

Abstract
We previously reported the establishment and characteristics of a DXM-resistant cell line (7TD1-DXM) generated from the IL6-dependent mouse B cell hybridoma, 7TD1 cell line. After withdrawing DXM from 7TD1-DXM cells over 90 days, DXM significantly inhibited the cell growth and induced apoptosis in the cells (7TD1-WD) compared with 7TD1-DXM cells. Additionally, IL-6 reversed while IL-6 antibody and AG490 enhanced the effects of growth inhibition and apoptosis induced by DXM in 7TD1-WD cells. Our study demonstrates that 7TD1-DXM cells become resensitized to DXM after DXM withdrawal, and IL-6 and JAK2/STAT3 pathways may regulate the phenomenon.
Keywords: multiple myeloma, dexamethasone resistance, dexamethasone withdrawal, interleukin-6, AG490, JAK2/STAT3 signaling pathway
1. Introduction
Multiple myeloma (MM) is a malignancy of terminally differentiated B-lymphocytes. It is characterized by the clonal proliferation of plasma cells within the bone marrow [1]. MM is the second most common blood cancer and compromised about 1% of all cancer types in the United States. It was estimated that there was about 21,700 new cases and 10,710 deaths of MM in 2012 in the United States [2]. Despite recent progress in understanding of the mechanism of MM, it is still currently incurable and the patients have poor prognosis [3].
The goals of treatment for patients with MM are to reduce symptoms, to slow disease progression and to prolong remissions. A variety of chemotherapy agents have been used to treat MM including melphalan, cyclophosphamide, doxorubicin, prednisone and dexamethasone (DXM) [4, 5]. Other options such as immunotherapy, radiation, bone marrow/stem cell transplant have also been used to treat MM [6]. Treatments options of MM depend on the stage of disease and the patients may be effectively response to the initial treatment but the disease may recur later. Patients with recurrence of the disease may develop refractory MM in which the patients no longer response to the treatments [4].
DXM, a synthetic glucocorticoid steroid hormone, has been commonly used in the treatment of MM. DXM acts via binding with the glucocorticoid receptor [7, 8]. DXM induces apoptosis in MM cells but the precise mechanism is still unknown. However, several possible mechanisms have been proposed. First, DXM may induce cell cycle arrest especially at the G1 phase and the cell cycle arrest may serve as an apoptotic signal [9, 10]. Second, DXM can induce apoptosis via activating the transcription of apoptotic genes such as Bim, Caspase-3, and Caspase-8 [11–14]. Third, DXM can also induce apoptosis via suppression of the Akt-PI3K pathway or glucocorticoid receptor involvement [15–18]. A major problem for DXM in treatment of MM is the occurrence of drug resistance due to MM cells resistance to DXM induced apoptosis besides drug induced side effects. Chronic exposure to DXM may lead to DXM resistance in patients with MM and DXM treatment is no longer beneficial. Several mechanisms have been discovered that count for the resistance to DXM induced apoptosis including insufficient ligand, glucocorticoid receptor mutations, abnormal GR translocation, or overexpression of Bcl-2 [17, 19, 20].
Interleukin 6 (IL-6), a 26 kDa protein, is a multifunctional cytokine secreted by many cell types such as activated monocytes and macrophages, endothelial cells, adipose cells and the Th-2 subset of T helper cells [21]. IL-6 belongs to the cytokine family that also includes IL-11, leukemia inhibitory factor, oncostatin M, cardiotrophin-1, cardiotrophin-1, and ciliary neurotrophic factor [22]. This cytokine family is characterized by a four-alpha-helix bundle structure. The different biological functions of IL-6 include immune response, hematopoiesis, and acute-phase reactions [21, 23]. Previous studies showed that the interaction between MM cells and bone marrow stromal cells triggered the production of IL-6 [24, 25]. IL-6 is essential for the survival and growth of myeloma cells, and also protects the cells from apoptosis caused by the chemotherapeutic agents such as DXM via various mechanisms [26–28]. Activated glucocorticoid receptors may repress the activity of NF-κB leading to the down-regulation of the transcription of IL-6 gene in various cells [29–32]. In the later stage of MM, the cells can proliferate without IL-6 stimulation [6, 33, 34]. Other studies also indicated the relationship between the endogenous production of IL-6 and the progress of malignant MM cells, suggesting the possible roles of IL-6 in resistance to DXM-induced apoptosis [35].
IL-6 acts via binding and activating the IL-6 receptor which is a single transmembrane receptor composing three main regions. The first is an outer immuunioglobulin-like domain, the second is a remaining extracellular protein known as the cytokine binding domain and the third is an intra-cytoplasmic domain [23]. The cytokine binding domain mediates the binding to IL-6 and gp130. The binding of IL-6 with its receptor recruits the binary complex with gp130 to form a high-affinity ternary complex, IL-6/IL-6 receptor/gp130 [36, 37]. The interaction of IL-6 and its receptors activates many signal transduction pathways. The gp130 forms a dimmer after the stimulation by IL-6 and leads to the activation of the Janus-activated kinase signal transducer and activator of transcription (STAT) signaling pathway [22, 38, 39]. The JAK/STAT signaling pathway is the major pathway of IL-6. Additionally, IL-6 also triggers the MAPK signaling pathway and the PI3K-AKT pathway [39–42].
In our previous studies, we demonstrated that cAMP and IL-6 dependent signaling pathway cross-talk regulated the proliferation and apoptosis of 7TD1 cells [43]. We generated DXM resistant 7TD1 cell line (name it as 7TD1-DXM cells) by chronic exposure of the parent 7TD1 cells to DXM and the 7TD1-DXM cells showed IL6-independent proliferation. We also indicated that the apoptotic resistance exhibited by 7TD1-DXM cells is regulated independently by IL-6 triggered signaling [44]. In order to further investigate the molecular mechanisms of DXM resistance in MM, we withdrew the continuously presence of DXM in the DXM resistant 7TD1-DXM cells (name the DXM withdrawn cells as 7TD1-WD cells) to see if it could reverse the resistance of the cells to DXM induced cell growth inhibition and apoptosis. We also investigate the effects of IL-6 and the JAK2 inhibitor AG490 on the cell proliferation and apoptosis in MM cells, as well as the possible involvement of signal transduction pathways in the DXM resistant and withdrawn 7TD1 cells.
2. Materials and methods
2.1. Reagents and antibody
DXM was purchased from Sigma-Aldrich (St Louis, MO) and dissolved in 100% ethanol at 1 mg/ml stock solution. AG490 was purchased from Promega (Madison, WI) and dissolved in DMSO at a stock concentration of 50 mM. The recombinant IL-6 was purchased from Upstate Biotechnology (Lake Placid, NY) and IL-6 antibody was purchased from Santa Cruz Biotechnology, Inc, (Santa Cruz, CA).
2.2. Cell culture
The parental 7TD1 cells were cultured in the media consisting of RPMI-1640 with glutamine (GIBCO, Invitrogen, Carlsbad, CA) and supplemented by 2 g/L sodium bicarbonate (Fisher Scientific), 20 mM HEPES (Sigma Aldrich, St. Louis), 1% (v/v) penicillin/streptomycin antibiotic (CELLGRO Mediatech Inc., Manassas, VA), 50 μg/ml gentamycin (MP Biomedicals, Aurora), 10% (v/v) FBS (Atlanta Biologicals, Lawrenceville, GA), and 50 mM 2-mercaptoethanol (Sigma-Aldrich, St. Louis, MO). The DXM resistant 7TD1 cells (7TD1-DXM) were established by chronically treated with 85 μM of DXM and continuously cultured in the absence of IL-6 over a period of 6 months as previously described [44]. The DXM withdrawn 7TD1 cells (7TD1-WD) were established from DXM resistant 7TD1 cells by continuously cultured in the absence of DXM and IL-6 over a period of 3–5 months.
2.3 MTT assay
The MTT assay is used to determine the effect of DXM, IL-6 or IL-6Ab on the cell growth and survival. A total of 1000 cells were added to a 96 well plate, and incubated with DXM, IL-6 and IL-6Ab in a 100 μl final volume for 72 hours, then 10 μl of MTT (Sigma Aldrich, St Louis) in PBS (5 mg/ml) was added into each well. Four hours later the reaction was stopped by adding 150 μl of acidified isopropanol. The blue precipitate was dissolved by repeated pipetting and the optical density (OD) was measured at 570 nm determined with Bio-Tek microtiter plate reader (Winooski, VT). Experiments were performed in triplicate and analyzed for statistical significance.
2.4. Detection of apoptosis
Apoptotic cells were quantified using the DNA fragmentation TUNEL assay (Molecular Probes, Eugene, OR). Briefly, 7TD1-DXM and 7TD1-WD cells were subjected to various specified treatments, incubated for 48 hours, washed with 1X PBS, fixed in a mixture of ice-cold 1X PBS with 70% ethanol, and kept at −20°C for 12–18 hour. The cells were washed with 1X PBS, stained using the APOBrdu TUNEL assay kit according to the manufacturer’s protocol, and analyzed using a flow cytometer (Becton Dickinson, Franklin Lakes, NJ).
2.5. Western blotting
As previously described [44], cells were harvested by centrifugation at 200 g for 6 min and whole cell lysates prepared using a lysis buffer containing 20 mM Tris-HCl (pH 7.6), 100 mM NaCl, 2% Triton X-100, 10 mM EDTA, 100 mM sodium fluoride, 60 mM sodium pyrophosphate, 0.2% sodium azide, 0.2 mM sodium orthovanadate, 1 mM PMSF, 20 μg/ml leupeptin, and 2 μg/ml aprotonin. Lysates were then sonicated for 5 seconds and centrifuged at 5,200 × g for 10 min; the resulting supernatants were mixed with 3X SDS loading buffer and incubated at 95°C for 3 min. The proteins were then separated on a 12% (w/v) polyacrylamide gel. Separated proteins were transferred to a PVDF membrane by electrophoresis at 100 V for 1 hour. After transferring, the PVDF membrane was blocked in a solution containing 5% milk powder and 2.5% bovine serum albumin (BSA) in 1X Tris buffered saline (TBS) with 0.05% tween (TBST) at 4°C for 6 hours and then washed with 1X TBS and 1X TBST for three to four times. After washing, the membrane was probed with primary antibody for total JAK2 and JAK2 phosphorylation using JAK2 and phosphor-JAK2 (Tyr1007/1008) antibodies, or for total STAT3 and STAT3 phosphorylation using STAT3 or phospho-STAT3 (Tyr705) antibodies (1:1,000 dilutions, Calbiochem, San Diego, CA) at 4°C overnight, followed by washing with 1X TBS and 1X TBST for three to four times. The membrane was then probed with Horse-Radish Peroxidase (HRP) labeled goat anti-rabbit IgG secondary antibody (1:2,500 dilutions, KPL, Gaithersburg, MD). Blots were then processed by Pierce chemiluminiscence kit as recommended by the commercial vendor and autoradiographed on an X-ray film (RPI corp., Beacon Falls, CT). Western blot analysis was carried out at least 3 times.
2.6. Statistical analysis
Statistical significance of the data was analyzed by one way univariate analysis of variance (ANOVA) followed by Tukey’s post-hoc test with a minimum significance level set at p < 0.05 (marked as *). The higher significance level was set at p < 0.01 (marked as **)
3. Results
3. 1. DXM resistance was reversed after withdrawing DXM from the 7TD1-DXM cells
We previously established and characterized a dexamethasone-resistant 7TD1 murine myeloma cell line phenotype 7TD1-DXM by chronic treatment with DXM (85 μM) and the cells are both resistant to inhibition of proliferation and DXM induced apoptosis [44]. To test whether withdrawing continuous DXM presence could reverse the resistance to DXM in 7TD1-DXM cells, and if so, how long duration of DXM withdrawal is needed for the reversion, we withdrew DXM from 7TD1-DXM cells for up to 150 days, and performed the MTT assay to compare the cell survival after 85 μM DXM treatment in the 7TD1-DXM cells and the DXM withdrawn (7TD1-WD) cells. We chose 85 μM concentration of DXM for the study because it was the IC50 for DXM against the parent 7TD1 cells ([44] and data not shown). There was no significant difference in cell survival after DXM treatment in both cell lines of 7TD1-DXM and 7TD1-WD-60 (DXM withdrawal for 60 days). The result indicates that withdrawing DXM for 60 days could not reverse the resistance to DXM in 7TD1-DXM cells. However, 85 μM of DXM significantly inhibited cell growth in the 7TD1-WD cells compared to 7TD1-DXM cells with statistical significance of p < 0.01 after DXM withdrawal for 90 and 150 days (Fig. 1A). After we determined that withdrawing DXM over 90 days could reverse the resistance of 7TD1-DXM cells to DXM, we further tested the sensitivity of the cells of 7TD1-DXM, 7TD1-WD-90 (DXM withdrawn for 90 days) and 7TD1-WD-150 (DXM withdrawn for 150 days) to DXM at various concentrations (10, 22, 43, 85, 128, 213 and 255 μM). The growth of all three cells was inhibited by DXM in a dose-dependent manner, however, DXM showed greater inhibitory effect against 7TD1-WD-90 and 7TD1-DW-150 cells compared with 7TD1-DXM cells (Fig. 1B) and the IC50 of DXM was 172 ± 10, 106 ± 6, and 86 ± 15 μM for 7TD1-DXM, 7TD1-WD-90 and 7TD1-WD-150, respectively (Fig. 1C). There was significantly statistical difference between 7TD1-DXM cells and 7TD1-WD-90 or 7TD1-WD-150 cells (p < 0.01). However, no significant cell growth inhibition was observed between the cells of 7TD1-WD-90 and 7TD1-WD-150 to DXM treatment with P > 0.05 and the data indicates that DXM withdrawal over 90 days is sufficient to reserve the resistance to DXM in 7TD1-DXM cells (Fig. 1C). DXM resistance in MM cells is mainly due to development of resistance to apoptosis induction. Therefore, we studied the apoptotic effects upon DXM treatment in both cells of 7TD-DXM and 7TD1-WD-90 with TUNEL assay. Various concentrations of DXM (85, 170 and 340 μM) were used to treat the cells of 7TD1-DXM and 7TD1-WD-90 and the percentage of apoptotic cells was measured. The data showed that the percentage of apoptosis was 10.3 ± 2.9 and 8.5 ± 3.1 in the negative controls (treated with media only) and 76.0 ± 15.6 and 72.3 ± 10.7 in the positive controls (treated with 10 μM of etoposide) for 7TD-DXM and 7TD1-WD cells, respectively. Treatment of 85 μM of DXM did not significantly induce apoptotic effects in both 7TD1-DXM and 7TD1-WD-90 cells compared to the media control with the percentage of apoptosis of 9.1 ± 3.7 and 12.5 ± 3.5, respectively (Fig. 1D), although 85 μM of DXM significantly inhibited the cell growth in 7TD1-WD-90 cells but not in 7TD1-DXM cells (Fig. 1A). However, higher concentration of DXM at 170 μM significantly induced apoptotic effect in 7TD1-WD-90 cells (34.3 ± 5.5) but not in 7TD1-DXM cells (12.8 ± 1.0) with significant difference (p < 0.05) between 7TD1-DXM and 7TD1-WD-90 cells. Further increasing the concentration of DXM to 340 μM dramatically induced apoptosis in both 7TD1-DXM and 7TD1-WD-90 cells compared the media control. However, the percentage of apoptotic cells was significantly higher in 7TD1-WD-90 cells (53.0 ± 10.4) compare to 7TD1-DXM (32.3 ± 8.1) with statistically significant difference (p < 0.05).
Fig. 1.

The effect of DXM on the cell growth inhibition (A), dose response curves (B), IC50 (C), and induction of apoptosis (D) in the cells of 7TD1-DXM and 7TD1-WD. 7TD1-WD-60: DXM withdrawal for 60 days; 7TD1-WD-90: DXM withdrawal for 90 days; and 7TD1-WD-150: DXM withdrawal for 150 days. Cell proliferation was determined by MTT assay after 72 hours treatment. The cell growth medium was used as a negative control. Values represent mean OD ± SD for triplicate assays. For apoptosis evaluation, the cells were treated with DXM (85, 170 or 340 μM) or 10 μM of etopside (positive control) and fixed after 48 hours treatment using the TUNEL assay. DNA fragments were analyzed by flow cytometry. Values were the average percent apoptotic cells from three independent experiments. *Denotes p < 0.05; **Denotes p < 0.01.
3.2. Effect of IL-6 and IL-6 Ab on DXM induced cytotoxicity and apoptosis in the cells of 7TD1-DXM and 7TD1-WD-90
7TD1 cell growth depends on IL-6 and the cells are sensitive to DXM- induced apoptosis. However, 7TD1-DXM cells are independent of exogenous IL-6 for proliferation and resistant to DXM induced cell growth inhibition and apoptosis [44]. To compared the effect of IL-6 on cell proliferation and apoptosis, we treated both cells of 7TD1-DXM and 7TD1-WD-90 with DXM (85 μM for cell survival and 170 μM for apoptosis) and IL-6 (4 μg/ml) alone or in combination. Cell survival was measured by MTT assay while apoptosis was measured by TUNEL assay and the results are summarized in Fig. 2. In 7TD1-DXM cells, IL-6 could not enhance the cell survival compared to the control and did not significantly affect the cell survival when combining with DXM (Fig. 2A). However, in 7TD1-WD-90 cells, IL-6 significantly reduced the inhibitory effect of DXM (p < 0.01) although IL-6 itself did not affect the cell survival (Fig. 2A). Similarly, IL-6 significantly reduced the apoptotic effects induced by DXM in 7TD1-WD-90 cells (p < 0.01) but had no effect in 7TD1-DXM cells (Fig. 2B). These data suggested that IL-6 may play a protective role in DXM induced toxicity and apoptosis in 7TD1-WD-90 cells. To further evaluate the effect of IL-6 on the cells of 7TD1-DXM and 7TD1-WD-90, we investigated the effect of DXM (85 μM for cell survival and 170 μM for apoptosis) and IL-6Ab (2 ng/ml) alone or in combination on the cell growth and apoptosis in the both cells. IL-6Ab inhibited the cell growth compared with the control group (p < 0.05) but did not further enhance the cell growth inhibition when in combined with DXM in 7TD1-DXM cells (Fig 2C). IL-6Ab profoundly inhibited the cell growth in 7TD1-WD-90 cells (p < 0.01 vs the control) compared with that in 7TD1-DXM cells with significant difference (p < 0.01). Additionally, IL-6Ab enhanced the cell growth inhibitory effects caused by DXM (p < 0.01) in 7TD1-WD-90 cells but not in 7TD1-DXM cells (Fig. 2C). For apoptosis study, the data showed that IL-6Ab and DXM alone or in combination did not induced significantly apoptotic effects compared to the control (p > 0.05) in 7TD-DXM cells. However, IL-6Ab significantly enhanced the apoptotic effects (p < 0.01) induced by DXM although IL-6Ab alone did not induce apoptosis compared to the control (p > 0.05) in 7TD1-WD-90 cells (Fig. 2D). These studies indicate that blocking the IL-6 effects by IL-6Ab may increase the cytotoxicity and apoptosis induced by DXM in 7TD1-WD-90 cells but not in 7TD1-DXM cells.
Fig. 2.
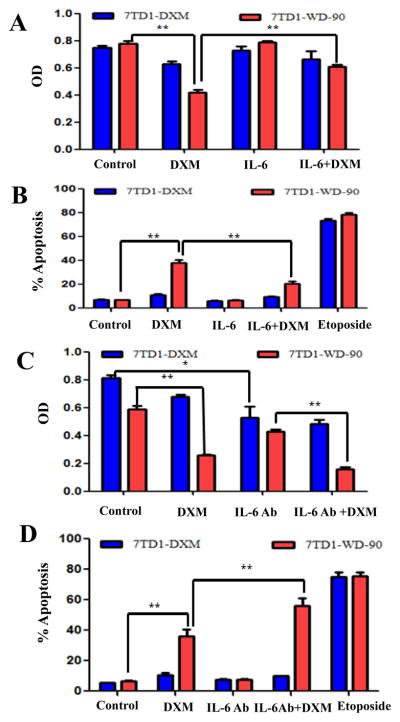
Effect of IL-6 and IL-6Ab alone or in combination on DXM induced cytotoxicity (A & C) and apoptosis (B & D) in the cells of 7TD1-DXM and 7TD1-WD-90. The cells were treated with IL-6 (4 μg/ml), IL-6Ab (2 ng/ml) or DXM (85 μM) alone or in combination for 72 hours. Cell proliferation was determined by MTT assay. Values represent mean OD ± SD with triplicate assays. For apoptosis study, the cells were treated with IL-6 (4 μg/ml), IL-6Ab (2 ng/ml), or DXM (170 μM) alone or in combination. The cells were fixed after 48 hours drug treatment, and evaluated for apoptosis using the TUNEL assay. DNA fragments were analyzed by flow cytometry. Etoposide (10 μM) was used as a positive control. The Values present Mean ± SD of percent apoptotic cells with triplicate experiments. *Denotes p < 0.05; **Denotes p < 0.01.
3.3. Effect of AG490 and DXM alone or in combination on cell growth and apoptosis in the cells of 7TD1-DXM and 7TD1-WD-90
AG490 is a JAK inhibitor and can inhibit the JAK/STAT signaling pathway. In order to explore the relationship between the JAK/STAT signaling pathway and DXM activity in the cells of 7TD1-DXM and 7TD1-WD-90, we investigated the effects of AG490 and DXM on cell growth and apoptosis by treated both cells with AG490 (10 μM for cytotoxicity study and 50 μM for apoptosis study) and DXM (85 μM for cytotoxicity study and 170 μM for apoptosis study) alone or in combination. The data showed that although AG490 inhibited the cell growth (p < 0.01 compared to the control), it could not further enhance the inhibitory effects of DXM in 7TD1-DXM cells. However, AG490 not only inhibited the cell growth (p < 0.01), but it also significantly (p < 0.01) enhanced the inhibitory effect of DXM in 7TD1-WD-90 cells (Fig. 3A). Additionally, AG490 and DXM alone or in combination did not significantly induce apoptosis (p > 0.05 compared to the control) in 7TD1-DXM cells. However, AG490 and DXM alone induced higher apoptotic effects compared with the control (p < 0.05 and p < 0.01, respectively) and the combination of AG490 and DXM could further significantly enhanced apoptosis compared to AG490 alone (p < 0.01) in 7TD1-WD-90 cells (Fig. 3B). The data indicate that AG490 could only enhance the effect of DXM in 7TD1-WD-90 cells but not in 7TD1-DXM cells.
Fig. 3.

Effects of AG490 and DXM alone or in combination on cell growth inhibition (A) and apoptosis (B) in the cells of 7TD1-DXM and 7TD1-WD-90. The cells were treated with AG490 (10 μM for cell growth inhibition and 50 μM for apoptosis) and DXM (85 μM for cell growth inhibition and 170 μM for apoptosis) alone or in combination. Cell growth was determined by MTT assay after 72 hours drug treatment. Values represent mean OD ± SD for triplicate assays. For apoptosis study, the cells were fixed after 48 hours drug treatment and evaluated for apoptosis by TUNEL assay, DNA fragments were analyzed by flow cytometry. The data present Mean ± SD from three independent experiments. *Denotes p < 0.05; **Denotes p < 0.01.
3.4. Effect of DXM, IL-6 or AG490 on JAK2/STAT3 signaling pathway in the cells of 7TD1-DXM and 7TD1-WD
The JAK2/STAT3 signaling pathway is the major pathway of IL-6 and is constitutively activated in 7TD1-DXM cells [44]. AG490 blocks the constitutive activation of STAT3 [45]. To examine the effects of DXM, IL-6 and AG490 on the JAK2/STAT3 signaling pathways in both cells of 7TD1-DXM and 7TD1-WD-90, we treated the cells with 170 μM of DXM, 4 μg/ml of IL-6 or 50 μM of AG490 and examined the expression level of total JAK2 and phosphorylated JAK2, total STAT3 and phosphorylated STAT3 using Western blotting analysis. The data showed that DXM or IL-6 did not cause significant changes in both of the total protein levels and phosphorylation of JAK2 and STAT3 in both cells (Fig. 4). AG490 also did not affect the protein levels of total JAK2 and STAT3 in both cells. However, AG490 significantly inhibited the phosphorylation of JAK2 and phosphorylation of STAT3 in 7TD1-WD-90 cells but not in 7TD1-DXM cells (Fig. 4). The data suggested that JAK2/STAT-3 signaling pathways may be changed in 7TD1-WD-90 cells compared with 7TD1-DXM cells.
Fig. 4.

Effects of DXM, IL-6 and AG490 on the JAK2/STAT3 signaling pathway in the cells of 7TD1-DXM and 7TD1-WD-90. Both cells were treated with 170 μM of DXM, 4 μg/ml of IL-6 or 50 μM of AG490 and incubated at 37°C for 6 hours, and then the cells were collected. Cell lysates were prepared and run on a SDS-PAGE following by Western blot analysis. Beta-actin was used as a loading control. The experiments were carried out at least three times.
4. Discussion
In order to investigate the possibility of reversing DXM resistance of myeloma cells, we have selected to use the DXM resistant 7TD1-DXM cell line which we previously established from a mouse myeloma 7TD1 cell line as the model system for our studies. Parental 7TD1 cell growth depends on IL-6 and is sensitive to DXM induced apoptosis [26, 28, 46]. Chronic exposure of 85 μM of DXM to 7TD1 cells over 6 months led the cells becoming resistant to DXM and growing independently of exogenous IL-6 [44]. The 85 μM of DXM is about the IC50 for 7TD1 cells (data not shown and [44]). After 7TD1 cells become resistant to DXM, the JAK/STAT pathway is constitutively activated [44]. In this study, we withdrew DXM from the DXM resistant 7TD1-DXM cells for 60, 90 and 150 days and tried to determine whether DXM withdrawal reverses the DXM resistance in 7TD1-DXM cells; and, if so, how long duration is needed for the reversal. Moreover, we investigated the effect of IL-6 and JAK/STAT pathway on 7TD1-DXM cells and DXM withdrawn cells.
First, we withdrew DXM for 60 days to test the cells growth inhibition with the treatment of 85 μM of DXM and compare to 7TD1-DXM cells, the results indicated there was no significant difference in cell growth inhibition between the cells of 7TD1-DXM and 7TD1-WD-60 (withdrew DXM for 60 days) (Fig. 1A). Next, we increased the DXM withdrawing time to 90 and 150 days, the data showed that the cells could be significantly inhibited and there was significantly statistical difference in the cell growth between the cells of 7TD1-DXM and the cells of 7TD1-WD-90 (p < 0.01) and 7TD1-WD-150 (P < 0.001) (Fig. 1A). Then, we treated the cells of 7TD1-DXM, 7TD1-WD-90 and 7TD1-WD-150 with various concentrations of DXM (10–255 μM) to determine the dose response curve and the IC50 of the DXM in each cells. The IC50 of the DXM was determined to be 172 ± 10, 106 ± 6 and 86 ± 15 μM, respectively (Fig. 1C). The data indicated that withdrawing DXM for 90 days almost completely reversed the sensitivity of the cell to DXM while withdrawing DXM for 150 days completely reversed the sensitivity of the cell to DXM compared to the parent 7TD1 cells (IC50 ~85 μM) (Fig. 1B and 1C).
DXM resistance 7TD1-DXM cells were resistant to apoptosis induced by DXM. Development of drug resistance of DXM in treatment of MM may be attributable to MM cells resistance to DXM induced apoptosis clinically. To investigate the effect of DXM withdrawal on DXM induced apoptosis, we detected the apoptotic effects induced by various concentrations of DXM (85–340 μM) in the cells of 7TD1-DXM and 7TD1-WD-90. Our data showed that low concentration of DXM (85 μM) did not induce significant apoptosis compared to the control in both cells. The data indicated that 85 μM of DXM could inhibit the cell proliferation, but was not able to induce apoptosis in 7TD1-WD-90 cells. However, when we increased the DXM concentration to 170 μM, significantly higher percentage of apoptotic cells were detected in the 7TD1-WD-90 cells but not in 7TD1-DXM cells (p < 0.01). Furthermore, high concentration (340 μM) of DXM significantly induced apoptosis in both cells compared to the control, however, the apoptotic effect was profoundly induced in 7TD1-WD-90 cells than that in 7TD1-DXM cells (Fig. 1D).
IL-6 plays an important role in regulating the proliferation and apoptosis of 7TD1 cells [43]. While DXM resistant 7TD1-DXM cells developed from 7TD1 cells are independent of exogenous IL-6 for proliferation. We want to know what role(s) of IL-6 may play in the DXM withdrawn cells. Our data showed that treatment of the 7TD1-WD-90 cells (withdrawing DXM over 90 days from 7TD1-DXM cells). with 4 μg/ml of IL-6 reversed the inhibitory and apoptotic effects caused by DXM but did not enhance the cell proliferation (Fig. 2A and 2B), suggesting that withdrawal of DXM may lead to the resistant cells becoming more sensitive to IL-6, and the growth of 7TD1-WD-90 cells was still IL-6 independent. We also used IL-6 antibody (IL-6Ab) to block the effects of IL-6, and compared the inhibitory and apoptotic effects caused by DXM in both cell lines. IL-6Ab alone inhibited the cell growth but did not further increase the cell growth inhibition when it combined with DXM in 7TD1-DXM cells. However, IL-6Ab not only inhibited the cell growth, but also significantly enhanced the inhibitory effects of DXM in 7TD1-WD-90 cells (Fig. 2C). We also investigate the apoptotic effects of IL-6Ab and DXM alone and in combination in the cells of 7TD1-DXM and 7TD1-WD-90. We observed that IL-6Ab significantly enhanced apoptosis induced by DXM in 7TD1-WD-90 cells but not in 7TD1-DXM cells (Fig. 2D). The data generated from IL-6 and IL-6Ab experiments indicate that IL-6 may be important to regulate the cell growth and survival as well as response to DXM induced apoptosis in 7TD1-WD-90 cells.
JAK/STAT pathway is the major signal transduction pathway in IL-6 signaling in multiple myeloma cells and this pathway can be inhibited by AG490. JAK/STAT pathway is constitutively activated in the DXM resistant 7TD1 cells [44]. AG490 is a member of the tyrphostin family of tyrosine kinase inhibitors and it inhibits the JAK/STAT, JAK/AP-1, and JAK/MAPK pathways [45]. AG490 selectively blocks leukemic cell growth in vitro and in vivo by inducing apoptosis with no deleterious effect on normal haematopoiesis [47]. Although AG490 could inhibit the cell growth of 7TD1-DXM cells, it could not induce apoptosis in these cells (Fig. 3). However, AG490 could inhibit cell growth and induce apoptosis in 7TD1-WD-90 cells. More importantly, it greatly potentiated the apoptotic effect induced by DXM in 7TD1-WD-90 cells but not in 7TD1-DXM cells (Fig. 3). The results suggested that JAK/STAT pathway may also be important in regulating cell growth and survival in 7TD1-WD-90 cells. Additionally, we examined the expression of total JAK2 and phosphorylated JAK2, and total STAT3 and phosphoryated STAT3 after DXM, IL-6 and AG490 treatment. We have previously showed that 50 μM of AG490 could completely block the phosphorylation in the parent 7TD1 cells while STAT3 was constitutively active in the DXM resistant 7TD1-DXM cells [44]. In the present study, we observed that AG490 (50 μM) significantly inhibited the expression of phosphorylated JAK2 and phosphorylated STAT3 in the 7TD1-WD-90 cells to some extends, but not in 7TD1-DXM cells. The data suggest that JAK2/STAT3 signaling pathway maybe changed in the 7TD1-WD-90 cells and IL-6 mediated JAK2/STAT3 signaling pathway may, at least in part, contribute to the reversion of DXM resistance following DXM withdrawal for 90 days in 7TD1-DXM cells (Fig. 4). In addition, we measured the expression of JAK2 and phosphorylated JAK2, STAT3 and phosphorylated STAT3 after treating both cell lines with the combination of DXM and AG490, and we could not detect significant change in the expression of these proteins compared with AG490 alone (data not shown). This suggested that DXM did not add any additional inhibitory effects on the JAK2/STAT3 signaling pathway compared with by AG490 alone.
Previous studies already showed the benefits of withdrawing therapeutic agents to prevent drug induced toxicity and/or to reverse drug resistance. Some chemotherapy agents such as paclitaxe, cisplatin and methotrexate could induce neurotoxicity, but neurons were able to recover after termination of drug treatment [48]. Macrolide antibiotics could reverse anticancer drug resistance such as vinblastine on leukemia cells [49]. Withdrawal of sex steroid reversed therapy related defects in bone marrow lymphopoiesis [50]. In this study, we demonstrate that withdrawing DXM for 90–150 days could near or completely reverse DXM resistant in 7TD1-DXM cells. We also revealed that IL-6 and JAK2/STAT3 pathway may play important roles in the DXM withdrawn cells. Microarray analysis showed that histone deacetylase 3 (HDAC3) was up-regulated in response to IL-6 treatment in 7TD1 cells. We evaluated the effects of two structurally different histone deacetylase inhibitors (HDACi), Suberoylanilide Hydroxamic Acid (SAHA) and Sodium Butyrate (NaB), on proliferation and apoptosis in dexamethasone sensitive, resistant, and withdrawn 7TD1 cell lines. We found that inhibition of HDAC3 can enhance the sensitivity of 7TD1 multiple myeloma cells to DXM. The data may suggest that the change of the sensitivity to chemotherapy agents may also due to some genetic changes (unpublished data).
For patients with MM, the average age of patients is about 70 years old and the 5 year survival rate is still relatively very low. DXM is one of the first line chemotherapy choices to treat MM, and the drug resistance is a major problem leading to the failure of the therapy. This study provides some information for DXM treatment in patients with MM. Patients with MM may discontinue DXM treatment for a period of time when DXM resistance occurred. The patients may become sensitive to DXM and resume DXM treatment again after termination of the treatment for certain times.
In brief, this study demonstrates that withdrawing DXM for 90 days or longer can restore the sensitivity of DXM in induction of cytotoxicity and apoptosis in DXM resistant 7TD1-DXM cells near to that of the parent 7TD1 cells. There are different effects of IL-6 and AG490 on cell growth inhibition and apoptosis in 7TD1-DXM and 7TD1-WD cells. It may have potential implication for DXM in treatment of MM clinically.
Acknowledgments
The authors would like to give our thanks to Saini Ashwani for technical assistance and to Dr. Matthew Ndonwi for his critical review of this manuscript.
Grants support
This work was supported by NIH Grant P20RR164454 from the IDeA Networks of Biomedical Research Excellence (INBRE) program of the National Center for Research Resources.
Footnotes
Conflict of interest
No conflict of interest relevant to this article is reported.
Authors’ contributions
CKD designed and guided the whole research and provided the funding for the research. TL designed and performed most of the experiments, ZF analyzed the flow cytometry data and most of other data, KJG performed the flow cytometry, SA studied the role of HDAC3 in 7TD1 cells. CKD, TL and SC systematically analyzed the data and wrote the manuscript. CKD critically reviewed the manuscript. All of the other authors contributed to review the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zaidi AA, Vesole DH. Multiple myeloma: an old disease with new hope for the future. CA Cancer J Clin. 2001;51:273–85. doi: 10.3322/canjclin.51.5.273. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 3.Gadóa K, Domjána G, Hegyesib H, Falus A. Role of interleukin-6 in the pathogensis of multiple myeloma. Cell Biol Int. 2000;24:195–209. doi: 10.1006/cbir.2000.0497. [DOI] [PubMed] [Google Scholar]
- 4.Kyle RA, Rajkumar SV. Multiple myeloma. Blood. 2008;111:2962–72. doi: 10.1182/blood-2007-10-078022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barlogie B, Shaughnessy J, Tricot G, Jacobson J, Zangari M, Anaissie E, et al. Treatment of multiple myeloma. Blood. 2004;103:20–32. doi: 10.1182/blood-2003-04-1045. [DOI] [PubMed] [Google Scholar]
- 6.Bruno B, Giaccone L, Rotta M, Anderson K, Boccadoro M. Novel targeted drugs for the treatment of multiple myeloma: from bench to bedside. Leukemia. 2005;19:1729–38. doi: 10.1038/sj.leu.2403905. [DOI] [PubMed] [Google Scholar]
- 7.Rousseau GG, Baxter JD, Tomkins GM. Glucocorticoid receptors: relations between steroid binding and biological effects. J Mol Biol. 1972;67:99–115. doi: 10.1016/0022-2836(72)90389-0. [DOI] [PubMed] [Google Scholar]
- 8.Okret S, Poellinger L, Dong Y, Gustafsson JA. Down-regulation of glucocorticoid receptor mRNA by glucocorticoid hormones and recognition by the receptor of a specific binding sequence within a receptor cDNA clone. Proc Natl Acad Sci U S A. 1986;83:5899–903. doi: 10.1073/pnas.83.16.5899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rogatsky I, Trowbridge JM, Garabedian MJ. Glucocorticoid receptor-mediated cell cycle arrest is achieved through distinct cell-specific transcriptional regulatory mechanisms. Mol Cell Biol. 1997;17:3181–93. doi: 10.1128/mcb.17.6.3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi SH, Kim SW, Choi DH, Min BH, Chun BG. Polyamine-depletion Induces p27Kip1 and enhances dexamethasone-induced G1 arrest and apoptosis in human T lymphoblastic leukemia cells. Leuk Res. 2000;24:119–27. doi: 10.1016/s0145-2126(99)00161-7. [DOI] [PubMed] [Google Scholar]
- 11.Nuutinen U, Suoranta S, Eeva J, Eray M, Pellinen R, Wahlfors J, et al. Dexamethasone-induced apoptosis and up-regulation of Bim is dependent on glycogen synthase kinase-3. Leuk Res. 2009;33:1714–7. doi: 10.1016/j.leukres.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 12.Marchetti MC, Marco BD, Cifone, Grazia, Migliorati G, Riccardi C. Dexamethasone-induced apoptosis of thymocytes: role of glucocorticoid receptor-associated Src kinase and caspase-8 activation. Blood. 2003;101:585–93. doi: 10.1182/blood-2002-06-1779. [DOI] [PubMed] [Google Scholar]
- 13.Chua CC, Chua BHL, Chen Z, Cathy L, Hamdy RC. Dexamethasone induces caspase activation in murine osteoblastic MC3T3-E1 cells. Biochimica et Biophysica Acta (BBA) -Molecular Cell Research. 2003;1642:79–85. doi: 10.1016/s0167-4889(03)00100-9. [DOI] [PubMed] [Google Scholar]
- 14.Abrams MT, Robertson NM, Yoon K, Wickstrom E. Inhibition of glucocorticoid-induced apoptosis by targeting the major splice variants of BIM mRNA with small interfering RNA and short hairpin RNA. J Biol Chem. 2004;279:55809–17. doi: 10.1074/jbc.M411767200. [DOI] [PubMed] [Google Scholar]
- 15.Chrysis D, Zaman F, Chagin AS, Takigawa M, Sävendahl L. Dexamethasone induces apoptosis in proliferative chondrocytes through activation of caspases and suppression of the Akt-phosphatidylinositol 3′-kinase signaling pathway. Endocrinology. 2005;146:1391–7. doi: 10.1210/en.2004-1152. [DOI] [PubMed] [Google Scholar]
- 16.Sharma S, Lichtenstein A. Dexamethasone-induced apoptotic mechanisms in myeloma cells investigated by analysis of mutant glucocorticoid receptors. Blood. 2008;112:1338–45. doi: 10.1182/blood-2007-11-124156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmidt S, Rainer J, Ploner C, Presul E, Riml S, Kofler R. Glucocorticoid-induced apoptosis and glucocorticoid resistance: molecular mechanisms and clinical relevance. Cell Death and Differentiation. 2004;11:S45–55. doi: 10.1038/sj.cdd.4401456. [DOI] [PubMed] [Google Scholar]
- 18.Marchetti MC, Marco BD, Santini MC, Bartoli A, Delfino DV, Riccardi C. Dexamethasone-induced thymocytes apoptosis requires glucocorticoid receptor nuclear translocation but not mitochondrial membrane potential transition. Toxicol Lett. 2003;139:175–80. doi: 10.1016/s0378-4274(02)00431-9. [DOI] [PubMed] [Google Scholar]
- 19.Bachmann PS, Gorman R, MacKenzie KL, Lutze-Mann L, Lock RB. Dexamethasone resistance in B-cell precursor childhood acute lymphoblastic leukemia occurs downstream of ligand-induced nuclear translocation of the glucocorticoid receptor. Blood. 2005;105:2519–26. doi: 10.1182/blood-2004-05-2023. [DOI] [PubMed] [Google Scholar]
- 20.Gazitt Y, Fey V, Thomas C, Alvarez R. Bcl-2 overexpression is associated with resistance to dexamethasone, but not melphalan, in multiple myeloma cells. Int J Oncol. 1998;13:397–405. doi: 10.3892/ijo.13.2.397. [DOI] [PubMed] [Google Scholar]
- 21.Kerr R, Stirling D, Ludlam CA. Interleukin 6 and haemostasis. Br J Haematol. 2001;115:3–12. doi: 10.1046/j.1365-2141.2001.03061.x. [DOI] [PubMed] [Google Scholar]
- 22.Heinrich PC, Behrmann I, Haan S, Hermanns HM, Müller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simpson RJ, Hammacher A, Smith DK, Matthews JM, Ward LD. Interleukin-6: structure-function relationships. Protein Science. 1997;6:929–55. doi: 10.1002/pro.5560060501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mitsiades CS, McMillin DW, Klippel S, Hideshima T, Chauhan D, Richardson PG, et al. The role of the bone marrow microenvironment in the pathophysiology of myeloma and its significance in the development of more effective therapies. Hematol Oncol Clin North Am. 2007;21:1007–34. doi: 10.1016/j.hoc.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 25.Chatterjee M, Hönemann D, Lentzsch S, Bommert K, Sers C, Herrmann P, et al. In the presence of bone marrow stromal cells human multiple myeloma cells become independent of the IL-6/gp130/STAT3 pathway. Blood. 2002;100:3311–8. doi: 10.1182/blood-2002-01-0102. [DOI] [PubMed] [Google Scholar]
- 26.Chauhan D, Pandey P, Hideshima T, Treon S, Raje N, Davies FE, et al. SHP2 mediates the protective effect of interleukin-6 against dexamethasone-induced apoptosis in multiple myeloma cells. J Biol Chem. 2000;275:27845–50. doi: 10.1074/jbc.M003428200. [DOI] [PubMed] [Google Scholar]
- 27.Urashima M, Teoh G, Chauhan D, Hoshi Y, Ogata A, Treon SP, et al. Interleukin-6 overcomes p21WAF1 upregulation and G1 growth arrest induced by dexamethasone and interferon-gamma in multiple myeloma cells. Blood. 1997;90:279–89. [PubMed] [Google Scholar]
- 28.Xu Fh, Sharma S, Gardner A, Tu Y, Raitano A, Sawyers C, et al. Interleukin-6-induced inhibition of multiple myeloma cell apoptosis: support for the hypothesis that protection is mediated via inhibition of the JNK/SAPK pathway. Blood. 1998;92:241–51. [PubMed] [Google Scholar]
- 29.Bosscher KD, Berghe WV, Vermeulen L, Plaisance S, Boone E, Haegeman G. Glucocorticoids repress NF-κB-driven genes by disturbing the interaction of p65 with the basal transcription machinery, irrespective of coactivator levels in the cell. Proc Natl Acad Sci U S A. 2000;97:3919–24. doi: 10.1073/pnas.97.8.3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ray A, Prefontaine KE. Physical association and functional antagonism between the p65 subunit of transcription factor NF-κB and the glucocorticoid receptor. Proc Natl Acad Sci U S A. 1994;91:752–6. doi: 10.1073/pnas.91.2.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.D’Acquisto F, May MJ, Ghosh S. Inhibition of nuclear factor kappa B (NF-κB): an emerging theme in anti-inflammatory therapies. Mol Interv. 2002;2:22–35. doi: 10.1124/mi.2.1.22. [DOI] [PubMed] [Google Scholar]
- 32.Boumpas D. A novel action of glucocorticoids--NF-kappa B inhibition. Br J Rheumatol. 1996;35:709–10. doi: 10.1093/rheumatology/35.8.709. [DOI] [PubMed] [Google Scholar]
- 33.Bruno B, Rotta M, Giaccone L, Massaia M, Bertola A, Palumbo A, et al. New drugs for treatment of multiple myeloma. Lancet Oncol. 2004;5:430–42. doi: 10.1016/S1470-2045(04)01511-6. [DOI] [PubMed] [Google Scholar]
- 34.Greenstein S, Krett NL, Kurosawa Y, Ma C, Chauhan D, Hideshima T, et al. Characterization of the MM.1 human multiple myeloma (MM) cell lines: a model system to elucidate the characteristics, behavior, and signaling of steroid-sensitive and -resistant MM cells. Exp Hematol. 2003;31:271–82. doi: 10.1016/s0301-472x(03)00023-7. [DOI] [PubMed] [Google Scholar]
- 35.Frassanito MA, Cusmai A, Iodice G, Dammacco F. Autocrine interleukin-6 production and highly malignant multiple myeloma: relation with resistance to drug-induced apoptosis. Blood. 2001;97:483–9. doi: 10.1182/blood.v97.2.483. [DOI] [PubMed] [Google Scholar]
- 36.Boulanger MJ, Chow DC, Brevnova EE, Garcia KC. Hexameric structure and assembly of the interleukin-6/IL-6α-receptor/gp130 complex. Science. 2003;300:2101–4. doi: 10.1126/science.1083901. [DOI] [PubMed] [Google Scholar]
- 37.Hibi M, Murakami M, Saito M, Hirano T, Taga T, Kishimoto T. Molecular cloning and expression of an IL-6 signal transducer, gp130. Cell. 1990;63:1149–57. doi: 10.1016/0092-8674(90)90411-7. [DOI] [PubMed] [Google Scholar]
- 38.Jakob U, Scheibel T, Bose S, Reinstein J, Buchner J. Assessment of the ATP binding properties of Hsp90. The Journal of Biological Chemistry. 1996;271:10035–41. doi: 10.1074/jbc.271.17.10035. [DOI] [PubMed] [Google Scholar]
- 39.Wegenka UM, Lutticken C, Jan B, Yuan J, Lottspeich F, Mueller-Esterl W, et al. The interleukin-6-activated acute-phase response factor is antigenically and functionally related to members of the signal transducer and activator of transcription (STAT) family. Mol Cell Biol. 1994;14:3186–96. doi: 10.1128/mcb.14.5.3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heinrich PC, Behrmann I, MÜller-Newen G, Schaper F, Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J. 1998;334:297–314. doi: 10.1042/bj3340297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lentzsch S, Chatterjee M, Gries M, Bommert K, Gollasch H, Doerken B, et al. PI3-K/AKT/FKHR and MAPK signaling cascades are redundantly stimulated by a variety of cytokines and contribute independently to proliferation and survival of multiple myeloma cells. Leukemia. 2004;18:1883–90. doi: 10.1038/sj.leu.2403486. [DOI] [PubMed] [Google Scholar]
- 42.Ogata A, Chauhan D, Teoh G, Treon SP, Urashima M, Schlossman RL, et al. IL-6 triggers cell growth via the Ras-dependent mitogen-activated protein kinase cascade. J Immunol. 1997;159:2212–21. [PubMed] [Google Scholar]
- 43.Irvina BJ, Hansona CL, Smithb LH, Daniels CK. Cyclic AMP- and IL6-signaling cross talk: comodulation of proliferation and apoptosis in the 7TD1 B cell hybridoma. Exp Cell Res. 2001;265:73–9. doi: 10.1006/excr.2001.5157. [DOI] [PubMed] [Google Scholar]
- 44.Gangavarapu KJ, Olbertz JL, Bhushan A, Lai JCK, Daniels CK. Apoptotic resistance exhibited by dexamethasone-resistant murine 7TD1 cells is controlled independently of interleukin-6 triggered signaling. Apoptosis. 2008;13:1394–400. doi: 10.1007/s10495-008-0265-y. [DOI] [PubMed] [Google Scholar]
- 45.Wang LH, Kirken RA, Erwin RA, Yu CR, Farrar WL. JAK3, STAT, and MAPK signaling pathways as novel molecular targets for the tyrphostin AG-490 regulation of IL-2-mediated T cell response. J Immunol. 1999;162:3897–904. [PubMed] [Google Scholar]
- 46.Raeve HRD, Vanderkerke K. The role of the bone marrow microenvironment in multiple myeloma. Histol Histopathol. 2005;20:1227–50. doi: 10.14670/HH-20.1227. [DOI] [PubMed] [Google Scholar]
- 47.Meydan N, Grunberger T, Dadi H, Shahar M, Arpaia E, Lapidot Z, et al. Inhibition of acute lymphoblastic leukaemia by a Jak-2 inhibitor. Nature. 1996;379:645– 8. doi: 10.1038/379645a0. [DOI] [PubMed] [Google Scholar]
- 48.James SE, Burden H, Burgess R, Xie Y, Yang T, Massa SM, et al. Anit-cancer drug Induced neurotoxicity and identification of Rho pathway signaling modulators as potential neuroprotectants. Neurotoxicology. 2008;29:605–12. doi: 10.1016/j.neuro.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang L, Kitaichi K, Hui CS, Takagi K, Takagi K, Sakai M, et al. Reversal of anticancer drug resistance by macrolide antibiotics in vitro and in vivo. Clin Exp Pharmacol Physiol. 2000;27:587–93. doi: 10.1046/j.1440-1681.2000.03308.x. [DOI] [PubMed] [Google Scholar]
- 50.Dudakov JA, Goldberg GL, Reiseger JJ, Chidgey AP, Boyd RL. Withdrawal of sex steroids reverses age- and chemotherapy-related defects in bone marrow lymphopoiesis. J Immunol. 2009;182:6247–60. doi: 10.4049/jimmunol.0802446. [DOI] [PubMed] [Google Scholar]