for homeostatic proliferation. The different conclusions from our study and the earlier study highlight the complex biological function of IL-21 and the problem of understanding the direct vs. indirect mechanisms in total knockout animals.
We found no difference in the GC B cell response between KRN WT and K/IL-21R−/− T cell transfer by antibody titers or GC B cell formation by flow cytometry or histology, demonstrating that there is no functional defect in TFH in the absence of IL-21R signaling. These results contrast with a study of lupus-like disease induced in a chronic graft-versus-host model, which found that GC B cells were less frequent and GCs were smaller when IL-21R−/− T cells were transferred (17). The differential dependence on IL-21R signaling may be related to the difference in frequency or affinity of alloreactive T cells, because it was shown that naïve antigen-specific helper T cells with TCRs of higher affinity preferentially differentiate into the CXCR5hi ‘resident’ TFH compartment (37).
Studies on the role of IL-21 in TFH differentiation and GC formation have led to different conclusions in mice immunized with protein antigens. Germinal center formation was relatively unaffected in IL-21−/− or IL-21R−/− mice in some studies (35, 38, 39) whereas a more profound effect on TFH and GCs was found in other studies (5, 8). It was suggested that the different results obtained in these studies might be explained by the different types of antigen, adjuvant, the avidity of TCR involved for peptide-MHC, or the timing of analysis (35, 39). Our conclusion that IL-21 is not required for TFH differentiation but rather acts on GC B cells is consistent with studies in mixed bone marrow chimeras (35, 39, 40). GC formation appears to be highly dependent on IL-21 in the K/BxN and other models of spontaneous autoimmune diseases (14, 41), although IL-21 does not play any role in the RoquinSanroque mouse model of lupus (42). Therapeutic intervention of the IL-21 signaling pathway has been explored in animal models (15, 16, 43). However, the efficacy was found to be variable and partial. For example, repeated treatment of BXSB-Yaa mice with a soluble IL-21R-Fc fusion protein had minimal effect on lupus symptoms and survival even though IL-21R−/− BXSB-Yaa have no sign of disease (14). This variation could be attributed to the partial effectiveness of IL-21R-Fc in blocking IL-21 signaling. The evidence that TFH development is not dependent on IL-21 in autoimmunity raises the possibility that an efficient inhibition of ongoing GC B cell response may be difficult to achieve in practice with partial effectiveness of IL-21 blockade. It is tempting to speculate that IL-21 blockade is more effective in cases where TFH is more dependent on the cytokine. IL-21 blockade together with a therapy targeting T cells might be most beneficial for treating certain antibody-mediated autoimmune diseases.
Arthritis development in K/BxN mice is dependent on gut microbiota, particularly the colonization of segmented filamentous bacteria. Because the KRN transfer model involves different strains of mice as source of donor cells and hosts, the potential difference in their gut microbiota might be a confounding factor in the interpretation of our experiments. However, we think it is not likely to be the case. In our experiments, we always used littermates for our cell transfer hosts, dividing the hosts housed in the same cage for different KRN genotype transfers. We used donors of different genotypes housed in the same cage when possible. Furthermore, the gut microbiota of K/IL-21−/− and K/IL-21R−/− donor mice should be very similar because they are defective in the same pathway.
TH17 cells are now recognized to interact with antigen-specific B cells as potential B cell helpers (12, 13). It was therefore important to investigate whether IL-21 required for disease induction was produced by TFH or from TH17 that enters the B cell follicle to initiate GC. By eliminating the TH17 subset through RORγtGFP/GFP, K/RORγtGFP/GFP T cells did not dramatically alter disease kinetics or severity, suggesting that IL-21 is derived from TFH cells but not TH17 cells.
At first glance, the result that TH17 cells is not essential for disease induction seems unexpected because TH17 cells have been suggested to play an important role in this disease model. TH17 cell induction correlated with disease onset, and neutralizing antibody against IL-17A prevented disease in K/BxN mice (34). IL-17R−/− B cells were defective in differentiating into GC B cells, suggesting that they are the targets of IL-17. However, other lymphocyte and innate-like cell populations, including γδ T cells and some recently characterized innate lymphoid cells are major producers of IL-17 (44, 45). The intriguing possibility that IL-17 produced constitutively by innate lymphoid cells play an important role is being investigated.
Acknowledgments
We thank Dr. Michael Grusby for IL-21R−/− mice; Drs. Diane Mathis and Christophe Benoist for KRN transgenic mice, B6.H2g7 congenic mice, Cα−/− B6, and Cα−/− NOD mice; Xiao Liu and Crystal Rayon for help with mice.
This work was supported by Grant R01 AI087645 (to H.H.) from the National Institutes of Health. K.E.B. was partially supported by National Institutes of Health/National Institute of Allergy and Infections Diseases Grant 2T32AI007090.
Abbreviations used in this article
- GPI
glucose-6-phosphate isomerase
- GC
germinal center
- TFH
follicular helper T cell
References
- 1.Spolski R, Leonard WJ. Interleukin-21: basic biology and implications for cancer and autoimmunity. Annu. Rev. Immunol. 2008;26:57–79. doi: 10.1146/annurev.immunol.26.021607.090316. [DOI] [PubMed] [Google Scholar]
- 2.Ettinger R, Kuchen S, Lipsky PE. The role of IL-21 in regulating B-cell function in health and disease. Immunol Rev. 2008;223:60–86. doi: 10.1111/j.1600-065X.2008.00631.x. [DOI] [PubMed] [Google Scholar]
- 3.Parrish-Novak J, Dillon SR, Nelson A, Hammond A, Sprecher C, Gross JA, Johnston J, Madden K, Xu W, West J, Schrader S, Burkhead S, Heipel M, Brandt C, Kuijper JL, Kramer J, Conklin D, Presnell SR, Berry J, Shiota F, Bort S, Hambly K, Mudri S, Clegg C, Moore M, Grant FJ, Lofton-Day C, Gilbert T, Rayond F, Ching A, Yao L, Smith D, Webster P, Whitmore T, Maurer M, Kaushansky K, Holly RD, Foster D. Interleukin 21 and its receptor are involved in NK cell expansion and regulation of lymphocyte function. Nature. 2000;408:57–63. doi: 10.1038/35040504. [DOI] [PubMed] [Google Scholar]
- 4.Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, Schluns K, Tian Q, Watowich SS, Jetten AM, Dong C. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- 5.Vogelzang A, Mcguire H, Yu D, Sprent J, Mackay C, King C. A Fundamental Role for Interleukin-21 in the Generation of T Follicular Helper Cells. Immunity. 2008;29:127–137. doi: 10.1016/j.immuni.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 6.Suto A, Kashiwakuma D, Kagami S-I, Hirose K, Watanabe N, Yokote K, Saito Y, Nakayama T, Grusby MJ, Iwamoto I, Nakajima H. Development and characterization of IL-21-producing CD4+ T cells. J Exp Med. 2008;205:1369–1379. doi: 10.1084/jem.20072057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Korn T, Bettelli E, Gao W, Awasthi A, Jäger A, Strom TB, Oukka M, Kuchroo VK. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nurieva RI, Chung Y, Hwang D, Yang XO, Kang HS, Ma L, Wang Y-H, Watowich SS, Jetten AM, Tian Q, Dong C. Generation of T Follicular Helper Cells Is Mediated by Interleukin-21 but Independent of T Helper 1, 2, or 17 Cell Lineages. Immunity. 2008;29:138–149. doi: 10.1016/j.immuni.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crotty S. Follicular helper CD4 T cells (TFH) Annu. Rev. Immunol. 2011;29:621–663. doi: 10.1146/annurev-immunol-031210-101400. [DOI] [PubMed] [Google Scholar]
- 10.King C, Tangye SG, Mackay CR. T follicular helper (TFH) cells in normal and dysregulated immune responses. Annu. Rev. Immunol. 2008;26:741–766. doi: 10.1146/annurev.immunol.26.021607.090344. [DOI] [PubMed] [Google Scholar]
- 11.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu. Rev. Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 12.Mitsdoerffer M, Lee Y, Jäger A, Kim H-J, Korn T, Kolls JK, Cantor H, Bettelli E, Kuchroo VK. Proinflammatory T helper type 17 cells are effective B-cell helpers. Proceedings of the National Academy of Sciences. 2010;107:14292–14297. doi: 10.1073/pnas.1009234107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsu H-C, Yang P, Wang J, Wu Q, Myers R, Chen J, Yi J, Guentert T, Tousson A, Stanus AL, Le T-VL, Lorenz RG, Xu H, Kolls JK, Carter RH, Chaplin DD, Williams RW, Mountz JD. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol. 2008;9:166–175. doi: 10.1038/ni1552. [DOI] [PubMed] [Google Scholar]
- 14.Bubier JA, Sproule TJ, Foreman O, Spolski R, Shaffer DJ, Morse HC, Leonard WJ, Roopenian DC. A critical role for IL-21 receptor signaling in the pathogenesis of systemic lupus erythematosus in BXSB-Yaa mice. Proceedings of the National Academy of Sciences. 2009;106:1518–1523. doi: 10.1073/pnas.0807309106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Young DA, Hegen M, Ma HLM, Whitters MJ, Albert LM, Lowe L, Senices M, Wu PW, Sibley B, Leathurby Y, Brown TP, Nickerson-Nutter C, Keith JC, Collins M. Blockade of the interleukin-21/interleukin-21 receptor pathway ameliorates disease in animal models of rheumatoid arthritis. Arthritis Rheum. 2007;56:1152–1163. doi: 10.1002/art.22452. [DOI] [PubMed] [Google Scholar]
- 16.Herber D, Brown TP, Liang S, Young DA, Collins M, Dunussi-Joannopoulos K. IL-21 has a pathogenic role in a lupus-prone mouse model and its blockade with IL-21R.Fc reduces disease progression. J Immunol. 2007;178:3822–3830. doi: 10.4049/jimmunol.178.6.3822. [DOI] [PubMed] [Google Scholar]
- 17.Nguyen V, Luzina I, Rus H, Tegla C, Chen C, Rus V. IL-21 Promotes Lupus-like Disease in Chronic Graft-versus-Host Disease through Both CD4 T Cell- and B Cell-Intrinsic Mechanisms. The Journal of Immunology. 2012;189:1081–1093. doi: 10.4049/jimmunol.1200318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Odegard JM, Marks BR, DiPlacido LD, Poholek AC, Kono DH, Dong C, Flavell RA, Craft J. ICOS-dependent extrafollicular helper T cells elicit IgG production via IL-21 in systemic autoimmunity. J Exp Med. 2008;205:2873–2886. doi: 10.1084/jem.20080840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sawalha AH, Kaufman KM, Kelly JA, Adler AJ, Aberle T, Kilpatrick J, Wakeland EK, Li Q-Z, Wandstrat AE, Karp DR, James JA, Merrill JT, Lipsky P, Harley JB. Genetic association of interleukin-21 polymorphisms with systemic lupus erythematosus. Annals of the Rheumatic Diseases. 2008;67:458–461. doi: 10.1136/ard.2007.075424. [DOI] [PubMed] [Google Scholar]
- 20.Webb R, Merrill JT, Kelly JA, Sestak A, Kaufman KM, Langefeld CD, Ziegler J, Kimberly RP, Edberg JC, Ramsey-Goldman R, Petri M, Reveille JD, Alarcón GS, Vilá LM, Alarcón-Riquelme ME, James JA, Gilkeson GS, Jacob CO, Moser KL, Gaffney PM, Vyse TJ, Nath SK, Lipsky P, Harley JB, Sawalha AH. A polymorphism within IL21R confers risk for systemic lupus erythematosus. Arthritis Rheum. 2009;60:2402–2407. doi: 10.1002/art.24658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spolski R, Kashyap M, Robinson C, Yu Z, Leonard WJ. IL-21 signaling is critical for the development of type I diabetes in the NOD mouse. Proceedings of the National Academy of Sciences. 2008;105:14028–14033. doi: 10.1073/pnas.0804358105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.King C, Ilic A, Koelsch K, Sarvetnick N. Homeostatic expansion of T cells during immune insufficiency generates autoimmunity. Cell. 2004;117:265–277. doi: 10.1016/s0092-8674(04)00335-6. [DOI] [PubMed] [Google Scholar]
- 23.Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D. Organ-specific disease provoked by systemic autoimmunity. Cell. 1996;87:811–822. doi: 10.1016/s0092-8674(00)81989-3. [DOI] [PubMed] [Google Scholar]
- 24.Matsumoto I, Staub A, Benoist C, Mathis D. Arthritis provoked by linked T and B cell recognition of a glycolytic enzyme. Science. 1999;286:1732–1735. doi: 10.1126/science.286.5445.1732. [DOI] [PubMed] [Google Scholar]
- 25.Korganow AS, Ji H, Mangialaio S, Duchatelle V, Pelanda R, Martin T, Degott C, Kikutani H, Rajewsky K, Pasquali JL, Benoist C, Mathis D. From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity. 1999;10:451–461. doi: 10.1016/s1074-7613(00)80045-x. [DOI] [PubMed] [Google Scholar]
- 26.Malherbe L, Mark L, Fazilleau N, McHeyzer-Williams LJ, McHeyzer-Williams MG. Vaccine adjuvants alter TCR-based selection thresholds. Immunity. 2008;28:698–709. doi: 10.1016/j.immuni.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.LaBranche TP, Hickman-Brecks CL, Meyer DM, Storer CE, Jesson MI, Shevlin KM, Happa FA, Barve RA, Weiss DJ, Minnerly JC, Racz JL, Allen PM. Characterization of the KRN cell transfer model of rheumatoid arthritis (KRN-CTM), a chronic yet synchronized version of the K/BxN mouse. Am J Pathol. 2010;177:1388–1396. doi: 10.2353/ajpath.2010.100195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kasaian MT, Whitters MJ, Carter LL, Lowe LD, Jussif JM, Deng B, Johnson KA, Witek JS, Senices M, Konz RF, Wurster AL, Donaldson DD, Collins M, Young DA, Grusby MJ. IL-21 limits NK cell responses and promotes antigen-specific T cell activation: a mediator of the transition from innate to adaptive immunity. Immunity. 2002;16:559–569. doi: 10.1016/s1074-7613(02)00295-9. [DOI] [PubMed] [Google Scholar]
- 29.Eberl G, Marmon S, Sunshine M-J, Rennert PD, Choi Y, Littman DR. An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nat Immunol. 2004;5:64–73. doi: 10.1038/ni1022. [DOI] [PubMed] [Google Scholar]
- 30.Luhder FF, Höglund PP, Allison JPJ, Benoist CC, Mathis DD. Cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) regulates the unfolding of autoimmune diabetes. J Exp Med. 1998;187:427–432. doi: 10.1084/jem.187.3.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, Lao C, Crotty S. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity. 2011;34:932–946. doi: 10.1016/j.immuni.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hickman-Brecks CL, Racz JL, Meyer DM, LaBranche TP, Allen PM. Th17 cells can provide B cell help in autoantibody induced arthritis. J Autoimmun. 2011;36:65–75. doi: 10.1016/j.jaut.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 34.Wu H-J, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y, Littman DR, Benoist C, Mathis D. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity. 2010;32:815–827. doi: 10.1016/j.immuni.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zotos DD, Coquet JMJ, Zhang YY, Light AA, D’Costa KK, Kallies AA, Corcoran LML, Godfrey DID, Toellner K-MK, Smyth MJM, Nutt SLS, Tarlinton DMD. IL-21 regulates germinal center B cell differentiation and proliferation through a B cell-intrinsic mechanism. J Exp Med. 2010;207:365–378. doi: 10.1084/jem.20091777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jang E, Cho S-H, Park H, Paik D-J, Kim JM, Youn J. A positive feedback loop of IL-21 signaling provoked by homeostatic CD4+CD25-T cell expansion is essential for the development of arthritis in autoimmune K/BxN mice. J Immunol. 2009;182:4649–4656. doi: 10.4049/jimmunol.0804350. [DOI] [PubMed] [Google Scholar]
- 37.Fazilleau N, McHeyzer-Williams LJ, Rosen H, McHeyzer-Williams MG. The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nat Immunol. 2009;10:375–384. doi: 10.1038/ni.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ozaki K, Spolski R, Feng CG, Qi C-F, Cheng J, Sher A, Morse HC, Liu C, Schwartzberg PL, Leonard WJ. A critical role for IL-21 in regulating immunoglobulin production. Science. 2002;298:1630–1634. doi: 10.1126/science.1077002. [DOI] [PubMed] [Google Scholar]
- 39.Linterman MA, Beaton L, Yu Di, Ramiscal RR, Srivastava M, Hogan JJ, Verma NK, Smyth MJ, Rigby RJ, Vinuesa CG. IL-21 acts directly on B cells to regulate Bcl-6 expression and germinal center responses. J Exp Med. 2010;207:353–363. doi: 10.1084/jem.20091738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bessa J, Kopf M, Bachmann MF. Cutting edge: IL-21 and TLR signaling regulate germinal center responses in a B cell-intrinsic manner. The Journal of Immunology. 2010;184:4615–4619. doi: 10.4049/jimmunol.0903949. [DOI] [PubMed] [Google Scholar]
- 41.Rankin AL, Guay H, Herber D, Bertino SA, Duzanski TA, Carrier Y, Keegan S, Senices M, Stedman N, Ryan M, Bloom L, Medley Q, Collins M, Nickerson-Nutter C, Craft J, Young D, Dunussi-Joannopoulos K. IL-21 receptor is required for the systemic accumulation of activated B and T lymphocytes in MRL/MpJ-Fas(lpr/lpr)/J mice. The Journal of Immunology. 2012;188:1656–1667. doi: 10.4049/jimmunol.1003871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Linterman MA, Rigby RJ, Wong RK, Yu D, Brink R, Cannons JL, Schwartzberg PL, Cook MC, Walters GD, Vinuesa CG. Follicular helper T cells are required for systemic autoimmunity. J Exp Med. 2009;206:561–576. doi: 10.1084/jem.20081886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bubier JA, BENNETT SM, SPROULE TJ, LYONS BL, OLLAND S, YOUNG DA, ROOPENIAN DC. Treatment of BXSB-Yaa mice with IL-21R-Fc fusion protein minimally attenuates systemic lupus erythematosus. Annals of the New York Academy of Sciences. 2007;1110:590–601. doi: 10.1196/annals.1423.063. [DOI] [PubMed] [Google Scholar]
- 44.Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol. 2010;10:479–489. doi: 10.1038/nri2800. [DOI] [PubMed] [Google Scholar]
- 45.Sutton CE, Mielke LA, Mills KHG. IL-17-producing γδ T cells and innate lymphoid cells. Eur J Immunol. 2012;42:2221–2231. doi: 10.1002/eji.201242569. [DOI] [PubMed] [Google Scholar]