

Abstract
Although the requirement for new protein synthesis in synaptic plasticity and memory has been well established, recent genetic, molecular, electrophysiological, and pharmacological studies have broadened our understanding of the translational control mechanisms that are involved in these processes. One of the critical translational control points mediating general and gene-specific translation depends on the phosphorylation of eukaryotic initiation factor 2 alpha (eIF2α) by four regulatory kinases. Here, we review the literature highlighting the important role for proper translational control via regulation of eIF2α phosphorylation by its kinases in long-lasting synaptic plasticity and long-term memory.
Keywords: protein synthesis, translation initiation, synaptic plasticity, learning, memory, knockout mouse, eIF2, GCN2, PERK, PKR, HRI
Introduction
One of the more remarkable features of the brain is the ability to acquire and store new information as lasting memory traces. This continuous capacity to learn and remember allows one to process changes in the environment, retain new information, and adapt to behavioral choices over time. A fundamental question remains that intrigues modern neuroscientists: how are memories formed and stored at the cellular and molecular level? Behavioral studies performed in mice treated with the protein synthesis inhibitor puromycin provided the first molecular clue that protein synthesis is required for long-term memory (LTM) formation, but not for task acquisition and short-term memory (STM) formation (Flexner et al., 1963). Since then, a plethora of pharmacological and genetic studies have highlighted the critical role for de novo gene expression and protein synthesis in LTM formation (Kandel, 2001; McGaugh, 2000).
Neurons can alter their molecular and physiological characteristics in response to temporaland activity-dependent changes in their environment. Synaptic plasticity refers to the ability of the brain to change the efficacy (strengthening or weakening) of synaptic connections between neurons and is hypothesized as the cellular basis for learning and memory (Bliss and Collingridge, 1993; Malenka and Nicoll, 1999). These persistent, activity-dependent, changes in synaptic strength are triggered by de novo protein synthesis (Klann and Sweatt, 2008). Evidence indicating a role for protein synthesis at local synaptic sites stem from observations that neuronal dendrites and their spines contain polyribosomes (Steward and Levy, 1982), translation factors (Tang and Schuman, 2002), and mRNA (Crino and Eberwine, 1996) that can be translated into proteins to support synaptic activity. Consistent with this notion, local protein synthesis was shown to be necessary for long-lasting increases in synaptic strength induced by brain-derived neurotrophic factor (BDNF; Kang and Schuman, 1996). Similarly, rapid, local protein synthesis also was required for long-lasting decreases in synaptic strength induced by activation of group I metabotropic glutamate receptors (mGluR; Huber et al., 2000). Together, these findings indicate that protein synthesis can be triggered locally at activated synapses and is required for persistent, activity-dependent forms of synaptic plasticity, which in turn is thought to be essential for memory formation.
Although the initial report fromFlexner et al. (1963) and other early studies identified new protein synthesis as a molecular requirement for memory formation, they offered little in the way of molecular translational control mechanisms because they relied mostly on the administration of general translation inhibitors into animals. In the last 10 years, however, a vast amount of genetic, biochemical, pharmacological, and physiological studies have increased our knowledge of the precise translational control mechanisms underlying long-lasting synaptic plasticity, memory formation, and cognitive function (Costa-Mattioli et al., 2009; Kelleher et al., 2004; Richter and Klann, 2009). In this review, we specifically discuss the functional role of eIF2α kinases and their regulation of activity-dependent synaptic plasticity and cognitive function, including learning and memory.
Translational Control by eIF2α Phosphorylation
Translational control can be defined as a change in either the efficiency or rate of mRNA translation. The process of mRNA translation can be divided into three main steps: initiation, elongation, and termination. Although regulation can occur at each step, translational control primarily occurs at the rate-limiting initiation step when the small 40S ribosomal subunit is recruited to the mRNA and positioned at the initiation codon (Jackson et al., 2010). Translation initiation itself can be further divided into three key steps. First, the initiator methionyl transfer RNA (Met-tRNAi Met) binds to the small 40S ribosomal subunit, forming the 43S preinitiation complex. This is followed by the binding of the 43S complex to the mRNA so that it can find the initiation codon, thereby forming the 48S complex. Finally, the large ribosomal subunit joins the 48S complex to generate an 80S translational-competent ribosome, which can subsequently proceed with elongation (Jackson et al., 2010; Pestova et al., 2007).
One highly conserved mechanism of translational control in eukaryotic cells involves phosphorylation of eukaryotic initiation factor 2 (eIF2). In this early step in translation initiation, eIF2, a heterotrimer consisting of α, β, and γ subunits, binds Met-tRNAi Met and GTP to form the stable 43S preinitiation complex (eIF2-GTP-Met-tRNAi Met). Exchange of GDP for GTP is promoted by eIF2B, a guanine-nucleotide exchange factor that is required to regenerate the active GTP-bound eIF2 that is required for new rounds of translation. The guanine-nucleotide exchange on eIF2 serves as a critical translational control point and is regulated via phosphorylation. Specifically, the phosphorylation of eIF2 on its α subunit at serine 51 (Ser51) converts eIF2 to a competitive inhibitor of eIF2B, which blocks the GDP/GTP-exchange and causes a decrease in general translation initiation (Pestova et al., 2007; Sonenberg and Dever, 2003).
Although eIF2α phosphorylation inhibits general translation, it also selectively increases the translation of a subset of mRNAs that contain upstream open reading frames (uORFs) in their 5’ untranslated region (UTR). uORFs are present in nearly half of rodent and human transcripts (Iacono et al., 2005; Matsui et al., 2007; Mignone et al., 2002), but despite their prevalence, they are less frequent than expected by chance (Iacono et al., 2005), and also are highly conserved (Neafsey and Galagan, 2007).
Probably the best characterized example of gene-specific translational control via eIF2α phosphorylation is that of the yeast transcriptional activator GCN4 (Hinnebusch and Natarajan, 2002). When general translation was inhibited by eIF2α phosphorylation, GCN4 translation, as well as the translation of the transcriptional modulator ATF4 (activating transcription factor 4; also termed CREB2) was enhanced (Harding et al., 2000; Vattem and Wek, 2004). Additional uORF-containing mRNAs have been shown to be translated under conditions resulting in enhanced eIF2α phosphorylation, including the CAAT/enhancer binding proteins C/EBPα and β (Calkhoven et al., 2000) and the β-site β-amyloid precursor protein (APP)-cleaving enzyme BACE1 (De Pietri Tonelli et al., 2004; Lammich et al., 2004). Notably, in multiple species ATF4 and its homologs act as repressors of cAMP-responsive element binding protein (CREB)-mediated gene expression, which is known to be required for long-lasting changes in synaptic plasticity and LTM (Abel et al., 1998; Bartsch et al., 1995; Chen et al., 2003). Thus, eIF2α phosphorylation controls both general and gene-specific translation that regulates CREB-mediated transcription, two distinct processes that are required for long-lasting synaptic plasticity and LTM formation.
eIF2α Kinases
eIF2α phosphorylation is regulated by four serine/threonine (Ser/Thr) protein kinases, each of which phosphorylate eIF2α on Ser51. The four eIF2α kinases are heme-regulated inhibitor (HRI), the double-stranded (ds) RNA activated protein kinase (PKR), the general control non-derepressible-2 (GCN2), and the PKR-like endoplasmic reticulum (ER) resident protein kinase (PERK). These four eIF2α kinases share a conserved kinase domain, but respond differentially to various cellular stressors due to divergent regulatory domains (Dever et al., 2007). For example, HRI is activated by conditions of heme deficiency (Mellor et al., 1994), PKR is activated by double-stranded RNA (dsRNA; Meurs et al., 1990), GCN2 is activated by amino acid deprivation as well as UV irradiation (Deng et al., 2002; Sood et al., 2000), and PERK is activated by an accumulation of misfolded proteins in the ER (Harding et al., 1999; Shi et al., 1998). Thus, depending on the particular cellular stimuli, specific eIF2α kinases become active and phosphorylate eIF2α to control both general and gene-specific translation (Fig. 1). There is very little information concerning whether these kinases are activated under normal physiological conditions, especially in neurons.
Fig. 1. Schematic representation of translational control by eIF2α kinases.
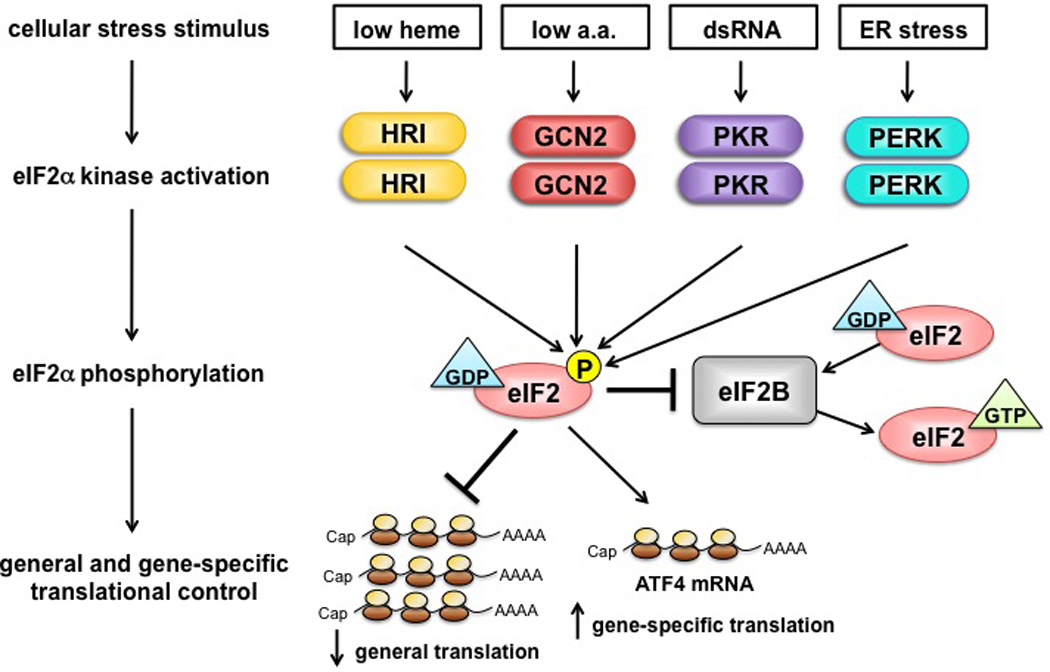
In response to distinct cellular stress stimuli, four eIF2α kinases become activated and phosphorylate the α subunit of eIF2. The guanine nucleotide exchange factor eIF2B catalyzes the exchange of inactive GDP for active GTP-bound eIF2, a process required for new rounds of translation initiation. Phosphorylation of eIF2 inhibits eIF2B activity, which blocks GDP/GTP-exchange, resulting in the reduction of general translation and the stimulation of gene-specific translation of uORF-containing mRNAs (for example, ATF4).
All eIF2α kinases are abundantly expressed in the mammalian brain (Berlanga et al., 1999;Harding et al., 1999; Meurs et al., 1990; Shi et al., 1998; Sood et al., 2000; Trinh et al., 2012;Zhu et al., 2011), with the exception of HRI whose expression is relatively low (Crosby et al., 1994; Mellor et al., 1994). We will describe the most salient aspects of GCN2, PKR, and PERK because they are known to play important roles in protein-synthesis dependent synaptic plasticity and cognitive function, including learning and memory.
GCN2 is present in all eukaryotes (Dever et al., 2007; Hinnebusch et al., 2004; Sood et al., 2000) and is activated in response to amino acid starvation via the accumulation of uncharged tRNAs. GCN2’s structure is complex and contains five domains: (1) an N-terminal domain that binds to GCN1 and is required for activation, (2) a pseudokinase domain, (3) an eIF2α kinase domain, (4) a regulatory domain resembling histidyl-tRNA synthetase (HisRS), containing a conserved sequence (motif 2), which is thought to bind all deacylated tRNAs with similar affinity, and (5) a carboxy-terminal domain that dimerizes, enhances tRNA binding, and mediates ribosomal binding (Dever et al., 2007). In contrast to the monomers PKR and PERK that require dimerization for activation, GCN2 exists constitutively as a dimer where the HisRS domain interacts with both the kinase domain and the carboxy-terminal domain to maintain it in inactive state. In response to amino acid starvation, uncharged tRNAs accumulate and bind to the HisRS domain, which results in the release of these inhibitory interactions and the subsequent activation of GCN2 (Dever et al., 2007). GCN2 also is activated by UV irradiation, high salinity, rapamycin, and glucose limitation (Deng et al., 2002; Hinnebusch, 2005). Interestingly, these stress stimuli could not activate a GCN2 mutant lacking a functional HisRS domain (Hinnebusch, 2005; Wek et al., 1995). All together, these findings indicate that uncharged tRNA is the main activator of GCN2.
PKR is expressed widely in vertebrates and activated in response to dsRNA produced during viral infection (Dever et al., 2007). Compared to GCN2 and PERK, the structure of PKR is relatively simple with an N-terminal dsRNA-binding domain (dsRBD), which consists of two dsRNA binding motifs (dsRBMs), and a carboxy-terminal kinase domain. Direct binding of dsRNA to the dsRBMs induces a conformational change exposing the kinase domain of PKR, which subsequently promotes dimerization and kinase activation (Carpick et al., 1997; Patel and Sen, 1998; Zhang et al., 2001). PKR and PERK regulate protein synthesis during viral infection and ER stress, respectively, but also regulate the levels of specific proteins via degradation (Baltzis et al., 2007; Raven et al., 2008). A growing body of evidence indicates that protein synthesis as well as degradation plays a critical role in synaptic plasticity and memory (Bingol and Sheng, 2011; Kaang and Choi, 2012). Thus, it seems likely that activation of eIF2α kinases control not only protein synthesis, but also other cellular processes that are important for changes in protein expression that regulate synaptic and cognitive function. Moreover, several neurological conditions including status epilepticus (Carnevalli et al., 2006), Alzheimer’s disease (Couturier et al., 2010; Dumurgier et al., 2013; Peel and Bredesen, 2003), Huntington’s disease (Bando et al., 2005; Peel et al., 2001), Parkinson’s disease (Bando et al., 2005), and Creutzfeldt-Jakob’s disease (Paquet et al., 2009) have been linked to activation of the PKR-eIF2α axis, suggesting a role for PKR-regulated translational control pathologies associated with neurological disorders.
PERK has been identified in both vertebrates and invertebrates and becomes activated in response to an accumulation of misfolded proteins in the ER (Harding et al., 1999; Shi et al., 1998; Wek and Cavener, 2007). PERK contains four domains, including a signal peptide, an N-terminal regulatory region, a transmembrane domain, and a cytoplasmic kinase domain. Normally, the ER chaperones BiP (GRP78) and GRP94 bind to PERK, keeping it inactive as a monomer (Bertolotti et al., 2000). During ER stress, BiP and GRP94 dissociate from PERK, which results in dimerization, autophosphorylation, and activation of the kinase. Previous studies showed that mutations in the human PERK gene (EIF2AK3) causes Wolcott-Rallison syndrome (WRS), a rare autosomal recessive disorder characterized by permanent neonatal diabetes, multiple epiphyseal dysplasia, liver dysfunction, and pancreas insufficiency (Delepine et al., 2000; Julier and Nicolino, 2010; Rubio-Cabezas et al., 2009). Consistent with these findings, molecular and genetic studies have shown that these pathologies are recapitulated in PERK-deficient mice (Harding et al., 2001; Wei et al., 2008; Zhang et al., 2002; Zhang et al., 2006). In some cases of WRS, patients have been reported to develop clinical features associated with mental retardation (Delepine et al., 2000; Reis et al., 2011; Senee et al., 2004; Thornton et al., 1997). In addition, cell lines lacking either of the tuberous sclerosis complex (TSC) proteins, TSC1 or TSC2, and both mouse and human tumors from TSC model mice and patients, respectively, exhibited activation of PERK-eIF2α signaling (Ozcan et al., 2008). Finally, a single nucleotide polymorphism in EIF2AK3 has been identified that is associated with progressive supranuclear palsy, a movement disorder with prominent tau neuropathology (Hoglinger et al., 2011). Collectively, these findings suggest that PERK is a key factor in severe pathologies associated with dysregulated protein synthesis.
In higher eukaryotes, the dynamic regulation of eIF2α phosphorylation is critical for cell survival. For example, PERK is rapidly activated by ER stress, but within minutes of the restoration of ER homeostasis, PERK is dephosphorylated and inactivated (Ma and Hendershot, 2003; Ron and Walter, 2007). Genetic screening studies in somatic cells have identified two phosphatase complexes that independently dephosphorylate eIF2α. The first complex consists of the eIF2α-specific regulatory subunit CReP (constitutive repressor of eIF2α phosphorylation) and PPIc (protein phosphatase I catalytic subunit; (Jousse et al., 2003). The second complex consists of the related regulatory subunit GADD34 (growth arrest and DNA damage-inducible gene 34) and PPIc (Connor et al., 2001; Jousse et al., 2003; Novoa et al., 2001). CReP is constitutively expressed and is thought to contribute to baseline eIF2α dephosphorylation, whereas GADD34 is transcriptionally induced by eIF2α phosphorylation and serves within a negative feedback loop to restore protein synthesis (Ma and Hendershot, 2003).
GCN2 Controls L-LTP and LTM
As previously mentioned, distinct eIF2α kinases phosphorylate eIF2α to control two fundamental processes that are crucial for the consolidation of long-term memories: de novo protein synthesis and CREB-mediated gene expression via the memory repressing factor ATF4. Evidence that regulation of eIF2α phosphorylation plays an important role in long-lasting synaptic plasticity and LTM was first provided by the observation that GCN2-deficient mice exhibit a lowered threshold for the induction of long-lasting late phase long-term potentiation (L-LTP) and the consolidation of long-term memory (Costa-Mattioli et al., 2005). The LTP phenotype observed in GCN2-deficient mice also has been observed in mice lacking other translational repressors such as 4E-BP2 (Banko et al., 2005) and TSC2 (Ehninger et al., 2008). Consistent with these findings, heterozygous eIF2α knockin mice with a mutation on Ser 51 of eIF2α also have a decreased threshold for the induction of L-LTP (Costa-Mattioli et al., 2007). Moreover, decreased eIF2α phosphorylation in GCN2-deficient mice and heterozygous eIF2α knockin mice was associated with enhanced LTM in multiple tasks using training paradigms that normally do not elicit LTM (Costa-Mattioli et al., 2005; Costa-Mattioli et al., 2007). In contrast, hippocampal infusion with Sal003, an inhibitor of eIF2α dephosphorylation (Boyce et al., 2005), prevented the induction of both L-LTP and LTM (Costa-Mattioli et al., 2007). Taken together, these findings indicate that GCN2-dependent phosphorylation of eIF2α is required for the expression of L-LTP and LTM.
A possible mechanism for gene-specific translation via GCN2-dependent phosphorylation of eIF2α in L-LTP and LTM involves the translation of ATF4 mRNA, which then would block the CREB-dependent transcription of synaptic plasticity-related genes and hence, L-LTP and LTM formation. Evidence supporting this hypothesis is provided by the observation that basal levels of eIF2α phosphorylation and ATF4 expression were decreased in GCN2-deficient mice, which were associated with a lower threshold for activation of CREB-mediated gene expression (Costa-Mattioli et al., 2005). Therefore, it is possible that the effect of GCN2 on long-lasting LTP and memory is mediated by the modulation of ATF4/CREB activity. Although the threshold for L-LTP is decreased in GCN2-deficient mice, normal L-LTP-inducing stimulation failed to elicit L-LTP (Costa-Mattioli et al., 2005), suggesting that additional inhibitory factors may be upregulated due to the lack of translational repression and consequently block L-LTP and LTM in these mice. For instance, L-LTP-inducing stimulation may increase the levels of IMPACT, a GCN2 inhibitor, whose expression is enriched in the mammalian brain (Pereira et al., 2005). Furthermore, although the threshold for inducing LTM is decreased in the GCN2-deficient mice, robust training paradigms that normally produce LTM fail to elicit it in these mice (Costa-Mattioli et al., 2005). Collectively, these findings suggest that GCN2-dependent regulation of ATF4 translation must be tightly controlled for the normal expression of L-LTP and LTM.
Genetic deletion of GCN2 appears to affect LTP, but not LTD. In contrast to LTP, mGluR-LTD, which was enhanced in 4E-BP2 knockout mice (Banko et al., 2006), was unaffected in GCN2-deficient mice (Costa-Mattioli et al., 2005). As will be discussed in more detail later, LTP, which has a lower threshold for induction in GCN2-deficient mice, was unaffected in mice with a forebrain specific disruption of PERK (PERK cKO; Trinh et al., 2013 in revision). Furthermore, mGluR-LTD, which is protein synthesis-dependent and unaffected in GCN2-deficient mice, was enhanced in PERK cKO mice (Trinh et al., 2013 in revision), suggesting the interesting possibility that distinct pools of mRNA may be differentially translated during protein synthesis-dependent forms of LTP and LTD. Thus, these findings suggest that the regulation of CREB-dependent gene expression via GCN2-dependent ATF4 translation is specific to LTP.
PKR Controls Gene-specific Translation, L-LTP, and LTM
Using a novel pharmacogenetic mouse model,Jiang et al. (2010) demonstrated that a selective increase in PKR-mediated eIF2α phosphorylation in CA1 hippocampal neurons impairs L-LTP and LTM. Accordingly, the increase in eIF2α phosphorylation was associated with an increase in ATF4 translation and the suppression of CREB-dependent gene expression (Jiang et al., 2010), including that of BDNF, a key protein involved in L-LTP and the consolidation of LTM (Bekinschtein et al., 2008). It should be noted that although general protein synthesis was not altered, ATF4 translation was increased in CA1 neurons from these mice, suggesting that gene-specific translation of ATF4 is required for the suppression of L-LTP and LTM. Consistent with these findings, Sal003, which increases eIF2α phosphorylation, failed to inhibit L-LTP in hippocampal slices from ATF4-deficient mice (Costa-Mattioli et al., 2007). Collectively, these findings indicate that precise control of specific mRNAs (such as ATF4 or BDNF), rather than general translation, is required for the proper expression of long-lasting synaptic plasticity and long-term memory.
Recent pharmacological and genetic studies confirm an important role for endogenous PKR in the control of neural networks and normal cognitive function. For example, pharmacological inhibition of PKR in the gustatory cortex of mice and rats enhanced both positive and negative forms of long-term taste memory (Stern et al., 2013). In addition, either genetic deletion or pharmacological inhibition of PKR results in hyperexcitability of cortical and hippocampal networks, facilitated L-LTP, and enhanced LTM (Zhu et al., 2011). It was proposed that PKR regulates these processes via interferon gamma (IFN-γ)-mediated disinhibition at the synapse. Indeed, IFNγ mRNA normally activates PKR in the cell to specifically inhibit its own translation (Ben-Asouli et al., 2002; Cohen-Chalamish et al., 2009). Subsequently, it was shown that when PKR activity was inhibited, IFNγ mRNA translation was increased, resulting in a loss of GABAergic transmission, enhanced neuronal excitability, and improved cognitive performance (Zhu et al., 2011). Thus, these findings indicate that PKR activation plays a key role in modulating activity-dependent changes in synaptic strength, network rhythmicity, and cognition.
PKR also has been proposed to be a cognitive decline biomarker in patients suffering from Alzheimer’s disease (AD) based on correlations between cognitive and memory test scores with activation and dysregulation of PKR-mediated translation (Damjanac et al., 2009). Moreover, recent studies showed that PKR activity and eIF2α phosphorylation was increased in mice overexpressing ApoE4, a model for sporadic AD, as well as in aging rodents (Segev et al., 2013).These findings suggest that PKR-eIF2α signaling may contribute to the aging-related cognitive decline associated with sporadic AD.
PERK Controls ATF4 Translation, mGluR-LTD, and Behavioral Flexibility
Using mouse behavioral genetics and biochemistry, it was demonstrated that PERK plays a crucial role in regulating behavioral flexibility (Trinh et al., 2012). Previous studies showed that global inactivation of PERK results in severe developmental abnormalities (Wei et al., 2008; Zhang et al., 2002). To rule out the possibility that the effects of PERK on synaptic and cognitive function occur during development, mice were generated in which PERK was selectively removed in the forebrain at approximately 3 weeks of age (Trinh et al., 2012). Although general protein synthesis was unaltered in the prefrontal cortex (PFC) of PERK cKO mice, both eIF2α phosphorylation and ATF4 expression were reduced significantly (Trinh et al., 2012). Interestingly, these molecular changes in the PERK cKO mice were associated with multiple phenotypes consistent with impaired information processing and behavioral flexibility (Trinh et al., 2012). For example, PERK cKO mice exhibited reduced paired pulse inhibition, a type of sensorimotor-gating that restricts the processing of sensory information (Bitsios et al., 2006; Braff et al., 2001). In addition, PERK cKO mice showed enhanced preference for the familiar object compared with the novel object when tested in the novel object recognition task (Trinh et al., 2012). Furthermore, although PERK cKO mice exhibited normal task acquisition and spatial memory in the Morris water maze, they displayed enhanced perseveration for the originally learned platform position, even after it has been moved to the opposite quadrant. Similarly, PERK cKO mice displayed severe behavioral inflexibility in the Y-water maze choice reversal task (Trinh et al., 2012) in which the mice are trained to shift their response pattern when the platform position is relocated to the other arm of the maze. This type of inhibitory learning is usually called reversal learning. One possible explanation for these results is that PERK plays an important role in regulating frontal and temporal cortex-dependent sensory information processing. Hence, in the absence of PERK, mice are unable to inhibit responses to sensory and cognitive information, which causes enhanced perseveration, impaired reversal learning, and behavioral inflexibility. Moreover, PERK cKO mice displayed impaired fear extinction (Trinh et al., 2012), indicating an equally important role for PERK in PFC-dependent updating of behavior. Collectively, these findings suggest that in the postnatal adult forebrain, PERK-directed translational control of ATF4 is critical for normal behavioral flexibility.
The behavioral studies with the PERK cKO mice suggest that eIF2α phosphorylation is normally altered during reversal learning. Indeed, wild-type mice exhibited reduced eIF2α phosphorylation 30 minutes after reversal learning (Trinh et al., 2012). Although basal eIF2α phosphorylation was reduced, there was no further reduction in the PERK cKO mice, suggesting that reversal learning normally is associated with decreased phosphorylation of eIF2α phosphorylation. However, unlike PERK cKO mice, GCN2-deficient mice exhibited normal reversal learning (Trinh et al., 2012), suggesting the interesting possibility that behavioral flexibility is controlled specifically by a pool of eIF2α that is normally phosphorylated by PERK. All together, these findings suggest that reversal learning normally decreases eIF2α phosphorylation and that PERK phosphorylates a specific pool of eIF2α, which stimulates the translation of ATF4, to regulate behavioral flexibility. It should be noted that in addition to ATF4, other genes recently have been identified that are preferentially translated by eIF2α phosphorylation, which suggests that additional factors may participate downstream of PERK to regulate cognitive control functions (Dey et al., 2010; Jackson et al., 2010).
Interestingly, it was shown that PERK and ATF4 expression are reduced in the frontal cortex of human schizophrenic patients (Trinh et al., 2012). In addition to schizophrenia, cognitive and information processing deficits have been implicated as core features of several other mental illnesses, including bipolar disorder, attention deficit/hyperactivity disorder (ADHD) and autism spectrum disorder (ASD) (Bora et al., 2009; Goos et al., 2009; Lesh et al., 2011; Solomon et al., 2009). In contrast to schizophrenic brains, PERK and ATF4 levels were unaltered in the frontal cortex of bipolar patients compared to normal control patients (Trinh et al., 2012). Together, these findings suggest that disruption of PERK-regulated translation of ATF4 in the frontal cortex contributes to the pathophysiology of schizophrenia. Future studies examining PERK and ATF4 expression in the frontal cortex of patients with other mental illnesses are needed to determine how broadly or specifically PERK-directed translation is involved in mental illnesses.
More recently, it was demonstrated that PERK regulates eIF2α phosphorylation and ATF4 translation during hippocampal mGluR-dependent LTD. Cre-mediated deletion of PERK in hippocampal area CA1 was associated with reduced eIF2α phosphorylation and ATF4 expression (Trinh et al., 2013 in revision). Interestingly, although PERK cKO mice exhibited enhanced mGluR-LTD, both E-LTP and L-LTP remained unchanged (Trinh et al., 2013 in revision). Moreover, in wild-type mice mGluR-LTD was associated with increased phosphorylation of eIF2α, which was absent in the PERK cKO mice. In contrast, L-LTP was associated with decreased phosphorylation of eIF2α. Together, these findings support the notion that bidirectional changes in synaptic transmission, namely LTP and LTD, use distinct mechanisms to regulate eIF2α phosphorylation and suggest that PERK-mediated phosphorylation of eIF2α may be specific to mGluR-LTD. Moreover, these results suggest that disruption of PERK-directed translational control following activation of group I mGluRs may serve as a possible mechanism for the behavioral phenotypes observed in the PERK cKO mice (Trinh et al., 2012).
Conclusions and Future Directions
It is now evident that eIF2α kinases and their ability to impact translational control via the phosphorylation of eIF2α are critical for long-lasting plasticity of synaptic connections and for cognitive functions that rely on such plasticity in the brain. One intriguing insight from the studies of the role of eIF2α kinases in synaptic plasticity thus far is that eIF2α phosphorylation is divergently regulated during LTP and LTD. Moreover, these studies suggest that eIF2α kinases, such as PERK and GCN2, modulate eIF2α phosphorylation in order to regulate the translation of distinct sets of mRNAs to enable the expression of either LTP or LTD. An important next step would be to establish the identity of the mRNAs whose translation is locally regulated by eIF2α kinases, perhaps with either microarray studies of polysome fractions or proteomic and bioinformatic analyses.
Studies investigating the role of eIF2α kinases in synaptic plasticity and memory have primarily focused on pyramidal neurons of the hippocampus and prefrontal cortex (in the case of PERK), but little is known about their function in other brain regions and cell types. For instance, mechanisms of translational control by eIF2α kinases in cortical or striatal GABAergic interneurons have not been explored. Thus, future studies characterizing the role of eIF2α kinases in different brain regions and cell types will be necessary to provide a comprehensive analysis of how proteins are synthesized throughout the brain in response to diverse stimuli. Furthermore, it will be important to investigate the biochemical signaling cascades that couple neurotransmitter and neurotrophin receptors to the activation/inactivation of eIF2α kinases. By identifying the upstream pathways that impinge on translational control by eIF2α kinases, one should be able to determine how these specific pathways couple receptor activation to trigger distinct forms of synaptic plasticity and multiple cognitive processes.
Research Highlights.
eIF2 kinases control general and gene-specific translation
eIF2 kinases are required for hippocampal synaptic plasticity
eIF2 kinases are required for various forms of long-term memory
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abel T, Martin KC, Bartsch D, Kandel ER. Memory suppressor genes: inhibitory constraints on the storage of long-term memory. Science. 1998;279:338–341. doi: 10.1126/science.279.5349.338. [DOI] [PubMed] [Google Scholar]
- Baltzis D, Pluquet O, Papadakis AI, Kazemi S, Qu LK, Koromilas AE. The eIF2alpha kinases PERK and PKR activate glycogen synthase kinase 3 to promote the proteasomal degradation of p53. J Biol Chem. 2007;282:31675–31687. doi: 10.1074/jbc.M704491200. [DOI] [PubMed] [Google Scholar]
- Bando Y, Onuki R, Katayama T, Manabe T, Kudo T, Taira K, Tohyama M. Double-strand RNA dependent protein kinase (PKR) is involved in the extrastriatal degeneration in Parkinson's disease and Huntington's disease. Neurochem Int. 2005;46:11–18. doi: 10.1016/j.neuint.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Banko JL, Hou L, Poulin F, Sonenberg N, Klann E. Regulation of eukaryotic initiation factor 4E by converging signaling pathways during metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2006;26:2167–2173. doi: 10.1523/JNEUROSCI.5196-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banko JL, Poulin F, Hou L, DeMaria CT, Sonenberg N, Klann E. The translation repressor 4E-BP2 is critical for eIF4F complex formation, synaptic plasticity, and memory in the hippocampus. J Neurosci. 2005;25:9581–9590. doi: 10.1523/JNEUROSCI.2423-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartsch D, Ghirardi M, Skehel PA, Karl KA, Herder SP, Chen M, Bailey CH, Kandel ER. Aplysia CREB2 represses long-term facilitation: relief of repression converts transient facilitation into long-term functional and structural change. Cell. 1995;83:979–992. doi: 10.1016/0092-8674(95)90213-9. [DOI] [PubMed] [Google Scholar]
- Bekinschtein P, Cammarota M, Katche C, Slipczuk L, Rossato JI, Goldin A, Izquierdo I, Medina JH. BDNF is essential to promote persistence of long-term memory storage. Proc Natl Acad Sci U S A. 2008;105:2711–2716. doi: 10.1073/pnas.0711863105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Asouli Y, Banai Y, Pel-Or Y, Shir A, Kaempfer R. Human interferon-gamma mRNA autoregulates its translation through a pseudoknot that activates the interferon-inducible protein kinase PKR. Cell. 2002;108:221–232. doi: 10.1016/s0092-8674(02)00616-5. [DOI] [PubMed] [Google Scholar]
- Berlanga JJ, Santoyo J, De Haro C. Characterization of a mammalian homolog of the GCN2 eukaryotic initiation factor 2alpha kinase. Eur J Biochem. 1999;265:754–762. doi: 10.1046/j.1432-1327.1999.00780.x. [DOI] [PubMed] [Google Scholar]
- Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- Bingol B, Sheng M. Deconstruction for reconstruction: the role of proteolysis in neural plasticity and disease. Neuron. 2011;69:22–32. doi: 10.1016/j.neuron.2010.11.006. [DOI] [PubMed] [Google Scholar]
- Bitsios P, Giakoumaki SG, Theou K, Frangou S. Increased prepulse inhibition of the acoustic startle response is associated with better strategy formation and execution times in healthy males. Neuropsychologia. 2006;44:2494–2499. doi: 10.1016/j.neuropsychologia.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bora E, Yucel M, Pantelis C. Cognitive endophenotypes of bipolar disorder: a meta-analysis of neuropsychological deficits in euthymic patients and their first-degree relatives. J Affect Disord. 2009;113:1–20. doi: 10.1016/j.jad.2008.06.009. [DOI] [PubMed] [Google Scholar]
- Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D, Yuan J. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005;307:935–939. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- Braff DL, Geyer MA, Light GA, Sprock J, Perry W, Cadenhead KS, Swerdlow NR. Impact of prepulse characteristics on the detection of sensorimotor gating deficits in schizophrenia. Schizophr Res. 2001;49:171–178. doi: 10.1016/s0920-9964(00)00139-0. [DOI] [PubMed] [Google Scholar]
- Calkhoven CF, Muller C, Leutz A. Translational control of C/EBPalpha and C/EBPbeta isoform expression. Genes Dev. 2000;14:1920–1932. [PMC free article] [PubMed] [Google Scholar]
- Carnevalli LS, Pereira CM, Jaqueta CB, Alves VS, Paiva VN, Vattem KM, Wek RC, Mello LE, Castilho BA. Phosphorylation of the alpha subunit of translation initiation factor-2 by PKR mediates protein synthesis inhibition in the mouse brain during status epilepticus. Biochem J. 2006;397:187–194. doi: 10.1042/BJ20051643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpick BW, Graziano V, Schneider D, Maitra RK, Lee X, Williams BR. Characterization of the solution complex between the interferon-induced, double-stranded RNA-activated protein kinase and HIV-I trans-activating region RNA. J Biol Chem. 1997;272:9510–9516. doi: 10.1074/jbc.272.14.9510. [DOI] [PubMed] [Google Scholar]
- Chen A, Muzzio IA, Malleret G, Bartsch D, Verbitsky M, Pavlidis P, Yonan AL, Vronskaya S, Grody MB, Cepeda I, Gilliam TC, Kandel ER. Inducible enhancement of memory storage and synaptic plasticity in transgenic mice expressing an inhibitor of ATF4 (CREB-2) and C/EBP proteins. Neuron. 2003;39:655–669. doi: 10.1016/s0896-6273(03)00501-4. [DOI] [PubMed] [Google Scholar]
- Cohen-Chalamish S, Hasson A, Weinberg D, Namer LS, Banai Y, Osman F, Kaempfer R. Dynamic refolding of IFN-gamma mRNA enables it to function as PKR activator and translation template. Nat Chem Biol. 2009;5:896–903. doi: 10.1038/nchembio.234. [DOI] [PubMed] [Google Scholar]
- Connor JH, Weiser DC, Li S, Hallenbeck JM, Shenolikar S. Growth arrest and DNA damage-inducible protein GADD34 assembles a novel signaling complex containing protein phosphatase 1 and inhibitor 1. Mol Cell Biol. 2001;21:6841–6850. doi: 10.1128/MCB.21.20.6841-6850.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Gobert D, Harding H, Herdy B, Azzi M, Bruno M, Bidinosti M, Ben Mamou C, Marcinkiewicz E, Yoshida M, Imataka H, Cuello AC, Seidah N, Sossin W, Lacaille JC, Ron D, Nader K, Sonenberg N. Translational 20 control of hippocampal synaptic plasticity and memory by the eIF2alpha kinase GCN2. Nature. 2005;436:1166–1173. doi: 10.1038/nature03897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Gobert D, Stern E, Gamache K, Colina R, Cuello C, Sossin W, Kaufman R, Pelletier J, Rosenblum K, Krnjevic K, Lacaille JC, Nader K, Sonenberg N. eIF2alpha phosphorylation bidirectionally regulates the switch from short- to long-term synaptic plasticity and memory. Cell. 2007;129:195–206. doi: 10.1016/j.cell.2007.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Sossin WS, Klann E, Sonenberg N. Translational control of long-lasting synaptic plasticity and memory. Neuron. 2009;61:10–26. doi: 10.1016/j.neuron.2008.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couturier J, Morel M, Pontcharraud R, Gontier V, Fauconneau B, Paccalin M, Page G. Interaction of double-stranded RNA-dependent protein kinase (PKR) with the death receptor signaling pathway in amyloid beta (Abeta)-treated cells and in APPSLPS1 knock-in mice. J Biol Chem. 2010;285:1272–1282. doi: 10.1074/jbc.M109.041954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crino PB, Eberwine J. Molecular characterization of the dendritic growth cone: regulated mRNA transport and local protein synthesis. Neuron. 1996;17:1173–1187. doi: 10.1016/s0896-6273(00)80248-2. [DOI] [PubMed] [Google Scholar]
- Crosby JS, Lee K, London IM, Chen JJ. Erythroid expression of the heme-regulated eIF-2 alpha kinase. Mol Cell Biol. 1994;14:3906–3914. doi: 10.1128/mcb.14.6.3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damjanac M, Page G, Ragot S, Laborie G, Gil R, Hugon J, Paccalin M. PKR, a cognitive decline biomarker, can regulate translation via two consecutive molecular targets p53 and Redd1 in lymphocytes of AD patients. J Cell Mol Med. 2009;13:1823–1832. doi: 10.1111/j.1582-4934.2009.00688.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Pietri Tonelli D, Mihailovich M, Di Cesare A, Codazzi F, Grohovaz F, Zacchetti D. Translational regulation of BACE-1 expression in neuronal and non-neuronal cells. Nucleic Acids Res. 2004;32:1808–1817. doi: 10.1093/nar/gkh348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delepine M, Nicolino M, Barrett T, Golamaully M, Lathrop GM, Julier C. EIF2AK3, encoding translation initiation factor 2-alpha kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat Genet. 2000;25:406–409. doi: 10.1038/78085. [DOI] [PubMed] [Google Scholar]
- Deng J, Harding HP, Raught B, Gingras AC, Berlanga JJ, Scheuner D, Kaufman RJ, Ron D, Sonenberg N. Activation of GCN2 in UV-irradiated cells inhibits translation. Curr Biol. 2002;12:1279–1286. doi: 10.1016/s0960-9822(02)01037-0. [DOI] [PubMed] [Google Scholar]
- Dever TE, Dar AC, Sicheri F. The eIF2 α Kinases. In: Michael NS, Matthews B, Hershey John WB, editors. Translational Control in Biology and Medicine. New York: Cold Spring Harbor Laboratory Press; 2007. pp. 319–344. [Google Scholar]
- Dey S, Baird TD, Zhou D, Palam LR, Spandau DF, Wek RC. Both transcriptional regulation and translational control of ATF4 are central to the integrated stress response. J Biol Chem. 2010;285:33165–33174. doi: 10.1074/jbc.M110.167213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumurgier J, Mouton-Liger F, Lapalus P, Prevot M, Laplanche JL, Hugon J, Paquet C. Cerebrospinal fluid PKR level predicts cognitive decline in Alzheimer's disease. PLoS One. 2013;8:e53587. doi: 10.1371/journal.pone.0053587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, Ramesh V, Silva AJ. Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat Med. 2008;14:843–848. doi: 10.1038/nm1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flexner JB, Flexner LB, Stellar E. Memory in mice as affected by intracerebral puromycin. Science. 1963;141:57–59. doi: 10.1126/science.141.3575.57. [DOI] [PubMed] [Google Scholar]
- Goos LM, Crosbie J, Payne S, Schachar R. Validation and extension of the endophenotype model in ADHD patterns of inheritance in a family study of inhibitory control. Am J Psychiatry. 2009;166:711–717. doi: 10.1176/appi.ajp.2009.08040621. [DOI] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, Sabatini DD, Ron D. Diabetes mellitus and exocrine pancreatic dysfunction in perk−/− mice reveals a role for translational control in secretory cell survival. Mol Cell. 2001;7:1153–1163. doi: 10.1016/s1097-2765(01)00264-7. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- Hinnebusch AG. Translational regulation of GCN4 and the general amino acid control of yeast. Annu Rev Microbiol. 2005;59:407–450. doi: 10.1146/annurev.micro.59.031805.133833. [DOI] [PubMed] [Google Scholar]
- Hinnebusch AG, Asano K, Olsen DS, Phan L, Nielsen KH, Valasek L. Study of translational control of eukaryotic gene expression using yeast. Ann N Y Acad Sci. 2004;1038:60–74. doi: 10.1196/annals.1315.012. [DOI] [PubMed] [Google Scholar]
- Hinnebusch AG, Natarajan K. Gcn4p, a master regulator of gene expression, is controlled at multiple levels by diverse signals of starvation and stress. Eukaryot Cell. 2002;1:22–32. doi: 10.1128/EC.01.1.22-32.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoglinger GU, Melhem NM, Dickson DW, Sleiman PM, Wang LS, Klei L, Rademakers R, de Silva R, Litvan I, Riley DE, van Swieten JC, Heutink P, Wszolek ZK, Uitti RJ, Vandrovcova J, Hurtig HI, Gross RG, Maetzler W, Goldwurm S, Tolosa E, Borroni B, Pastor P, Cantwell LB, Han MR, Dillman A, van der Brug MP, Gibbs JR, Cookson MR, Hernandez DG, Singleton AB, Farrer MJ, Yu CE, Golbe LI, Revesz T, Hardy J, Lees AJ, Devlin B, Hakonarson H, Muller U, Schellenberg GD. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet. 2011;43:699–705. doi: 10.1038/ng.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Kayser MS, Bear MF. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science. 2000;288:1254–1257. doi: 10.1126/science.288.5469.1254. [DOI] [PubMed] [Google Scholar]
- Iacono M, Mignone F, Pesole G. uAUG and uORFs in human and rodent 5'untranslated mRNAs. Gene. 2005;349:97–105. doi: 10.1016/j.gene.2004.11.041. [DOI] [PubMed] [Google Scholar]
- Jackson RJ, Hellen CU, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol. 2010;11:113–127. doi: 10.1038/nrm2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Belforte JE, Lu Y, Yabe Y, Pickel J, Smith CB, Je HS, Lu B, Nakazawa K. eIF2alpha Phosphorylation-dependent translation in CA1 pyramidal cells impairs hippocampal memory consolidation without affecting general translation. J Neurosci. 2010;30:2582–2594. doi: 10.1523/JNEUROSCI.3971-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jousse C, Oyadomari S, Novoa I, Lu P, Zhang Y, Harding HP, Ron D. Inhibition of a constitutive translation initiation factor 2alpha phosphatase, CReP, promotes survival of stressed cells. J Cell Biol. 2003;163:767–775. doi: 10.1083/jcb.200308075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julier C, Nicolino M. Wolcott-Rallison syndrome. Orphanet J Rare Dis. 2010;5:29. doi: 10.1186/1750-1172-5-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaang BK, Choi JH. Synaptic protein degradation in memory reorganization. Adv Exp Med Biol. 2012;970:221–240. doi: 10.1007/978-3-7091-0932-8_10. [DOI] [PubMed] [Google Scholar]
- Kandel ER. The molecular biology of memory storage: a dialog between genes and synapses. Biosci Rep. 2001;21:565–611. doi: 10.1023/a:1014775008533. [DOI] [PubMed] [Google Scholar]
- Kang H, Schuman EM. A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science. 1996;273:1402–1406. doi: 10.1126/science.273.5280.1402. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ, 3rd, Govindarajan A, Tonegawa S. Translational regulatory mechanisms in persistent forms of synaptic plasticity. Neuron. 2004;44:59–73. doi: 10.1016/j.neuron.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Klann E, Sweatt JD. Altered protein synthesis is a trigger for long-term memory formation. Neurobiol Learn Mem. 2008;89:247–259. doi: 10.1016/j.nlm.2007.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammich S, Schobel S, Zimmer AK, Lichtenthaler SF, Haass C. Expression of the Alzheimer protease BACE1 is suppressed via its 5'-untranslated region. EMBO Rep. 2004;5:620–625. doi: 10.1038/sj.embor.7400166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesh TA, Niendam TA, Minzenberg MJ, Carter CS. Cognitive control deficits in schizophrenia: mechanisms and meaning. Neuropsychopharmacology. 2011;36:316–338. doi: 10.1038/npp.2010.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Hendershot LM. Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. J Biol Chem. 2003;278:34864–34873. doi: 10.1074/jbc.M301107200. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. Long-term potentiation--a decade of progress? Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- Matsui M, Yachie N, Okada Y, Saito R, Tomita M. Bioinformatic analysis of post-transcriptional regulation by uORF in human and mouse. FEBS Lett. 2007;581:4184–4188. doi: 10.1016/j.febslet.2007.07.057. [DOI] [PubMed] [Google Scholar]
- McGaugh JL. Memory--a century of consolidation. Science. 2000;287:248–251. doi: 10.1126/science.287.5451.248. [DOI] [PubMed] [Google Scholar]
- Mellor H, Flowers KM, Kimball SR, Jefferson LS. Cloning and characterization of cDNA encoding rat hemin-sensitive initiation factor-2 alpha (eIF-2 alpha) kinase. Evidence for multitissue expression. J Biol Chem. 1994;269:10201–10204. [PubMed] [Google Scholar]
- Meurs E, Chong K, Galabru J, Thomas NS, Kerr IM, Williams BR, Hovanessian AG. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell. 1990;62:379–390. doi: 10.1016/0092-8674(90)90374-n. [DOI] [PubMed] [Google Scholar]
- Mignone F, Gissi C, Liuni S, Pesole G. Untranslated regions of mRNAs. Genome Biol. 2002;3:REVIEWS0004. doi: 10.1186/gb-2002-3-3-reviews0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neafsey DE, Galagan JE. Dual modes of natural selection on upstream open reading frames. Mol Biol Evol. 2007;24:1744–1751. doi: 10.1093/molbev/msm093. [DOI] [PubMed] [Google Scholar]
- Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol. 2001;153:1011–1022. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan U, Ozcan L, Yilmaz E, Duvel K, Sahin M, Manning BD, Hotamisligil GS. Loss of the tuberous sclerosis complex tumor suppressors triggers the unfolded protein response to regulate insulin signaling and apoptosis. Mol Cell. 2008;29:541–551. doi: 10.1016/j.molcel.2007.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquet C, Bose A, Polivka M, Peoc'h K, Brouland JP, Keohane C, Hugon J, Gray F. Neuronal phosphorylated RNA-dependent protein kinase in Creutzfeldt-Jakob disease. J Neuropathol Exp Neurol. 2009;68:190–198. doi: 10.1097/NEN.0b013e318196cd7c. [DOI] [PubMed] [Google Scholar]
- Patel RC, Sen GC. Requirement of PKR dimerization mediated by specific hydrophobic residues for its activation by double-stranded RNA and its antigrowth effects in yeast. Mol Cell Biol. 1998;18:7009–7019. doi: 10.1128/mcb.18.12.7009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peel AL, Bredesen DE. Activation of the cell stress kinase PKR in Alzheimer's disease and human amyloid precursor protein transgenic mice. Neurobiol Dis. 2003;14:52–62. doi: 10.1016/s0969-9961(03)00086-x. [DOI] [PubMed] [Google Scholar]
- Peel AL, Rao RV, Cottrell BA, Hayden MR, Ellerby LM, Bredesen DE. Double-stranded RNA-dependent protein kinase, PKR, binds preferentially to Huntington's disease (HD) transcripts and is activated in HD tissue. Hum Mol Genet. 2001;10:1531–1538. doi: 10.1093/hmg/10.15.1531. [DOI] [PubMed] [Google Scholar]
- Pereira CM, Sattlegger E, Jiang HY, Longo BM, Jaqueta CB, Hinnebusch AG, Wek RC, Mello LE, Castilho BA. IMPACT, a protein preferentially expressed in the mouse brain, binds GCN1 and inhibits GCN2 activation. J Biol Chem. 2005;280:28316–28323. doi: 10.1074/jbc.M408571200. [DOI] [PubMed] [Google Scholar]
- Pestova TV, Lorsh JR, Hellen CUT. The Mechanism of Translation Initiation in Eukaryotes. In: Mathews MB, Sonenberg N, Hershey JWB, editors. Translational Control in Biology and Medicine. New York: Cold Spring Harbor Laboratory Press; 2007. pp. 87–128. [Google Scholar]
- Raven JF, Baltzis D, Wang S, Mounir Z, Papadakis AI, Gao HQ, Koromilas AE. PKR and PKR-like endoplasmic reticulum kinase induce the 24 proteasome-dependent degradation of cyclin D1 via a mechanism requiring eukaryotic initiation factor 2alpha phosphorylation. J Biol Chem. 2008;283:3097–3108. doi: 10.1074/jbc.M709677200. [DOI] [PubMed] [Google Scholar]
- Reis AF, Kannengiesser C, Jennane F, Manna TD, Cheurfa N, Oudin C, Savoldelli RD, Oliveira C, Grandchamp B, Kok F, Velho G. Two novel mutations in the EIF2AK3 gene in children with Wolcott-Rallison syndrome. Pediatr Diabetes. 2011;12:187–191. doi: 10.1111/j.1399-5448.2010.00679.x. [DOI] [PubMed] [Google Scholar]
- Richter JD, Klann E. Making synaptic plasticity and memory last: mechanisms of translational regulation. Genes Dev. 2009;23:1–11. doi: 10.1101/gad.1735809. [DOI] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Rubio-Cabezas O, Patch AM, Minton JA, Flanagan SE, Edghill EL, Hussain K, Balafrej A, Deeb A, Buchanan CR, Jefferson IG, Mutair A, Hattersley AT, Ellard S. Wolcott-Rallison syndrome is the most common genetic cause of permanent neonatal diabetes in consanguineous families. J Clin Endocrinol Metab. 2009;94:4162–4170. doi: 10.1210/jc.2009-1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segev Y, Michaelson DM, Rosenblum K. ApoE epsilon4 is associated with eIF2alpha phosphorylation and impaired learning in young mice. Neurobiol Aging. 2013;34:863–872. doi: 10.1016/j.neurobiolaging.2012.06.020. [DOI] [PubMed] [Google Scholar]
- Senee V, Vattem KM, Delepine M, Rainbow LA, Haton C, Lecoq A, Shaw NJ, Robert JJ, Rooman R, Diatloff-Zito C, Michaud JL, Bin-Abbas B, Taha D, Zabel B, Franceschini P, Topaloglu AK, Lathrop GM, Barrett TG, Nicolino M, Wek RC, Julier C. Wolcott-Rallison Syndrome: clinical, genetic, and functional study of EIF2AK3 mutations and suggestion of genetic heterogeneity. Diabetes. 2004;53:1876–1883. doi: 10.2337/diabetes.53.7.1876. [DOI] [PubMed] [Google Scholar]
- Shi Y, Vattem KM, Sood R, An J, Liang J, Stramm L, Wek RC. Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol Cell Biol. 1998;18:7499–7509. doi: 10.1128/mcb.18.12.7499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon M, Ozonoff SJ, Ursu S, Ravizza S, Cummings N, Ly S, Carter CS. The neural substrates of cognitive control deficits in autism spectrum disorders. Neuropsychologia. 2009;47:2515–2526. doi: 10.1016/j.neuropsychologia.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonenberg N, Dever TE. Eukaryotic translation initiation factors and regulators. Curr Opin Struct Biol. 2003;13:56–63. doi: 10.1016/s0959-440x(03)00009-5. [DOI] [PubMed] [Google Scholar]
- Sood R, Porter AC, Olsen DA, Cavener DR, Wek RC. A mammalian homologue of GCN2 protein kinase important for translational control by phosphorylation of eukaryotic initiation factor-2alpha. Genetics. 2000;154:787–801. doi: 10.1093/genetics/154.2.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern E, Chinnakkaruppan A, David O, Sonenberg N, Rosenblum K. Blocking the eIF2alpha kinase (PKR) enhances positive and negative forms of cortex-dependent taste memory. J Neurosci. 2013;33:2517–2525. doi: 10.1523/JNEUROSCI.2322-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward O, Levy WB. Preferential localization of polyribosomes under the base of dendritic spines in granule cells of the dentate gyrus. J Neurosci. 1982;2:284–291. doi: 10.1523/JNEUROSCI.02-03-00284.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang SJ, Schuman EM. Protein synthesis in the dendrite. Philos Trans R Soc Lond B Biol Sci. 2002;357:521–529. doi: 10.1098/rstb.2001.0887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton CM, Carson DJ, Stewart FJ. Autopsy findings in the Wolcott-Rallison syndrome. Pediatr Pathol Lab Med. 1997;17:487–496. [PubMed] [Google Scholar]
- Trinh MA, Kaphzan H, Wek RC, Pierre P, Cavener DR, Klann E. Brain-specific disruption of the eIF2alpha kinase PERK decreases ATF4 expression and impairs behavioral flexibility. Cell Rep. 2012;1:676–688. doi: 10.1016/j.celrep.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vattem KM, Wek RC. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci U S A. 2004;101:11269–11274. doi: 10.1073/pnas.0400541101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Sheng X, Feng D, McGrath B, Cavener DR. PERK is essential for neonatal skeletal development to regulate osteoblast proliferation and differentiation. J Cell Physiol. 2008;217:693–707. doi: 10.1002/jcp.21543. [DOI] [PubMed] [Google Scholar]
- Wek RC, Cavener DR. Translational control and the unfolded protein response. Antioxid Redox Signal. 2007;9:2357–2371. doi: 10.1089/ars.2007.1764. [DOI] [PubMed] [Google Scholar]
- Wek SA, Zhu S, Wek RC. The histidyl-tRNA synthetase-related sequence in the eIF-2 alpha protein kinase GCN2 interacts with tRNA and is required for activation in response to starvation for different amino acids. Mol Cell Biol. 1995;15:4497–4506. doi: 10.1128/mcb.15.8.4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Romano PR, Nagamura-Inoue T, Tian B, Dever TE, Mathews MB, Ozato K, Hinnebusch AG. Binding of double-stranded RNA to protein kinase PKR is required for dimerization and promotes critical autophosphorylation events in the activation loop. J Biol Chem. 2001;276:24946–24958. doi: 10.1074/jbc.M102108200. [DOI] [PubMed] [Google Scholar]
- Zhang P, McGrath B, Li S, Frank A, Zambito F, Reinert J, Gannon M, Ma K, McNaughton K, Cavener DR. The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol Cell Biol. 2002;22:3864–3874. doi: 10.1128/MCB.22.11.3864-3874.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Feng D, Li Y, Iida K, McGrath B, Cavener DR. PERK EIF2AK3 control of pancreatic beta cell differentiation and proliferation is required for postnatal glucose homeostasis. Cell Metab. 2006;4:491–497. doi: 10.1016/j.cmet.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Zhu PJ, Huang W, Kalikulov D, Yoo JW, Placzek AN, Stoica L, Zhou H, Bell JC, Friedlander MJ, Krnjevic K, Noebels JL, Costa-Mattioli M. Suppression of PKR Promotes Network Excitability and Enhanced Cognition by Interferon-gamma-Mediated Disinhibition. Cell. 2011;147:1384–1396. doi: 10.1016/j.cell.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **.Trinh MA, Ma T, Kaphzan H, Antion MD, Cavener DR, Hoeffer CA, Klann E. PERK-dependent eIF2α phosphorylation limits the expression of hippocampal metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2013 doi: 10.1101/lm.032219.113. (in revision) [DOI] [PMC free article] [PubMed] [Google Scholar]