

Summary
Removal of the 5′ cap structure by Dcp2 is a major step in several 5′–3′ mRNA decay pathways. The activity of Dcp2 is enhanced by Dcp1 and bound coactivators, yet the details of how these interactions are linked to chemistry are poorly understood. Here we report three crystal structures of the catalytic Nudix hydrolase domain of Dcp2 that demonstrate binding of a catalytically essential metal ion, and enzyme kinetics are used to identify several key active site residues involved in acid/base chemistry of decapping. Using NMR and molecular dynamics, we find that a conserved metal binding loop on the catalytic domain undergoes conformational changes during the catalytic cycle. These findings describe key events during the chemical step of decapping, suggest local active site conformational changes are important for activity, and provide a framework to explain stimulation of catalysis by the regulatory domain of Dcp2 and associated coactivators.
Introduction
Eukaryotic mRNAs contain a 7-methylguanosine cap at the 5′ end that promotes different steps in the mRNA lifecycle, including splicing, export, translation and decay (Moore, 2005). Removal of this 5′-cap is the committed step in 5′-to-3′ mRNA decay, resulting in exposure of the 5′ terminal phosphate on an RNA that is recognized and degraded by a conserved 5′–3′ exonuclease (Stevens and Maupin, 1987). Decapping is carried out by several enzymes (Chang et al., 2012; Ghosh et al., 2004; Jiao et al., 2010; Liu et al., 2002; Song et al., 2010; Wang and Kiledjian, 2001), including the Nudix hydrolase Dcp2 and its obligate in vivo activator Dcp1. Dcp2 activity is involved in bulk mRNA decay (Beelman et al., 1996; Dunckley and Parker, 1999; Wang et al., 2002) as well as multiple specific pathways including nonsense mediated decay (Amrani et al., 2004; Isken and Maquat, 2007), miRNA-induced decay (Behm-Ansmant et al., 2006; Chen et al., 2009; Eulalio et al., 2007), AU-rich element-mediated decay (Chen et al., 1995; Fenger-Gron et al., 2005), and 3′-uridylation (Heo et al., 2009; Rissland and Norbury, 2009; Shen and Goodman, 2004; Song and Kiledjian, 2007). Dcp2 is at the core of a large network of protein-protein interactions involving multiple decapping factors that can act in either a generic or pathway specific manner (e.g. Edc 1–3, Upf1, Lsm 1–7, Pat1, Dhh1) (Arribas-Layton et al., 2012). These decapping complexes have been implicated in many cellular processes including stress response (Hilgers et al., 2006), development (Schier, 2007), transcription (Brannan et al., 2012), translation (Hu et al., 2009), immune response (Li et al., 2012), differentiation (Sweet et al., 2012), and transcript quality control (Lykke-Andersen, 2002).
Dcp2 has two conserved domains that exist as a bilobed structure which is thought to undergo an open to closed transition to form a composite active site upon activation of the enzyme (Floor et al., 2012; Floor et al., 2010; She et al., 2006; She et al., 2008). The decapping activity of Dcp2 is located in its catalytic Nudix domain, which is functionally active in vitro, although addition of the N-terminal regulatory domain of Dcp2 enhances decapping activity dramatically, in part by binding the essential activator Dcp1 (Deshmukh et al., 2008; She et al., 2006; She et al., 2008). Coactivators of decapping Edc1 and Edc2 can enhance Dcp2 activity in vitro by binding to Dcp1, possibly promoting the closed active form of Dcp2 within the decapping holoenzyme (Borja et al., 2011; Floor et al., 2012). Other Dcp1 interaction partners such as Xrn1 (Braun et al., 2012) and PNRC2 (Lai et al., 2012) may activate decapping in a similar manner. In addition, Dcp2 binding partners can also enhance decapping (Fromm et al., 2012; Harigaya et al., 2010). However, the mechanistic details about how coactivators can accelerate decapping by coupling conformational changes to the chemical step remains unclear.
Some clues about the chemical mechanism of Dcp2 can be taken from studies of related Nudix enzymes (Mildvan et al., 2005). Nudix enzymes catalyze hydrolysis of a nucleoside diphosphate from another chemical group and contain the catalytic Nudix motif GX5EX7REUXEEXGU, where X is any residue and U is a bulky hydrophobic residue (Mildvan et al., 2005). This motif folds into a characteristic loop-helix-loop structure that forms the active site. Hydrolysis typically occurs by nucleophilic substitution at the substrate phosphorus and requires a variable number of divalent cations. Several conserved glutamates within the Nudix motif coordinate metal, and one usually serves as a general base in the catalytic cycle, though the identity and location of the general base varies between family members. A general acid near the active site often enhances catalysis by stabilizing departure of the leaving group. Additional insertions or domains outside the canonical Nudix hydrolase fold can make specific contacts with the substrate and position it for catalysis (Gabelli et al., 2002). However, due to the large variation in substrates for this family of enzymes, these features are tailored to each family member to provide specificity in substrate recognition, and the potential for regulation by protein interactions in the case of Dcp2.
Cap hydrolysis by Dcp2 results in formation of an m7GDP product and 5′ monophosphate RNA body. Four conserved glutamates are crucial for decapping activity both in vivo and in vitro (Deshmukh et al., 2008; Dunckley and Parker, 1999; She et al., 2006). Three of these glutamates are located on the Nudix helix (S. cerevisiae E149, E152, and E153), while a fourth (E198) is located on a loop near the active site that changes conformation in the open, unliganded and closed, liganded Dcp1/Dcp2 co-crystal structure containing an ATP crystallization additive (She et al., 2006). The latter finding echoes observations made for other Nudix enzymes that conformational rearrangements of metal binding loops or the general base can be coupled to substrate binding (Gabelli et al., 2002). However, it is unknown how these residues contribute to cap hydrolysis by Dcp2.
Here, we provide insight into the catalytic cycle of Dcp2 by integrating data from X-ray crystallography, pH-dependent enzyme kinetics and NMR spectroscopy, and molecular dynamics simulations. The crystal structure of the catalytic domain of S. cerevisiae Dcp2 with bound Mg2+, along with structures of two mutants, implicate distinct glutamate residues in metal binding and general base activity, including a glutamate located on a loop near the active site. Kinetic studies are employed to determine which glutamate is the general base and further identify a cationic general acid important for substrate positioning and leaving-group stabilization. Methyl-NMR spectroscopy and molecular dynamics simulations suggest that in a loop containing the fourth essential glutamate changes conformation depending on protonation of the general base. These findings provide roles for conserved catalytic residues of the Nudix domain of Dcp2, document how a local conformational change in a metal binding loop is coupled to protonation of the general base, and suggest a mechanism of how protein interactions with Dcp2 could be linked to efficient decapping chemistry.
Results
Essential metal binding site of Dcp2 revealed by X-ray crystallography
To determine which residues in the active site of Dcp2 are involved in metal binding and acid/base chemistry, we determined crystal structures of the wild-type, and E198Q and E153Q catalytic glutamate mutants of the Dcp2 Nudix domain (S. cerevisiae 100–245). These structures were solved to 2.1, 1.8 and 1.7 Å, respectively (Table 1 and Fig. 1). For the wild-type Nudix domain, there is one molecule of Dcp2 in the asymmetric unit, which has an overall conformation very similar to the previously reported structure (She et al., 2006; She et al., 2008). Density for an octahedrally coordinated metal ion was clearly visible in the wild-type structure (Fig 1B). Since the crystallization buffer contained magnesium formate as the sole source of divalent metal and Dcp2 is known to be a Mg-dependent enzyme (Steiger et al., 2003), we conclude the metal ion bound to Dcp2 is magnesium. Three conserved glutamates coordinate the magnesium ion through a water mediated contact (E152, E153, and E198), while a fourth (E149) directly contacts the metal, implying E149 plays a crucial role in metal binding (Figs. 1A,B).
Table 1. Crystallographic Data and Refinement Statistics.
Statistics for highest-resolution shell are shown in parentheses
| Wild-type | E198Q | E153Q | |
|---|---|---|---|
| Wavelength (Å) | 0.95372 | 1.11587 | 1.11587 |
| Resolution range (Å) | 50–2.1 (2.14–2.1) | 50–1.8 (1.83–1.8) | 50-1.7 (1.75–1.7) |
| Space group | P 62 | P 1 21 1 | C 121 |
| Unit cell (Å) | 85.054 85.054 48.749 90 90 120 | 57.352 47.804 60.957 90 97.29 90 | 140.789 49.27 84.022 90 91.37 90 |
| Molecules/ASU | 1 | 2 | 3 |
| Total Reflections | 86873 | 53880 | 81472 |
| Unique reflections | 11889 | 29454 | 54478 |
| Multiplicity | 7.3 (7.3) | 1.8 (1.8) | 10.4 (11.0) |
| Completeness (%) | 99.97 (99.92) | 96.48 (94.68) | 85.69 (60.92) |
| I/σ | 26.8 (3.5) | 28.6(2.7) | 20.5 (1.4) |
| Wilson B-factor | 28.33 | 27.42 | 20.84 |
| R-merge | 0.081 (0.557) | 0.04 (0.383) | 0.099 (0.447) |
| R-work | 0.1530 (0.1903) | 0.1871 (0.2359) | 0.1770 (0.3545) |
| R-free | 0.1910 (0.2442) | 0.2140 (0.2836) | 0.2127 (0.4151) |
| RMS (bonds) | 0.007 | 0.007 | 0.007 |
| RMS (angles) | 1.09 | 1.16 | 1.11 |
| Ramachandran favored (%) | 97 | 95 | 97 |
| Ramachandran outliers (%) | 0.7 | 1.1 | 0.24 |
| Clashscore | 3.33 | 3.89 | 3.23 |
| Average B-factor | 43.30 | 41.80 | 40.5 |
Figure 1. Mg2+ coordination in the Dcp2 catalytic domain.

(A) Ribbon diagram of Mg2+ coordination in the wild-type S. cerevisiae Dcp2 catalytic Nudix domain at the catalytic helix. Four conserved glutamates (E149, E152, E153, E198) and K135 are shown as sticks in green. Water molecules are shown as blue spheres, and the Mg2+ is shown as a black sphere. Colored in grey is the backbone carbonyl of N243 in a symmetry related molecule. Distances between atoms are shown in Å.
(B) Fo−Fc difference electron density map of wild-type Dcp2 depicted as black mesh at an I/σ cutoff of 2.5.
(C) Fo−Fc difference electron density map for the E198Q mutant at an I/σ cutoff of 2.0.
(D) Fo−Fc difference electron density map of E153Q mutant at an I/sigma cutoff of 3.0.
In contrast to the structure of wild-type Nudix domain, the E198Q mutant lacked clear density for a metal ion in the active site and failed to crystallize in the presence of any divalent cation (Fig. 1C). The final structure has two molecules of Dcp2 in the asymmetric unit that differ in the position of loops near the catalytic Nudix helix, including a loop harboring E198Q (Fig. S1A,C; henceforth known as the 190’s loop). The overall structures of both E198Q chains superimpose to an average all-atom 0.374 Å root-mean-square deviation (RMSD) as compared to the wild-type structure. The E198Q 190’s loops in isolation have an elevated average all-atom root-mean-square deviation of 0.519 Å as compared to the wild-type 190’s loop, and importantly, this change in conformation is not mediated by crystal contacts. These observations suggest that E198 is important for metal binding and that the conformation of the 190’s loop is altered in the absence of metal.
In contrast, the crystal structure of the E153Q mutant of Dcp2 had 3 molecules in the asymmetric unit, which superimpose to an average all-atom 0.253 Å RMSD as compared to the wild-type structure (Fig. S1B,D). Interestingly, there is clear electron density for an octahedrally coordinated Mg2+ in the E153Q structure, similar to wild-type (Fig. 1D), suggesting E153 is not necessary for metal binding and may instead serve as the general base in the decapping reaction.
Discovery of the catalytic general base and general acid of Dcp2
To determine the function of residues known to be essential to catalysis by Dcp2, we performed kinetic studies for wild-type and mutant Dcp1/Dcp2 complexes as a function of pH. This approach has been used to identify and determine the pKa of both the general acid and general base in the related Nudix enzyme MutT (Harris et al., 2000; Waley, 1975). Single-turnover experiments were performed under kmax conditions ([E] ≫ KM) to monitor the effect of pH on all steps that occur after substrate binding but before product release. Because product release is fast, kmax corresponds to kcat measured under multiple-turnover conditions (Deshmukh et al., 2008). As expected, decapping activity is dramatically affected by pH (Fig. 2A). Between pH 5.5 and 7.5, log(kmax) increases linearly by approximately two log-units (Fig. 2B), with a maximum occurring between pH 7.5 and 8.0. This is typical behavior for an enzyme utilizing a general base, which should have a near-neutral pKa in order to efficiently abstract a proton from water during catalysis. The decrease in decapping rates between pH 8.0–9.5 indicates the presence of a general acid: a positively charged residue that is deprotonated at high pH and unable to stabilize the negatively charged leaving group, resulting in decreased decapping activity. Fitting the log(kmax)-versus-pH dependence of wild type Dcp2 yields a general base pKa of 7.7 ± 0.5 and a general acid pKa of 8.1 ± 0.7, in good agreement with other Nudix enzymes (Harris et al., 2000).
Figure 2. Decapping rates for wild-type and mutant Dcp1/Dcp2 are affected by pH.
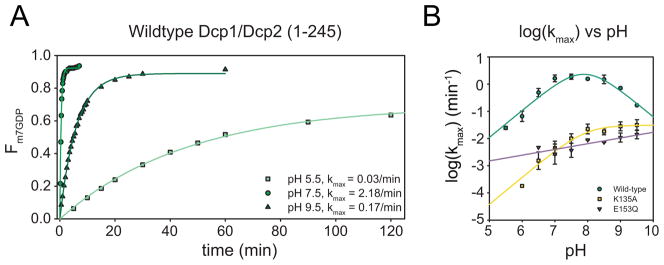
(A) Representative time courses of the fraction m7GDP released and the corresponding first-order exponential fits to obtain kobs over a range of pH values for wild-type Dcp1/Dcp2 decapping complex.
(B) Plot of log (kmax) versus pH for wild-type decapping complex (green), K135A (yellow) and E153Q (purple). Symbols are mean of at least 3 independent experiments and error bars shown are standard deviation. Wild-type and K135A are fit using the 4-parameter equation used to model the dependence of kmax on pH (Harris et al., 2000). E153Q is fit to a line due to the linear dependence of kmax on pH.
Our wild-type and mutant Dcp2 crystal structures led us to hypothesize that E153 serves as a general base in the decapping reaction, and so we first examined the pH dependence of kmax for the E153Q mutant (Fig. 2B). Not surprisingly, E153Q was severely catalytically compromised, with a reduction in kmax of approximately three log-units at pH 7.5 compared to wild-type. Notably, E153Q exhibited multiple products (Fig. 3A–C). Incubation of E153Q decapping reaction products with Nucleoside Diphosphate Kinase (NDPK) identified the major product as m7GTP (as compared to the wild-type product m7GDP) and the two other products as m7GDP and m7GMP (Fig. S2). Such loss of specificity of the cleavage site is consistent with a severe positioning defect of either the substrate or the attacking nucleophile. Because each product formed at the same rate and the endpoint ratios did not vary with pH or enzyme concentration, data were analyzed for total product formed over time (Table S1, S2; see Methods and Discussion). The E153Q mutant displayed a linear dependence on pH over the range studied (pH 7–9.5). Decapping data for E153Q at pH values lower than pH 7.0 were not consistent, most likely due to enzyme instability over the longer time courses needed for the slower reaction. However, the linear behavior observed from pH 7 to 9.5 for E153Q is consistent with E153 functioning as the general base. A similar analysis was performed for the E198Q mutant, which also gave rise to multiple products, consistent with its altered metal binding properties (Fig S2). However, decapping rates were extremely variable, likely due to protein instability under the assay conditions used.
Figure 3. Active site mutations in Dcp1/2 complex result in multiple decapping reaction products.

(A) Representative TLC plate decapping assay data for Dcp1/2 K135A decapping complex at pH 8.0. Arrows point out the location of RNA substrate (black), m7GTP (green), m7GDP (blue) and m7GMP (red) as identified by incubation with NDPK (Fig. S2E).
(B) Representative time course of the fraction of each K135A product formed as a function of time. m7GTP, m7GDP, and m7GMP are shown in green, blue, and red respectively. Total product formed is shown in black. Single exponential fits were used to obtain the individual kobs for each product (see methods and Tables S1, S2).
(C) Distribution of each product formed in the decapping reactions of wild-type, K135A, and E153Q variants, calculated by an average ratio of endpoints for each product across all pH values fit using a single exponential (Table S1).
(D) Representative TLC plate for E153Q showing multiple products. Arrows point out the location of RNA substrate (black), m7GTP (green), m7GDP (blue) and m7GMP (red) as identified by incubation with NDPK (Fig. S2E).
(E) Representative time course for the fraction of each E153Q product formed as a function of time. Coloring is the same as in (B). Single exponential fits were used to obtain the individual kobs for each product (see methods and Tables S1, S2).
Additionally, we sought to use this same approach to identify the general acid of the Dcp2 chemical step. K135 of S. cerevisiae Dcp2 is conserved, and is important for decapping in yeast and in vitro, but its role in catalysis is unclear (Deshmukh et al., 2008; She et al., 2008). Therefore, we suspected it might function as a catalytic general acid. To definitively identity the general acid, we reasoned that mutating this residue should abolish the descending limb of the kmax vs pH plot at the higher pHs. To this end, we examined the pH dependence of kmax for the K135A mutant, which was previously shown to reduce catalysis by 300-fold in vitro (Fig. 2B) (Deshmukh et al., 2008). The K135A mutant also displayed multiple products, and was analyzed as total product formed (Fig. 3C–E; Tables S1, S2) (Deshmukh et al., 2008). The K135A mutation abolished the descending limb of the log(kmax) versus pH profile, indicating it plays the role of general acid, likely by stabilizing the departure of the 5′ monophosphate RNA leaving group as suggested previously (Deshmukh et al,. 2008). The formation of multiple products by the K135A variant suggest K135 may also play a role in positioning substrate relative to the attacking water and general base within the active site of Dcp2.
Measurement of catalytic glutamate pKa values by NMR spectroscopy
To directly monitor the electrostatic environment in the active site of Dcp2 during a pH titration, we turned to NMR spectroscopy. NMR allows pKa measurements to be determined with site-specific resolution (Markley, 1975; Oda et al., 1994; Zhang and Vogel, 1993). Typically, side chain chemical shifts of acidic or basic amino acids are monitored as a function of pH but indirect methods such as monitoring the effect of backbone amide chemical shifts are also employed. We used methyl group 13C NMR spectroscopy to indirectly detect titration of protonatable groups in Dcp2, since these probes allow for sensitive and robust detection of NMR signals in proteins at low concentration, and relevant regions of Dcp2 are unresolved by backbone nitrogen HSQC experiments (data not shown; Floor et al 2010). We reasoned that during a pH titration, methyl groups near the general base should exhibit a change in chemical shift due to changes in the protonation state of the general base. In Dcp2, Ile, Leu and Val (ILV) residues are well distributed near the active site, enabling indirect monitoring of the chemical environment using 13C methyl NMR spectroscopy (Fig. 4A). We focused on the catalytic Nudix domain of Dcp2 to observe chemical shift changes due to protonation events only and eliminate confounding effects of large scale domain reorientation in solution (Floor et al., 2012). At neutral pH, the Dcp2 Nudix domain 13C-ILV HSQC spectrum is well-resolved with homogenous peak intensity (Fig. S3A) with the exception of I199, which is addressed below. Multiple peaks show chemical shift changes as a function of pH, including those corresponding to residues V121, I136, V195, and I199 (Fig. 4B). That only a small set of residues are perturbed over five pH units indicates the fold of the enzyme is relatively insensitive to pH. To determine the apparent pKa (pKaapp) of the environment surrounding I199 and V121 as representative residues near the active site, the total chemical shift change was plotted as a function of pH and fitted to a sigmoidal function, yielding pKaapp values of 7.3 ± 0.02 and 7.2 ± 0.02 respectively (Fig. 4C). Given the only histidine on this domain is approximately 30 Å from these residues in our wild-type crystal structure and the titration was qualitatively similar in the presence of excess magnesium (data not shown), these data suggest that the titratable group belongs to the catalytic general base.
Figure 4. Methyl NMR pH titrations confirm E153 is the general base.

(A) Select Ile, Leu and Val residues on the catalytic domain of S. cerevisiae Dcp2 are highlighted in blue with catalytic glutamates E149, E152, E153 and E198 highlighted in green and the Nudix helix in red. Residues are visualized on PDB 2JVB.
(B) A superposition of 13C-1H HSQC spectra of wild-type scDcp2 (100–245) at pH 4.5 (red), 5.5 (orange), 6.5 (lime), 7.1 (green), 7.6 (cyan), 8.0 (magenta) and 9.0 (purple).
(C) Quantification of the normalized chemical shift change (Δδ) for L120, V121 and I199 across the pH titration. Note that the sidechain of L120 points in the opposite direction from V121. Solid lines are sigmoidal fits, with pKaapp values of 7.3 ± 0.02 and 7.2 ± 0.02 for I199 and V121, respectively. Standard error of the fit parameter is indicated.
(D) 13C-1H HSQC spectra of the E198Q mutant Dcp2 catalytic domain collected at pH values of 5 (red), 5.6 (orange), 6.0 (lime), 6.6 (green), 7.2 (cyan), 8.1 (magenta) and 8.9 (purple). Weak peaks in (B) are likely from residual contamination by the GB1 solubility tag.
(E) 13C-1H HSQC spectra of the E153Q mutant Dcp2 catalytic domain collected at pH values of 5.5 (orange), 6.6 (lime), 7.0 (green), 7.6 (cyan) and 8.5 (purple).
To determine which residues are responsible for the observed chemical shift changes, we systematically mutated the Nudix motif glutamates 149, 152, 153 and 198 of Dcp2 to glutamine and repeated the pH titration. Mutation of E149Q had little effect on pH-sensitive chemical shift changes, with the exception of abrogating the perturbation at I136 (data not shown). Interestingly, despite its proximity to I199 and conservation across species (Fig. S3B), mutation at E198 had little effect besides marginal reduction of the magnitude of chemical shift change (Fig 4D, ~3ppm to ~2ppm in 13C). Mutation of either glutamate 152 or 153 to glutamine strongly attenuated all observed pH-dependent chemical shift changes (Fig. 4E; data not shown for E152). Since either E149 or E198 mutation have little effect, and E152 is not required for decapping by Dcp2 in vivo (Dunckley and Parker, 1999), we conclude that E153 is the general base, as predicted by our kinetic data.
General base protonation remodels a conserved metal binding loop
The pH dependent chemical shift changes in the 13C HSQC depend on the general base (E153) and are consistent with rapid exchange between protonated and deprotonated forms of this residue (Fig. 4). One residue (I199) has a large normalized chemical shift change of around 1.5 ppm (~3ppm in 13C; Fig. 4B), which is consistent with a conformational change in the 190’s loop. Another significantly perturbed residue (V121) is in the beta core of the protein and close to E153. Linewidth changes result from fluctuations in the magnetic environment that occur on the chemical shift timescale (ms-μsec). The only residue with significant pH dependent linewidth changes is I199 (Fig. 5). Collinear chemical shift changes of I199 suggest protonation of E153 by lowering the pH or by removing a titratable group by conservative mutation to glutamine could bias the conformation of the 190s loop into one state (Fig. S4). This suggests the dynamics of the 190s loop is coupled to protonation of E153, although we can not exclude the possibility that the linewidth effects are due to structural dynamics of nearby residues that are not detected in our ILV labeling scheme. Accordingly, the pH dependent spectral changes could be due to change in electrostatic environment from protonation of E153, a coupled conformational transition or a combination of both effects.
Figure 5. Change in pH Induces Changes in Dynamics.

(A) The 13C linewidth (FWHM) for residues I199, V121 and L115 across the pH titration. (B–E) Example fitted 13C linewidths of I199 at pH values indicated. The experimental data is in points with the mixed Gaussian-Lorentzian fit as a line. Intensity is in units of signal-to-noise (S:N) with noise measured in NMRPipe.
To explore the structural consequences of protonation of E153 we performed a series of molecular dynamics simulations of the catalytic domain of Dcp2 from both S. cerevisiae and S. pombe. We simulated wild-type Dcp2 with E153 (S. pombe E147) deprotonated to emulate high pH, E153 with one of the terminal oxygens protonated to emulate low pH, and E153 mutated to glutamine (for S. pombe only). The starting structure for the S. cerevisiae simulations was the NMR structure of Dcp2 100–245 (PDB 2JVB), which partially unfolded during the simulations; we focused instead on the results from S. pombe (started from the crystal structure PDB 2A6T), which were qualitatively similar (Fig. S5A,B). Residue numbers for the remainder of this section therefore refer to S. pombe. When E147 (S. cerevisiae E153) was protonated or mutated to glutamine we observed increased association of the 190’s loop with a beta strand adjacent to the catalytic Nudix helix (Fig. 6). Specifically, snapshots from the simulations show that I193 and V114 (S. cerevisiae I198 and V121) pack close together when E147 is protonated or mutated (Fig. 6A). Meanwhile, I193 had a higher chance of being exposed when E147 was charged (Fig. 6B). The simulations suggest that uncharged E147 hydrogen bonds with the backbone amide of K127, while charged E147 can alternatively interact with cationic residues in the 190’s loop, leading to a conformational change in the 190’s loop.
Figure 6. Molecular dynamics simulations show qualitative and quantitative conformational changes between S. pombe Dcp2 with charged or protonated E147.

(A) Simulations with protonated E147 to mimic the low pH state show relatively little flexibility in the 190’s loop. I193 (I199 in S. cerevisiae) is shown in blue sticks, V114 in red sticks, and the Nudix helix is colored green.
(B) Simulations with charged E147 to mimic the high pH state show increased flexibility with I193 solvent exposed in the final state of two out of six simulations. Colors are the same as in (A).
(C) Histograms of the distance between the terminal methyl groups of V114 and I193 when E147 is protonated to mimic the low pH state (C), when E147 is mutated to glutamine (D), or when E147 is charged to mimic the high pH state (E). The ordinate of all three histograms is probability density per bin; n ≈ 600,000 per histogram. Residues I193, V114 and E147 in S. pombe are S. cerevisiae I199, V121 and E153, respectively.
To quantitatively assess the effect of protonation of E147 on the conformation of the 190’s loop, the distance between the terminal methyl groups of V114 and I193 was measured for each of three states: E147 protonated (Fig. 6C), E147Q (Fig. 6D), and E147 charged (Fig. 6E). These two residues were selected as they exhibit large chemical shift changes across NMR pH titrations (Fig. 4A; V121 and I199). For both E147 protonated and E147Q, the distance histogram shows a single peak corresponding to close packing of V114 and I193 for most of the simulation, with few snapshots exhibiting longer distances (Figs. 6C,D). In contrast, when E147 is negatively charged, a second peak emerges in the distance histogram corresponding to a second conformation of the 190’s loop with greater separation of V114 and I193 (Fig. 6E). The observed conformational changes are not sensitive to the choice of reference residue used to calculate distance histograms (Fig. S5C,D). We conclude that the conformation of the 190’s loop is coupled to the protonation state of the general base E147 (E153 in S. cerevisiae).
Discussion
Using a suite of complementary techniques we have defined the role of multiple conserved glutamate residues in the mRNA decapping enzyme Dcp2, and identified loop motions that are coupled to protonation of the general base, a key step that occurs during the catalytic cycle. NMR spectroscopy and pH-dependent kinetics show that E153 is the catalytic general base, while a trio of crystal structures demonstrate that metal is coordinated by E149, E152 and E198. Additionally, using molecular dynamics simulations and NMR, we show that protonation of the general base influences the catalytic 190’s loop, which was previously shown to be involved in collective, interdomain motions in Dcp2 (Floor et al., 2012). This work describes the role of catalytic residues in Dcp2 for the first time, uncovers dynamics associated with the catalytic cycle, and suggests an alternative mechanism by which closure of Dcp2 could stimulate enzyme activity.
Several observations suggest E153 plays the role of general base in Dcp2. First, wild-type and the E153Q mutant bind metal in the same manner, yet decapping complexes harboring substitutions at E153 are still somehow severely catalytically compromised both in vitro and in vivo (Dunckley and Parker, 1999; She et al., 2006). Second, the E153Q mutation abolishes the ascending limb of the log(kmax) versus pH profile. Third, NMR pH titrations detect a residue with a near neutral pKa that is abolished by the E153Q mutation. Furthermore, the change in major product from m7GDP to m7GTP of the E153Q variant is consistent with a general base mutation, leaving the enzyme dependent on ambient hydroxide ions in the active site to serve as a nucleophile. Taken together, these data identify E153 as the general base in decapping, and explain why E153 has been shown to be critical for decapping both in vitro and in vivo (Dunckley and Parker, 1999; She et al., 2006).
The identification of K135 as a cationic residue responsible for stabilizing the leaving group is reminiscent of other Nudix enzymes that utilize a general acid to promote catalysis (Harris et al., 2000; Legler et al., 2002; Maksel et al., 2001; Mildvan et al., 2005). This offers an explanation for its previously reported effect on kmax (Deshmukh et al., 2008) and the measured pKa of 8.0 is similar to catalytic acids for other Nudix enzymes (Harris et al., 2000). In the S. pombe Dcp1/Dcp2 closed crystal structure, K129 (K135 in S. cerevisiae) is poised near the phosphates of the crystallization additive ATP, suggesting it may play a role in contacting one of the phosphates of cap (She et al., 2008), consistent with the multiple products formed in the K135A decapping reaction (Deshmukh et al., 2008).
The 190’s loop of Dcp2, and E198 in particular, exhibits a spectrum of conformations: in the crystal structure of S. pombe Dcp2, E192 (the S. cerevisiae E198 equivalent) is pointed towards solvent, whereas in the crystal structure of the ATP-bound S. pombe Dcp1:Dcp2 complex, this side chain is pointed towards the catalytic center (She et al., 2006; She et al., 2008). Additionally, in the open form of the S. pombe Dcp1:Dcp2 complex crystal structure, the 190’s loop is unresolved (She et al., 2008). Our results are consistent with the observed flexibility of the 190’s loop and specifically E198. Apparently, ILV methyl labeling can be used to detect protonation states of titratable residues in proteins in addition to reflecting slower exchange processes. This approach may be of broad utility for investigating pH dependent processes in enzymes, molecular machines and ion channels because methyl-TROSY NMR can be applied on assemblies as large as 0.5 MDa (Sprangers and Kay, 2007).
What is the functional role of flexibility in the 190’s loop? Loop conformational changes involving a catalytic residue are reminiscent of the mechanism of Nudix hydrolase ADP-ribose pyrophosphatase (ADPRP), which contains a loop that undergoes an open-to-closed transition of a full 10 Å over the course of the catalytic cycle, bringing the general base into position (Gabelli et al., 2002). Though our experiments show Dcp2 differs from ADPRP in the location of the general base, changing conformations of the 190’s loop in Dcp2 could aid in product release following catalysis. Alternatively, motions of E198 and the 190’s loop could contribute to metal coordination changes over the catalytic cycle, analogous to the metal hand-off role E98 plays in E. coli MutT (Harris et al., 2000).
It was proposed that the closed form of Dcp2 is the more active form of the enzyme because the regulatory domain and catalytic domain both bind cap using a composite active site, and that coactivators can affect this domain closure to stimulate decapping (Floor et al., 2010; She et al., 2008). The results reported here are consistent with this notion, but also suggest two possible mechanisms for how Dcp2 activity might be fine-tuned by coactivators affecting conformational dynamics. If inter-domain closure stimulates catalysis by Dcp2 via substrate positioning, then mutations in both the regulatory and catalytic domains should result in aberrant cleavage events; indeed, we observe multiple products upon mutation of catalytic residues, and deletion of the regulatory domain gives rise to multiple products (Piccirillo et al., 2003). There is a second possible route for catalytic rate enhancement: coupled inter-domain and catalytic loop motions of Dcp2. Regions adjacent to the catalytic helix are involved in collective motions in apo Dcp2 (Floor et al., 2012), and here we find that the conformation of the 190’s loop is influenced by the catalytically essential glutamates E153 and E198. While it is possible that that global conformational changes may be coupled to both changes in substrate positioning and local rearrangements of the 190’s loop, definitive proof requires future structural work with substrate RNA.
Experimental Procedures
Protein expression and purification
Wild type and mutant S. cerevisiae Nudix domain constructs were expressed with an N-terminal GB1 tag (Card and Gardner, 2005) followed by a hexahistidine affinity tag, a TEV protease site and the coding region of S. cerevisiae Dcp2 residues 100–245 in Escherichia coli BL21-Star cells (Invitrogen). Proteins for crystallography were purified from cells grown in LB media with an induction lasting 18 hours at 20°C. Cells were harvested at 5000g, sonicated, clarified at 25,000g, purified using Ni-NTA affinity chromatography, and cleaved overnight at room temperature with TEV protease. Purification proceeded by Ni-NTA backpass to remove the His-GB1 tag followed by gel filtration chromatography into NMR buffer: 21.1 mM NaH2PO4 and 28.8 mM Na2HPO4 (pH 7.0), 200 mM NaCl, 100 mM Na2SO4, and 5 mM DTT in H2O. Samples were used promptly following purification and were > 95% pure as judged by coomassie staining. Protein samples for NMR were obtained from cells expressed in H2O M9 minimal media supplemented with 10 mg biotin per liter, and forty minutes prior to induction by 1mM IPTG, 50 mg L−1 of 13C-methyl alpha-ketobutyric acid and 100 mg L−1 of 13C-dimethyl alpha-ketoisovaleric acid (Cambridge Isotopes) were added directly to the media. Purification proceeded as above. For kinetic assays, the wild-type and mutant S. cerevisiae Dcp1/Dcp2 decapping complex constructs GB1-t-Dcp1/Dcp2(1-245) were expressed in BL21(DE3)-Rosetta cells, purified by nickel affinity chromatography and treated with 10 mM EDTA for 1 hour, followed by gel filtration into Dcp2 storage buffer (50 mM HEPES, 100 mM NaCl, 20 % glycerol, 5 mM DTT pH 7.5), flash frozen and stored at −80°C.
X-ray Crystallography
After purification S. cerevisiae Dcp2 (100–245) was buffer exchanged into 10 mM HEPES pH 7.0, 1 mM DTT, 50 mM NaCl, 100 mM Na2SO4 and then mixed with an equal volume of crystallization conditions and grown at room temperature by the hanging drop vapor diffusion method. Wild-type protein was mixed with 0.1 M Mg Formate, 0.1 M sodium acetate pH 4.4, 20% PEG 3350; E198Q protein was mixed with 0.25 M Na Acetate, 0.1 M Na cacodylate pH 6.5, 20% PEG 3350; and E153Q was mixed with 0.1 Mg Formate, 0.1 M HEPES pH 7.4, 20% PEG 3350. The wild-type data was collected at room temperature at a wavelength of 0.95372 Å. The E198Q and E153Q crystals were transferred to the well solution containing 20% glycerol and then flash frozen before data collection at a wavelength of 1.11587 Å and 100 K. All data was collected at ALS (Advanced Light Source) 8.3.1, and indexed, integrated and scaled using HKL2000 (Otwinowski and Minor, 1997). Phasing was done via molecular replacement using the previously solved S. pombe structure with loops 189–202 and 213–219 removed (PDB 2A6T) (She et al., 2006) using Phaser in PHENIX, model building was performed in Coot (Adams et al., 2010; Emsley and Cowtan, 2004; McCoy et al., 2007) and the structures were refined in PHENIX (Adams et al., 2010) with the use of TLS.
Kinetic Assays
Decapping reactions were carried out at 4 °C on wild-type or mutant budding yeast Dcp1/2 complexes as previously described (Jones et al., 2008) under saturating single-turnover conditions for all constructs tested. Saturation was verified by doubling the enzyme concentration at pH values 5.5, 7.5, and 9.5. The final buffer concentration used was 50 mM NH4Cl, 0.01% NP-40, 1 mM DTT, 5 mM MgCl2 and either 50 mM Bis-tris methane (pH 5.5–6.5), 50 mM Tris-HCl (pH 7.0–8.0), or 50 mM Bis-Tris propane (pH 8.5–9.5). Stored protein (see protein purification) was diluted to 3X final concentration in 1X decapping reaction buffer, incubated at 4 °C for 15 minutes to allow for complete temperature and pH equilibration, and then the reaction was initiated by addition of RNA substrate in 1X reaction buffer. Kinetic data were processed and fit as previously described (Jones et al., 2008). The data for the K135A mutation were analyzed as fraction total product formed (see text, Fig. 3C–E). For pH values 7.5–9.5, K135A data were fit to a single exponential curve yielding kobs (Fig. S2A). For pH values lower than 7.5, the reaction rates were too slow to reliably obtain data that could be fit exponentially and instead initial rates were measured (Fig. S2B). The initial linear rate of the reaction (no more than 30% complete) was divided by an endpoint of 0.75, which was an average of all endpoints for K135A timecourses fit with a single exponential to obtain kobs. E153Q was also analyzed as fraction total product formed (see text, Fig. 3A–E), by single exponential fit for pH values 8.0–9.5 (Fig. S2C), and by initial rates for pH values lower than 8.0 (Fig. S2D). For wild-type and K135A decapping complexes, the kmax dependence on pH was fit using previously established methods (Harris et al., 2000) to obtain the general acid and general base pKa’s. Fitting theE153Q data in this manner was not successful, while a linear model of E153Q kmax pH dependence had better agreement with the experimental results.
NMR spectroscopy
NMR experiments were performed using the gradient-enhanced 13C-1H HSQC (Kay et al., 1992) on either a Varian Inova 600 MHz or Bruker Avance 800 MHz spectrometer, both outfitted with cryogenic four-channel probes. Assignments are BMRB entry 7325 (Deshmukh et al., 2008). Changes in pH were performed by direct addition of NaOH or HCl to the sample while monitoring pH with a micro-electrode. Titrations were performed with protein concentrations between 100 μM and 200 μM at 298K. A mild amount of precipitation was observed at pH extrema. Experiments over pH 9 or less than pH 5.5 that showed global chemical shift changes, indicative of unfolding, were discarded. Structure figures were generated with PyMOL (http://pymol.org) and NMR spectra figures were generated with Sparky (T.D. Goddard and D.G. Kneller, UCSF). Changes in chemical shift were calculated according to the total chemical shift change: NMR data were processed using NMRPipe (Delaglio et al., 1995) with apodization, linear prediction and low-frequency deconvolution to remove residual H2O. Peak shapes were fit using FuDA (D.F. Hansen; http://www.biochem.ucl.ac.uk/hansen/fuda) by a mixed Lorentzian and Gaussian curve.
Molecular dynamics
100 ns simulations of S. cerevisiae Dcp2 wild-type and with protonated E153 from PDB 2JVB, along with S. pombe Dcp2 wild-type, E153 protonated, and E153Q from PDB 2A6T were performed with the OPLS forcefield (Kaminski et al., 2001) and SPC water (H.J.C. Berendsen, 1981) as previously described, except with 0.5 M NaCl (Rapp et al., 2013). Additional details can be found in the supplemental info.
Supplementary Material
Highlights.
Glu153 serves as the general base in Dcp2 decapping reaction
Methyl group NMR can report on protonation states in proteins
Protonation state of general base E153 controls conformation of a catalytic loop
This conformational change may be coupled to protein interactions with coactivators
Acknowledgments
We thank Mark Kelly for NMR support; Pascal Wassam for computer support; Jamie Fraser for help with x-ray data collection and refinement, and D. Flemming Hansen for the FuDA software. The Advanced Light Source is supported by the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. This work was supported by US National Institutes of Health grant R01GM078360 to J.D.G., fellowships from the Sandler Family Foundation for Basic Sciences and the Achievement Awards for College Scientists Foundation to S.N.F, supercomputer time provided by the National Science Foundation Teragrid program at the Texas Advanced Supercomputing Center (Project TG-MCB090109 to M.P.J.), and funding from the UCSF integrated Program in Quantitative Biology fellowship and the UCSF Cancer Research Coordinating Committee for C.L.M. M.P.J. is a consultant to Schrödinger, L.L.C.
Footnotes
Acession Numbers
The coordinates and structure-factor amplitudes of the wild-type, E198Q and E153Q crystal structures of Dcp2 have been deposited in the Protein Data Bank under accession codes under 4K6E (wild-type), 4KG4 (E198Q), and 4KG3 (E153Q).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrani N, Ganesan R, Kervestin S, Mangus DA, Ghosh S, Jacobson A. A faux 3′-UTR promotes aberrant termination and triggers nonsense-mediated mRNA decay. Nature. 2004;432:112–118. doi: 10.1038/nature03060. [DOI] [PubMed] [Google Scholar]
- Arribas-Layton M, Wu D, Lykke-Andersen J, Song H. Structural and functional control of the eukaryotic mRNA decapping machinery. Biochim Biophys Acta. 2012;1829:580–589. doi: 10.1016/j.bbagrm.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beelman CA, Stevens A, Caponigro G, LaGrandeur TE, Hatfield L, Fortner DM, Parker R. An essential component of the decapping enzyme required for normal rates of mRNA turnover. Nature. 1996;382:642–646. doi: 10.1038/382642a0. [DOI] [PubMed] [Google Scholar]
- Behm-Ansmant I, Rehwinkel J, Doerks T, Stark A, Bork P, Izaurralde E. mRNA degradation by miRNAs and GW182 requires both CCR4:NOT deadenylase and DCP1:DCP2 decapping complexes. Genes Dev. 2006;20:1885–1898. doi: 10.1101/gad.1424106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brannan K, Kim H, Erickson B, Glover-Cutter K, Kim S, Fong N, Kiemele L, Hansen K, Davis R, Lykke-Andersen J, et al. mRNA decapping factors and the exonuclease Xrn2 function in widespread premature termination of RNA polymerase II transcription. Mol Cell. 2012;46:311–324. doi: 10.1016/j.molcel.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun JE, Truffault V, Boland A, Huntzinger E, Chang CT, Haas G, Weichenrieder O, Coles M, Izaurralde E. A direct interaction between DCP1 and XRN1 couples mRNA decapping to 5′ exonucleolytic degradation. Nat Struct Mol Biol. 2012;19:1324–1331. doi: 10.1038/nsmb.2413. [DOI] [PubMed] [Google Scholar]
- Card PB, Gardner KH. Identification and optimization of protein domains for NMR studies. Methods Enzymol. 2005;394:3–16. doi: 10.1016/S0076-6879(05)94001-9. [DOI] [PubMed] [Google Scholar]
- Chang JH, Jiao X, Chiba K, Oh C, Martin CE, Kiledjian M, Tong L. Dxo1 is a new type of eukaryotic enzyme with both decapping and 5′–3′ exoribonuclease activity. Nat Struct Mol Biol. 2012;19:1011–1017. doi: 10.1038/nsmb.2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Xu N, Shyu AB. mRNA decay mediated by two distinct AU-rich elements from c-fos and granulocyte-macrophage colony-stimulating factor transcripts: different deadenylation kinetics and uncoupling from translation. Mol Cell Biol. 1995;15:5777–5788. doi: 10.1128/mcb.15.10.5777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Zheng D, Xia Z, Shyu AB. Ago-TNRC6 triggers microRNA-mediated decay by promoting two deadenylation steps. Nat Struct Mol Biol. 2009;16:1160–1166. doi: 10.1038/nsmb.1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Deshmukh MV, Jones BN, Quang-Dang DU, Flinders J, Floor SN, Kim C, Jemielity J, Kalek M, Darzynkiewicz E, Gross JD. mRNA decapping is promoted by an RNA-binding channel in Dcp2. Mol Cell. 2008;29:324–336. doi: 10.1016/j.molcel.2007.11.027. [DOI] [PubMed] [Google Scholar]
- Dunckley T, Parker R. The DCP2 protein is required for mRNA decapping in Saccharomyces cerevisiae and contains a functional MutT motif. EMBO J. 1999;18:5411–5422. doi: 10.1093/emboj/18.19.5411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Eulalio A, Rehwinkel J, Stricker M, Huntzinger E, Yang SF, Doerks T, Dorner S, Bork P, Boutros M, Izaurralde E. Target-specific requirements for enhancers of decapping in miRNA-mediated gene silencing. Genes Dev. 2007;21:2558–2570. doi: 10.1101/gad.443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenger-Gron M, Fillman C, Norrild B, Lykke-Andersen J. Multiple processing body factors and the ARE binding protein TTP activate mRNA decapping. Mol Cell. 2005;20:905–915. doi: 10.1016/j.molcel.2005.10.031. [DOI] [PubMed] [Google Scholar]
- Floor SN, Borja MS, Gross JD. Interdomain dynamics and coactivation of the mRNA decapping enzyme Dcp2 are mediated by a gatekeeper tryptophan. Proc Natl Acad Sci U S A. 2012;109:2872–2877. doi: 10.1073/pnas.1113620109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floor SN, Jones BN, Hernandez GA, Gross JD. A split active site couples cap recognition by Dcp2 to activation. Nat Struct Mol Biol. 2010;17:1096–1101. doi: 10.1038/nsmb.1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromm SA, Truffault V, Kamenz J, Braun JE, Hoffmann NA, Izaurralde E, Sprangers R. The structural basis of Edc3- and Scd6-mediated activation of the Dcp1:Dcp2 mRNA decapping complex. EMBO J. 2012;31:279–290. doi: 10.1038/emboj.2011.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabelli SB, Bianchet MA, Ohnishi Y, Ichikawa Y, Bessman MJ, Amzel LM. Mechanism of the Escherichia coli ADP-ribose pyrophosphatase, a Nudix hydrolase. Biochemistry. 2002;41:9279–9285. doi: 10.1021/bi0259296. [DOI] [PubMed] [Google Scholar]
- Ghosh T, Peterson B, Tomasevic N, Peculis BA. Xenopus U8 snoRNA binding protein is a conserved nuclear decapping enzyme. Mol Cell. 2004;13:817–828. doi: 10.1016/s1097-2765(04)00127-3. [DOI] [PubMed] [Google Scholar]
- Berendsen HJC, van Gunsteren JPW, Hermans J. Intermolecular Forces. Reidel; Dordrecht: 1981. [Google Scholar]
- Harigaya Y, Jones BN, Muhlrad D, Gross JD, Parker R. Identification and analysis of the interaction between Edc3 and Dcp2 in Saccharomyces cerevisiae. Mol Cell Biol. 2010;30:1446–1456. doi: 10.1128/MCB.01305-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris TK, Wu G, Massiah MA, Mildvan AS. Mutational, kinetic, and NMR studies of the roles of conserved glutamate residues and of lysine-39 in the mechanism of the MutT pyrophosphohydrolase. Biochemistry. 2000;39:1655–1674. doi: 10.1021/bi9918745. [DOI] [PubMed] [Google Scholar]
- Heo I, Joo C, Kim YK, Ha M, Yoon MJ, Cho J, Yeom KH, Han J, Kim VN. TUT4 in concert with Lin28 suppresses microRNA biogenesis through pre-microRNA uridylation. Cell. 2009;138:696–708. doi: 10.1016/j.cell.2009.08.002. [DOI] [PubMed] [Google Scholar]
- Hilgers V, Teixeira D, Parker R. Translation-independent inhibition of mRNA deadenylation during stress in Saccharomyces cerevisiae. RNA. 2006;12:1835–1845. doi: 10.1261/rna.241006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Sweet TJ, Chamnongpol S, Baker KE, Coller J. Co-translational mRNA decay in Saccharomyces cerevisiae. Nature. 2009;461:225–229. doi: 10.1038/nature08265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isken O, Maquat LE. Quality control of eukaryotic mRNA: safeguarding cells from abnormal mRNA function. Genes Dev. 2007;21:1833–1856. doi: 10.1101/gad.1566807. [DOI] [PubMed] [Google Scholar]
- Jiao X, Xiang S, Oh C, Martin CE, Tong L, Kiledjian M. Identification of a quality-control mechanism for mRNA 5′-end capping. Nature. 2010;467:608–611. doi: 10.1038/nature09338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones BN, Quang-Dang DU, Oku Y, Gross JD. A kinetic assay to monitor RNA decapping under single- turnover conditions. Methods Enzymol. 2008;448:23–40. doi: 10.1016/S0076-6879(08)02602-5. [DOI] [PubMed] [Google Scholar]
- Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. Journal of Physical Chemistry B. 2001;105:6474–6487. [Google Scholar]
- Kay LE, Keifer P, Saarinen T. Pure Absorption Gradient Enhanced Heteronuclear Single Quantum Correlation Spectroscopy with Improved Sensitivity. Journal of the American Chemical Society. 1992;114:10663–10665. [Google Scholar]
- Lai T, Cho H, Liu Z, Bowler MW, Piao S, Parker R, Kim YK, Song H. Structural basis of the PNRC2-mediated link between mrna surveillance and decapping. Structure. 2012;20:2025–2037. doi: 10.1016/j.str.2012.09.009. [DOI] [PubMed] [Google Scholar]
- Legler PM, Massiah MA, Mildvan AS. Mutational, kinetic, and NMR studies of the mechanism of E. coli GDP-mannose mannosyl hydrolase, an unusual nudix enzyme. Biochemistry. 2002;41:10834–10848. doi: 10.1021/bi020362e. [DOI] [PubMed] [Google Scholar]
- Li Y, Dai J, Song M, Fitzgerald-Bocarsly P, Kiledjian M. Dcp2 decapping protein modulates mRNA stability of the critical interferon regulatory factor (IRF) IRF-7. Mol Cell Biol. 2012;32:1164–1172. doi: 10.1128/MCB.06328-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Rodgers ND, Jiao X, Kiledjian M. The scavenger mRNA decapping enzyme DcpS is a member of the HIT family of pyrophosphatases. EMBO J. 2002;21:4699–4708. doi: 10.1093/emboj/cdf448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykke-Andersen J. Identification of a human decapping complex associated with hUpf proteins in nonsense-mediated decay. Mol Cell Biol. 2002;22:8114–8121. doi: 10.1128/MCB.22.23.8114-8121.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maksel D, Gooley PR, Swarbrick JD, Guranowski A, Gange C, Blackburn GM, Gayler KR. Characterization of active-site residues in diadenosine tetraphosphate hydrolase from Lupinus angustifolius. Biochem J. 2001;357:399–405. doi: 10.1042/0264-6021:3570399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markley JL. Observation of Histidine Residues in Proteins by Means of Nuclear Magnetic Resonance Spectroscopy. Accounts of Chemical Research. 1975;8:70–80. [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mildvan AS, Xia Z, Azurmendi HF, Saraswat V, Legler PM, Massiah MA, Gabelli SB, Bianchet MA, Kang LW, Amzel LM. Structures and mechanisms of Nudix hydrolases. Arch Biochem Biophys. 2005;433:129–143. doi: 10.1016/j.abb.2004.08.017. [DOI] [PubMed] [Google Scholar]
- Oda Y, Yamazaki T, Nagayama K, Kanaya S, Kuroda Y, Nakamura H. Individual ionization constants of all the carboxyl groups in ribonuclease HI from Escherichia coli determined by NMR. Biochemistry. 1994;33:5275–5284. doi: 10.1021/bi00183a034. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallography, Pt A. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Piccirillo C, Khanna R, Kiledjian M. Functional characterization of the mammalian mRNA decapping enzyme hDcp2. RNA. 2003;9:1138–1147. doi: 10.1261/rna.5690503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapp C, Klerman H, Levine E, McClendon CL. Hydrogen bond strengths in phosphorylated and sulfated amino acid residues. PLoS One. 2013;8:e57804. doi: 10.1371/journal.pone.0057804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissland OS, Norbury CJ. Decapping is preceded by 3′ uridylation in a novel pathway of bulk mRNA turnover. Nat Struct Mol Biol. 2009;16:616–623. doi: 10.1038/nsmb.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schier AF. The maternal-zygotic transition: death and birth of RNAs. Science. 2007;316:406–407. doi: 10.1126/science.1140693. [DOI] [PubMed] [Google Scholar]
- She M, Decker CJ, Chen N, Tumati S, Parker R, Song H. Crystal structure and functional analysis of Dcp2p from Schizosaccharomyces pombe. Nat Struct Mol Biol. 2006;13:63–70. doi: 10.1038/nsmb1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She M, Decker CJ, Svergun DI, Round A, Chen N, Muhlrad D, Parker R, Song H. Structural basis of dcp2 recognition and activation by dcp1. Mol Cell. 2008;29:337–349. doi: 10.1016/j.molcel.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, Goodman HM. Uridine addition after microRNA-directed cleavage. Science. 2004;306:997. doi: 10.1126/science.1103521. [DOI] [PubMed] [Google Scholar]
- Song MG, Kiledjian M. 3′ Terminal oligo U-tract-mediated stimulation of decapping. RNA. 2007;13:2356–2365. doi: 10.1261/rna.765807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MG, Li Y, Kiledjian M. Multiple mRNA decapping enzymes in mammalian cells. Mol Cell. 2010;40:423–432. doi: 10.1016/j.molcel.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprangers R, Kay LE. Quantitative dynamics and binding studies of the 20S proteasome by NMR. Nature. 2007;445:618–622. doi: 10.1038/nature05512. [DOI] [PubMed] [Google Scholar]
- Stevens A, Maupin MK. A 5′----3′ exoribonuclease of Saccharomyces cerevisiae: size and novel substrate specificity. Arch Biochem Biophys. 1987;252:339–347. doi: 10.1016/0003-9861(87)90040-3. [DOI] [PubMed] [Google Scholar]
- Sweet T, Kovalak C, Coller J. The DEAD-box protein Dhh1 promotes decapping by slowing ribosome movement. PLoS Biol. 2012;10:e1001342. doi: 10.1371/journal.pbio.1001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waley SG. The pH-dependence and group modification of beta-lactamase I. Biochem J. 1975;149:547–551. doi: 10.1042/bj1490547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Jiao X, Carr-Schmid A, Kiledjian M. The hDcp2 protein is a mammalian mRNA decapping enzyme. Proc Natl Acad Sci U S A. 2002;99:12663–12668. doi: 10.1073/pnas.192445599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Kiledjian M. Functional link between the mammalian exosome and mRNA decapping. Cell. 2001;107:751–762. doi: 10.1016/s0092-8674(01)00592-x. [DOI] [PubMed] [Google Scholar]
- Zhang M, Vogel HJ. Determination of the side chain pKa values of the lysine residues in calmodulin. J Biol Chem. 1993;268:22420–22428. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.