

Abstract
Activating mutations in PTPN11 (encoding SHP2), a protein tyrosine phosphatase (PTP) that plays an overall positive role in growth factor and cytokine signaling, are directly associated with the pathogenesis of Noonan syndrome and childhood leukemias. Identification of SHP2 selective inhibitors could lead to the development of new drugs that ultimately serve as treatments for PTPN11-associated diseases. As the catalytic core of SHP2 shares extremely high homology to those of SHP1 and other PTPs that play negative roles in cell signaling, to identify selective inhibitors of SHP2 using computer-aided drug design, we targeted a protein surface pocket that is adjacent to the catalytic site, is predicted to be important for binding to phosphopeptide substrates, and has structural features unique to SHP2. From computationally selected candidate compounds, #220–324 effectively inhibited SHP2 activity with an IC50 of 14 μM. Fluorescence titration experiments confirmed its direct binding to SHP2. This active compound was further verified for its ability to inhibit SHP2-mediated cell signaling and cellular function with minimal off-target effects. Furthermore, mouse myeloid progenitors with the activating mutation (E76K) in PTPN11 and patient leukemic cells with the same mutation were more sensitive to this inhibitor than wild-type cells. This study provides evidence that SHP2 is a “druggable” target for the treatment of PTPN11-associated diseases. Since the small molecule SHP2 inhibitor identified has a simple chemical structure, it represents an ideal lead compound for the development of novel anti-SHP2 drugs.
Keywords: PTPN11 (SHP2), Phosphatase, Inhibitor, Drug development, Leukemia
Introduction
SHP2 (encoded by PTPN11), a ubiquitously expressed SH2 domain-containing protein tyrosine phosphatase (PTP), is involved in multiple cell signaling processes, such as the Ras-Erk, PI3K-Akt, Jak-Stat, NF-κB, and mTOR pathways (1–3). It contains two tandem SH2 domains at the N-terminus and a PTP domain at the C-terminus, with flexible polypeptide linker regions connecting the three domains. The SH2 domains, in particular, the N-terminal SH2 (N-SH2) domain, mediate the binding of SHP2 to other signaling proteins via phosphorylated tyrosine residues in a sequence-specific fashion (4, 5). This directs SHP2 to the appropriate subcellular location and helps determine the specificity of substrate-enzyme interactions. The X-ray crystal structure of SHP2 reveals the formation of an intramolecular interface between the N-SH2 domain and the PTP domain (6–8). This self-interaction blocks substrate access to the catalytic site, and polypeptide ligands with phospho-tyrosine (pY) residues activate SHP2 by binding the SH2 domains (primarily the N-SH2 domain), thereby disrupting the self-inhibitory interaction between N-SH2 and PTP domains and exposing the phosphatase catalytic site (6–8). Intriguingly, despite its direct function in protein dephosphorylation, SHP2 plays an overall positive role in transducing signals initiated from growth factors/cytokines and extracellular matrix proteins (1–3). However, the signaling mechanisms of SHP2 are still not completely understood. In particular, the molecular basis for the positive role of its catalytic activity in the Ras-Erk pathway as well as other pathways remains elusive.
Germline or somatic mutations in PTPN11 (encoding SHP2) that cause hyperactivation of SHP2 catalytic activity have been identified in the developmental disorder Noonan syndrome and various childhood leukemias. Specifically, heterozygous PTPN11 mutations have been found in 50% of patients with Noonan syndrome, as well as in 35% of juvenile myelomonocytic leukemia (JMML), 10% of myelodysplastic syndrome, 7% of B cell acute lymphoblastic leukemia/lymphoma, and 4% of acute myeloid leukemia cases (9–13). Additionally, activating mutations of PTPN11 have been identified in sporadic solid tumors (14). Such mutations disrupt the inhibitory intramolecular interaction of SHP2, leading to gain-of-function by allowing constitutive access to the phosphatase catalytic site on the enzyme (12, 15). PTPN11 mutations and other JMML-associated Ras, c-CBL, or Neurofibromatosis1 mutations are mutually exclusive (9–13, 16, 17). Remarkably, recent studies have demonstrated that single PTPN11 gain-of-function mutations are sufficient to induce Noonan syndrome, JMML-like myeloproliferative disease, and acute leukemias in mice (18–23), suggesting that the PTPN11 mutations play a causal role in the development of these diseases.
The direct connection between hyperactivation of SHP2 and human diseases points to SHP2 as a potential target for mechanism-based therapeutics. Selective and potent SHP2 inhibitors are needed. Discovery of SHP2-specific inhibitors would not only facilitate research on SHP2 signaling in model systems but could also lead to the development of new drugs for PTPN11-associated diseases. However, little progress has been made in this field because the discovery of SHP2-specific inhibitors is complicated by the extremely high homology of the catalytic core of SHP2 with those of SHP1 and other PTPs that play negative roles in growth factor and cytokine signaling. To overcome this we previously applied computer-aided drug design (CADD) to screen a virtual database of compounds targeting a protein surface pocket that is adjacent to the catalytic site of SHP2, is predicted to be important for binding to phosphopeptide substrates, and has structural features unique to SHP2. This effort led to the identification of several low-molecular weight compounds capable of inhibiting SHP2 (24). In the present study we expand that effort by using CADD similarity searching to identify analogs of the previously discovered active compounds that may have improved activity, leading to the identification of a new compound (#220–324) that is superior to the previously reported parent inhibitors in terms of selectivity and potency. This new compound selectively inhibited SHP2 catalytic activity without significant off-target effects. Moreover, it was efficacious in blocking SHP2-mediated cellular functions, even in PTPN11 mutated mouse and human leukemia cells, providing evidence that SHP2 is a “druggable” target for the treatment of PTPN11-associated diseases.
Materials and Methods
Reagents and Chemicals
RPMI 1640, IMDM, DMEM, and the penicillin-streptomycin stock solution were purchased from Thermo Scientific/HyClone Laboratories (Logan, UT). Fetal bovine serum (FBS) was obtained from Invitrogen (Grand Island, NY). The CellTiter 96® AQueous One Solution Cell Proliferation Assay kit was purchased from Promega (Madison, WI). Antibodies against p-Erk (E-4), Erk (C-16), Jak2 (C-20), and SHP2 (C-18) were purchased from Santa Cruz Biotechnology, Inc (Santa Cruz, CA). Antibodies against p-Akt (Ser473), Akt, and p-Stat5 (Y694) were obtained from Cell Signaling Technology (Beverly, MA). Antibodies against p-Jak2 (Tyr1007/1008) and phospho-tyrosine were purchased from Millipore Corporation (Temecula, CA). Antibody against Stat5 was purchased from BD Biosciences (San Jose, CA). Goat-anti-rabbit and anti-mouse IgG horseradish peroxidase (HRP) conjugates were purchased from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA). DMSO and other chemicals used in buffer solutions were purchased from Fisher Scientific (Pittsburgh, PA). #220–324 was obtained from Specs (Wakefield, RI) and dissolved in DMSO to prepare stock solutions (20 mM) for subsequent experiments.
CADD Similarity Searching
Identification of compounds similar to the published lead compounds used the program MOE (Chemical Computing Group, Inc., Montreal, Canada). An in house database of 1.3 million commercially-available compounds was screened using the BIT MACCS structural fingerprints, with the extent of similarity quantified using the Tanimoto coefficient (25). For each active compound the top 50 scoring compounds were considered for experimental evaluation.
Computational Modeling of the Binding Pose of #220–324
#220–324 was built into the previously-predicted binding site (24) using coordinates of analogous atoms from the –previously-docked pose of the parent compound #220 (24) and maintaining SHP2 coordinates as per this previous docked complex. The protein-ligand system was represented using the CHARMM36 protein force field (26) and the CHARMM General Force Field (CGenFF) (27) for the ligand in combination with the GBMV Generalized Born implicit solvent model (28, 29), with automatic assignment of atom types and missing parameters for #220–324 generated via the CGenFF engine (30, 31) as implemented in the ParamChem web portal. Molecular dynamics (MD) simulations were done with a 1.5 fs integration timestep, the SHAKE algorithm (32) to constrain bonds to hydrogen atoms to their equilibrium values, an 18-Å spherical cutoff for non-bonded interactions, with a switching function (33) applied in the 16–18 Å interval, and Langevin thermostatting (34) at 298 K. The CHARMM software (35) was used for all system construction and simulation. Images were generated using VMD (36).
In Vitro Phosphatase Activity Assay
Glutathione S-transferase (GST) fusion proteins of SHP2 purified in-house were used as the enzyme and a phosphopeptide corresponding to the surrounding sequence of pTyr1146 in the insulin receptor (Thr-Arg-Asp-Ile-Tyr[PO3H2]-Glu-Thr-Asp-Tyr-Tyr) was used as the substrate. The assay determines free phosphate generated by dephosphorylation of the substrate using the Malachite Green reagent (Sigma, St. Louis, MO). Briefly, 0.5 μg of GST-SHP2 PTP was incubated in 40 μL assay buffer (25 mM Tris-HCl, pH 7.4, 50 mM NaCl, 5 mM DTT, and 2.5 mM EDTA) with test compounds at various concentrations at room temperature for 30 min. The substrate was then added to a final concentration of 0.2 mM. The system was incubated at 30°C for 30 min. Finally, 50 μL of Malachite Green solution was added and OD620 was measured after 10 min. The protocols for the phosphatase assays for SHP1, CD45, LAR, MEG2, and TC-PTP were similar, with the exception that GST-SHP1, GST-CD45 cytoplasmic domain, GST-LAR, GST-MEG2, and GST-TC-PTP enzymes purchased from Biomol International, L.P. (Plymouth Meeting, PA), were used in the respective assays. For IC50 determinations, 5 concentrations of #220–324 were tested. Each experiment was performed in triplicate.
Fluorescence Titrations
For all experiments, purified SHP2 PTP domain GST-fusion protein was diluted into 20 mM Tris-HCl, pH 7.5. Fluorescence spectra were recorded with a Luminescence Spectrometer LS50 (Perkin-Elmer, Boston, MA). Titrations were performed by increasing the test compound concentrations while maintaining the SHP2 protein concentration at 3 μM. Contributions from background fluorescence of the inhibitor were accounted for by subtracting the fluorescence of the inhibitor alone from the protein-inhibitor solution. The excitation wavelength was 295 nm and fluorescence was monitored from 360 to 500 nm. All reported fluorescence intensities were relative values and were not corrected for wavelength variations in detector response.
Cell Proliferation Assay
Ba/F3 cells and mouse embryonic fibroblasts (MEFs) were seeded into 96-well plates at a density of 1×104 cells/well (Ba/F3) and 5×103 cells/well (MEFs) in RPMI-1640 containing 10% FBS plus recombinant mouse IL-3 (1.0 ng/mL) and DMEM containing 10% FBS, respectively. Cells were grown overnight and then treated with either test compounds or the same concentrations of DMSO. Three days later, the number of viable cells was determined using a CellTiter 96® AQueous One Solution Cell Proliferation Assay kit.
Western Blotting
Ba/F3 cells were starved in serum and cytokine-free RPMI1640 overnight. Cells were then treated with #220–324 for 3 hrs prior to IL-3 stimulation. Stimulated cells were harvested and lysed on ice with RIPA buffer containing 50 mM Tris-HCl, pH 7.4; 1% NP-40; 0.25% Na-deoxycholate; 150 mM NaCl; 1 mM EDTA; 1 mM NaF; 1 mM Na3VO4; 1 mM PMSF and protease inhibitor cocktail (Roche, Indianapolis, IN). Equivalent amounts of protein (50 μg) were resolved on 10% SDS-PAGE and transferred to nitrocellulose membranes (Millipore, Bedford, MA). Membranes were blocked with 2% BSA in TBS-T [20 mM Tris-HCl (pH 7.4), 150 mM NaCl, and 0.1% Tween 20] for 1 hr at room temperature and probed with primary antibodies overnight at 4°C. Blots were washed with TBS-T and exposed to HRP conjugated goat-anti-mouse or goat-anti-rabbit secondary antibodies for 1 hr at room temperature. Immunoreactive bands were detected by using ECL Plus Reagents (GE Healthcare, Piscataway, NJ).
Colony-Forming Unit Assay
Freshly harvested mouse bone marrow cells or patient splenocytes (2×104 cells/mL) were assayed for colony forming units (CFUs) in 0.9% methylcellulose IMDM containing 30% FBS, glutamine (10−4 M), β-mercaptoethanol (3.3×10−5 M), GM-CSF (1.0 ng/mL), and varying doses of #220–324 or the same concentrations of DMSO. After 7 days (mouse bone marrow cells) or 14 days (patient cells) of culture at 37°C in a humidified 5% CO2 incubator, myeloid colonies (CFU-GM and CFU-M) were counted under an inverted microscope.
Results
Identification of #220–324 as a novel selective SHP2 inhibitor
As reported in our previous study, by targeting a putative phosphopeptide binding site adjacent to the catalytic site that has structural features unique to SHP2, nine active compounds were identified by CADD chemical database screening combined with experimental assays (24). To identify more potent and selective SHP2 inhibitors, in the present work we extend those efforts by performing a similarity search of a chemical database of 1.3 million compounds based on the structural characteristics of the 9 active compounds following the procedures previously described (37). From the top scoring compounds sixty-two candidate compounds were obtained and subjected to biological screening. Twenty of these compounds inhibited SHP2 catalytic activity with various efficiencies in biochemical assays (Supplementary Fig. 1A and 1B). Among these inhibitors, #220–324 was found to be the most potent and most selective in subsequent cell-based screenings that test the effects of the compounds on IL-3-induced cell proliferation (data not shown). This compound inhibited SHP2 catalytic activity with an IC50 of 13.88 μM (Fig. 1A). To determine functional specificities of #220–324, its inhibitory activity was assessed against several mammalian PTPs, in particular those phosphatases that are highly expressed in hematopoietic cells. As shown in Fig. 1B, #220–324 was approximately 3-fold more selective for SHP2 compared to SHP1, and 13-, 10-, 13-, 3-fold more selective for SHP2 compared to CD45, LAR, MEG2, and TC-PTP, respectively.
Figure 1. #220–324 selectively inhibited the phosphatase activity of SHP2.

(A) Chemical structure of #220–324. (B) Phosphatase assays were carried out using the indicated phosphatases as enzymes and the phospho-insulin receptor peptide as a substrate in the presence of various concentrations of #220–324, as described in Materials and Methods. Experiments were performed three times with similar results obtained. Representative results from one experiment are shown. (C) PTP activities of the SHP2 PTP domain, WT full length SHP2, and full length SHP2 E76K were determined in the absence or presence of #220–324 at the indicated concentrations using the in vitro phosphatase assay. Experiments were repeated three times. Similar results were obtained in each. Data shown are mean±S.E.M. from one experiment. (D) Inhibitory effects of #220–324 (25 μM) on the enzymatic activities of the SHP2 PTP domain, WT SHP2, and SHP2 E76K were determined using the phospho-insulin receptor peptide or pNPP as the substrates. Experiments were repeated three times. Similar results were obtained in each. Data shown are mean±S.E.M. from one experiment. (E) Fluorescence titration of SHP2 was performed by increasing the concentrations of #220–324 while maintaining the SHP2 protein concentration. The fluorescence is plotted against the log concentration in mol/L (Log [M]) for the test compound. Experiments were repeated twice. Similar results were obtained in each. Data shown are mean±S.E.M. from one experiment.
Molecular dynamics (MD) simulations were performed on the SHP2:#220–324 complex to test the plausibility of a non-catalytic site binding pose for this inhibitor. Using the previously-docked coordinates of the parent compound #220 as the basis for a starting conformation, the SHP2:#220–324 complex was first minimized for 1000 steps and then allowed to evolve unrestrained for 70,000 steps (105 ps) of generalized Born MD at 298 K. Like the parent compound in its docked pose (24), after the MD refinement #220–324 makes extensive hydrophobic contact with SHP2 (Supplementary Fig. 2). Additionally, the predicted binding site of SHP2 undergoes conformational changes to accommodate some relaxation of this compound relative to its initial conformation, such that after MD refinement, the new SHP2 conformation results in unfavorable steric clashes with the original docked pose of the parent compound #220. These results suggest a conformationally-flexible binding pocket adjacent to and distinct from the catalytic site that is capable of binding drug-like small molecules with substantial hydrophobic moieties, like #220–324. The results also provide a potential explanation for the increased activity of #220–324 over the parent compound #220.
#220–324 preferentially binds to the activated form of SHP2
In the basal state, SHP2 is self-inhibited by hydrogen bonding of the N-SH2 domain loop to the deep pocket of the PTP domain. Tumor-associated mutations of PTPN11 result in amino acid changes at the interface formed by the N-SH2 and PTP domains, disrupting the inhibitory intramolecular interaction, leading to hyperactivation of SHP2 catalytic activity (12, 15). The E76K mutation, localized in the N-SH2 domain is the most common and most active mutation found in leukemias (38, 39). To further characterize the mechanisms of action of #220–324, we purified GST fusion proteins of the SHP2 PTP domain, wild-type (WT) full-length SHP2 and the full-length SHP2 E76K mutant, and compared the sensitivities of these SHP2 recombinant proteins to #220–324. #220–324 inhibited the SHP2 E76K mutant in a dose-dependent manner, with an IC50 of 23.81 μM that is 1.5-fold higher than that for the SHP2 PTP domain (13.88 μM). However, the IC50 for SHP2 E76K is 2.7 fold lower than that for full-length WT SHP2 (64.74 μM) (Fig. 1C). The observation that full-length SHP2 is less sensitive to #220–324 than the SHP2 PTP domain and that the SHP2 E76K mutant is more sensitive than WT SHP2 to this inhibitor indicates that the N-SH2-PTP domain interaction interferes with #220–324 binding to SHP2 and that this compound preferentially inhibits the activated form of SHP2. The activity of #220–324 was further characterized by testing its effect on SHP2 catalytic activity using para-Nitrophenylphosphate (pNPP), a general small compound substrate for all PTPs. As demonstrated in Fig. 1D, #220–324 did not inhibit the SHP2 dephosphorylation of this small substrate regardless of which form of the SHP2 protein was used as the enzyme. This result suggests that #220–324 inhibits SHP2 dephosphorylation of the phosphopeptide substrate by disrupting binding of the phosphopeptide to SHP2 at a location other than the catalytic core.
To validate that #220–324 was binding directly to the SHP2 protein, we determined whether binding of the inhibitor altered the fluorescence of the SHP2 PTP domain using the fluorescence quenching assay taking advantage of the four tryptophans in the PTP domain. Of these residues, Trp248 is located 15 Å from the drug docking pocket and Trp423 located 8 Å from the phosphatase active site. As shown in Fig. 1E, #220–324 exhibited strong quenching of SHP2 fluorescence in a dose-dependent manner. These fluorescence quenching experiments indicate that #220–324 binds directly to SHP2, thereby attenuating its activity toward the phosphopeptide substrate.
Effects of #220–324 on SHP2-mediated cellular function
Given the potency and selectivity of #220–324 toward SHP2 in vitro, we proceeded to evaluate its ability to inhibit SHP2-mediated signaling and cellular function. PTPN11-associated leukemia is characterized by hypersensitivity of myeloid progenitors to GM-CSF and IL-3 (18, 20–22). Our previous studies have demonstrated that SHP2, in contrast to other PTPs, especially SHP1, promotes IL-3-triggered cell proliferation and activation of Erk, Akt, and Jak2-Stat5 pathways (40). To verify the selectivity of #220–324 in cells, it was critical to assess the cellular activities of this compound in GM-CSF or IL-3-induced cellular responses. IL-3-dependent Ba/F3 cells were therefore used. As shown in Fig. 2A, treatment of Ba/F3 cells with the compound for 72 hrs greatly decreased cell growth in a concentration-dependent manner, with an IC50 of 37.26 μM, consistent with the overall positive role of SHP2 catalytic activity in cellular response to IL-3 (40). Furthermore, as the SHP2 signaling is known to play a positive role in the cell cycle progression (41), we performed cell cycle analysis in #220–324 treated Ba/F3 cells. We found that this compound inhibited cell cycle progression, specifically the G1-S transition, in a dose-dependent manner (Fig. 2B). To evaluate whether #220–324 inhibited SHP2-dependent cell signaling, we examined the effects of this inhibitor on IL-3-induced Erk, Akt, Jak2, and Stat5 activation. Ba/F3 cells were serum-starved overnight followed by pretreatment with #220–324 or DMSO for 3 hrs, and then stimulated with IL-3. As shown in Fig. 2C, IL-3 induced activation of Erk, Akt, Jak2, and Stat5 determined by their phosphorylation levels was greatly suppressed by #220–324 at all time points. Furthermore, Ba/F3 cells were preincubated with #220–324 at various concentrations, followed by IL-3 stimulation for 10 min. We observed that #220–324 inhibited IL-3 induced Erk, Akt, and Stat5 activation in a dose-dependent manner (Supplementary Fig. 3A), in agreement with previous findings that SHP2 catalytic activity is required for optimal activation of these signaling pathways elicited by IL-3 (40). In addition, we found that tyrosyl phosphorylation of Gab2 (carried out by Jak2 kinase), an important intermediary of the PI3K-Akt pathway (42, 43), and the interaction between SHP2 and phosphorylated Gab2 was markedly decreased by #220–324 (Supplementary Fig. 3B). Taken together, these results imply that IL-3 signaling, including Ras-Erk, PI3K-Akt, and Jak2-Stat5 pathways, was down-regulated through a mechanism involving the inhibition of SHP2 activity, and point to the possibility that #220–324 mainly inhibits the catalytic activity of SHP2, not those of other PTPs.
Figure 2. #220–324 inhibited SHP2-mediated cell signaling and cellular proliferation.

(A) Ba/F3 cells were cultured in IL-3 (1.0 ng/mL) containing medium supplemented with #220–324 at the indicated concentrations or control DMSO. Cell numbers were determined 72 hrs later using the One Solution Cell Proliferation Assay kit. Experiments were performed three times with similar results obtained. Data shown are mean±S.E.M. from one experiment. (B) The cell cycle of Ba/F3 cells was analyzed 24 hrs after the treatment of #220–324 at the indicated concentrations. Percentages of the cells at G1, S, and G2/M phases were quantified by flow cytometric analyses. Experiments were repeated three times. Results shown are mean±S.E.M. from one experiment. (C) Ba/F3 cells were deprived of IL-3 overnight. Cells were treated with #220–324 (30 μM) for 3 hrs and then stimulated with IL-3 (2.0 ng/mL) for the indicated times. Whole cell lysates were prepared. The levels of p-Erk, p-Akt, p-Stat5, p-Jak2 were determined by immunoblotting analysis. Blots were striped and reprobed with anti-Erk, anti-Akt, and anti-Jak2, and anti-Stat5 antibodies to check for protein loading. Experiments were repeated three times. Representative results from one experiment are shown.
#220–324 has minimal off-target effects
To validate that the effects of #220–324 on cell growth were mediated through inhibiting SHP2, we tested #220–324 in PTPN11 knockout cells. PTPN11 conditional knockout (PTPN11Fl/Fl/Ade-Cre+) and WT control (PTPN11+/+/Ade-Cre+) mouse embryonic fibroblasts (MEFs) were treated with #220–324 at various concentrations or control DMSO for 72 hrs, and cell growth rates were determined. As shown in Fig. 3A, proliferation of WT cells was significantly decreased by the SHP2 inhibitor, consistent with the positive role of SHP2 in growth factor signaling (1–3). However, SHP2-depleted MEFs showed only minimal inhibitory effects. We also treated knockout and WT control cells with #220–324 at 10 μM or DMSO for various times and obtained similar results (Fig. 3B). The observation that PTPN11 deficient cells were insensitive to #220–324 strongly suggests that this compound is an agent capable of inhibiting SHP2 in cells without significant off-target effects.
Figure 3. #220–324 had minimal effects in PTPN11 knockout cells.

PTPN11 knockout (PTPN11Fl/Fl/Ad-Cre+) and WT (PTPN11+/+/Ad-Cre+) MEFs were cultured in DMEM with 10% FBS and #220–324 at the indicated concentrations for 72 hrs (A) or at 20 μM for the indicated periods of time (B). DMSO-treated cells were included as negative controls. Cell numbers were determined using the One Solution Cell Proliferation Assay kit. Experiments were performed twice with similar results. Data shown are mean±S.E.M. from one experiment.
Mutant cells harboring hyperactivated SHP2 are more sensitive to #220–324
We next assessed effects of #220–324 on mutant cells with leukemia-associated PTPN11E76K mutation. As illustrated in Fig. 4A, treatment of both WT and PTPN11E76K/+ MEFs with the compound for 72 hrs decreased cell growth in a dose-dependent manner. Intriguingly, PTPN11E76K/+ mutant cells were more sensitive to SHP2 inhibition. We also compared the effects of #220–324 in WT and PTPN11E76K/+ myeloid progenitors using CFU assays. In the presence of GM-CSF, the colony-forming capabilities of PTPN11E76K/+ myeloid progenitors were more sensitive to #220–324 compared to those of WT counterparts (Fig. 4B). Additionally, we found that the cytokine-independent colony formation of PTPN11E76K mutant myeloid progenitors, a characteristic of PTPN11-associated JMML and other myeloid malignancies, was significantly suppressed by this inhibitor (Fig. 4C). Additionally, we tested for growth inhibitory function of #220–324 in human lung cancer cell lines. Both H661 and H596 cell lines are WT in K-Ras and epidermal growth factor receptor. However, H661 cells carry an activating mutation (N58S) in PTPN11 while H596 cells have WT PTPN11. As demonstrated in Fig. 5A and 5B, #220–324 showed a marked inhibitory effect in H661 cells, but it was less effective in H596 cells. Together, these results further support that #220–324 is capable of decreasing proliferative advantages associated with gain-of-functions in SHP2 and that PTPN11 mutated cells are more sensitive to SHP2 inhibition.
Figure 4. Mutant MEFs and myeloid progenitors with the PTPN11E76K/+ mutation were more sensitive to #220–324 than WT cells.

(A) WT and PTPN11E76K/+ MEFs were cultured in 10% FBS-containing DMEM supplemented with #220–324 at the indicated concentrations. DMSO-treated cells were included as negative controls. Cell numbers were determined using the One Solution Cell Proliferation Assay kit 72 hrs later. Experiments were repeated three times. Similar results were obtained in each. Data shown are mean±S.E.M. from one experiment. (B) Bone marrow cells (2×104 cells/mL) harvested from PTPN11E76K/+/Mx1-Cre+ and PTPN11+/+/Mx1-Cre+ mice were plated in methylcellulose medium containing GM-CSF (1.0 ng/mL) and #220–324 at the indicated concentrations or control DMSO. Colonies were enumerated 7 days later and normalized against the number of colonies derived from WT control cells without #220–324 treatment. Representative results from two independent experiments are shown. Data are presented as mean±S.E.M. (C) Bone marrow cells (5×104 cells/mL) harvested from PTPN11E76K/+/Mx1-Cre+ mice were plated in methylcellulose medium (without GM-CSF) supplemented with #220–324 at the indicated concentrations or control DMSO. Colonies derived in the cytokine-free medium were enumerated 7 days later and normalized against the number of colonies derived from the cells without #220–324 treatment. Representative results from two independent experiments are shown. Data are presented as mean±S.E.M.
Figure 5. Lung cancer cells with the PTPN11N58S/+ mutation were more sensitive to #220–324 than those without the PTPN11N58S/+ mutation.
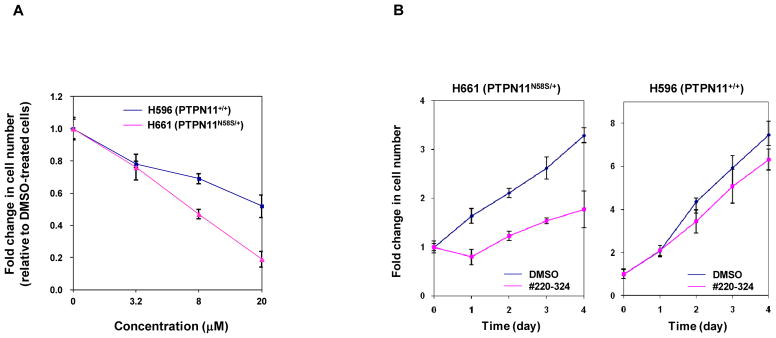
Human lung cancer cell lines H596 and H661 were cultured in DMEM supplemented with 10% FBS and #220–324 at the indicated concentrations for 72 hrs (A) or the cells were treated with #220–324 at 20 μM for the indicate times (B). DMSO-treated cells were included as negative controls. Cell numbers were determined using the One Solution Cell Proliferation Assay kit. Experiments were repeated three times. Similar results were obtained in each. Data are presented as mean±S.E.M. from one representative experiment.
#220–324 corrects the hypersensitive growth pattern of JMML myeloid progenitors with the PTPN11E76K mutation
Bone marrow cells from patients with JMML display a characteristic hypersensitive growth pattern, resulting in greater production of granulocyte-macrophage colonies in response to GM-CSF (44, 45). As shown in Fig. 6A, in the presence of GM-CSF, colony formation of myeloid blast cells from JMML patients with the PTPN11E76K/+ mutation was sensitive to #220–324 (IC50 = 7.24 μM). Consistent with this data, the total cell number of JMML cells cultured in GM-CSF-containing liquid medium was greatly decreased by the treatment of this inhibitor, with an IC50 = 3.09 μM (Fig. 6B). By contrast, we did not observe significant inhibition of healthy donor myeloid progenitors by #220–324 under 100 μM (Fig. 6C). The finding that #220–324 is able to selectively overcome the dominant effects of oncogenic SHP2 E76K in primary human leukemia cells provides strong support for the notion that SHP2 is a useful therapeutic target for the treatment of PTPN11 mutation-associated JMML.
Figure 6. #220–324 abrogated the growth of JMML myeloid progenitors with PTPN11E76K/+ mutation.

(A) Splenocytes (2×104 cells/mL) from a JMML patient with the PTPN11E76K/+ mutation were plated in methylcellulose medium containing GM-CSF (1.0 ng/mL) and #220–324 at the indicated concentrations or control DMSO. Colonies were enumerated 14 days later and normalized against the number of colonies derived from the cells without #220–324 treatment. Three patient samples were tested in three independent experiments. Similar results were obtained in each. Data are presented as mean±S.E.M. from one patient sample. (B) Splenocytes from a JMML patient with the PTPN11E76K/+ mutation were cultured in RPMI 1640 medium containing GM-CSF (1.0 ng/mL) and #220–324 at the indicated concentrations or control DMSO. Total cell numbers were determined and normalized against the number of the cells in the untreated group. Three patient samples were tested in three independent experiments. Similar results were obtained in each. Data are presented as mean±S.E.M. from one patient sample. (C) Apheresis peripheral blood cells (2×104 cells/mL) from normal donors were plated in methylcellulose medium containing GM-CSF (1.0 ng/mL) and #220–324 at the indicated concentrations. Colonies were enumerated 14 days later and normalized against the number of colonies derived from the cells treated with control DMSO. Three samples were tested in three independent experiments. Similar results were obtained in each. Data are presented as mean±S.E.M. from one sample.
Discussion
Identification of a lead inhibitor of SHP2 would greatly facilitate the development of anti-SHP2 therapeutic drugs. Mutations in PTPN11 (encoding SHP2) that cause enhanced catalytic activity of SHP2 have been found in the patients with Noonan syndrome, childhood leukemias, and solid tumors (9, 12–14), and more importantly, these mutations appear to play a causative role in the development of JMML and acute leukemias (18–23). Selective and potent SHP2 inhibitors have the potential to be further developed into therapeutic drugs for these diseases. Previous studies have reported several inhibitors of SHP2 (46–49). However, none of those compounds have been well characterized, for example, their potential off-target effects and their functions in suppressing GM-CSF/IL-3 –induced signaling and cellular responses (that are known to be associated with the pathogenesis of PTPN11-associated JMML) have not been determined. More importantly, it is not clear whether those compounds have any potential therapeutic benefits in PTPN11 mutated human or mouse leukemia cells. In this study, we have now identified and extensively characterized a novel small molecule SHP2 inhibitor.
Targeting the phospho-peptide binding site in SHP2 appears to be a feasible approach to developing SHP2 selective inhibitors. Despite a great need, little progress has been made in the development of SHP2 inhibitors. One of the challenges is the specificity of potential inhibitors. The active site of SHP2, which catalyzes de-phosphorylation of phospho-tyrosine (pY) in pY-containing peptides, is almost identical to that of the SHP1 phosphatase and shares high homology to those of other PTPs, such that compounds that target the catalytic core of SHP2 often inhibit other phosphatases. Therefore, our initial structure-based approach was based on targeting a non-homologous pocket in the pY-peptide binding cleft in SHP2 adjacent to the homologous catalytic site (24). This non-homologous pocket has unique features in both amino acid composition and 3D structure. Using CADD database screening targeting this pocket in combination with experimental assays, we previously identified 9 SHP2 selective inhibitors (24) although the activities of these inhibitors were low. In the present study, we have used CADD to identify compounds that are structurally similar to those 9 active compounds. Of these, 20 were shown to be active, of which #220–324 was shown to have a much improved potency and selectivity over the previously identified compounds. This small-molecule inhibitor selectively inhibited SHP2 over SHP1 and other PTPs. Most notably, it does not seem to have significant off-target effects as it only had minimal effects in PTPN11 knockout cells. Additional biological analyses validated that it was effective in blocking SHP2-mediated signaling and cellular function. This inhibitor was verified to directly bind to the SHP2 protein by the fluorescence quenching assay. Since it only inhibited SHP2 enzymatic activity toward the pY-peptide substrate but not the small molecule substrate pNPP, it is likely that it functions as designed by disrupting the interaction between pY-peptide substrates and SHP2 at a location distinct from the catalytic core.
This study also provides proof of concept that SHP2 is a “druggable” target for the treatment of PTPN11-associated malignancies. SHP2 phosphatase plays an overall positive role in cell signaling and cellular function (1–3). Loss of SHP2 function leads to severe consequences in embryogenesis and hematopoietic cell development (50–52). A prevalent question is whether SHP2 can be used as a drug target for therapeutic purposes. Our studies have now provided evidence that mutant MEFs or lung cancer cells with activating mutations of PTPN11 were more sensitive to SHP2 inhibition than control cells with WT PTPN11, and the sensitivity of mutant mouse myeloid progenitors with the PTPN11E76K mutation was greater to SHP2 inhibition than WT counterparts. The same is true in human JMML cells. Leukemic cells from JMML patients with the PTPN11E76K mutation were more sensitive to the SHP2 inhibitor than healthy donor cells. These observations suggest that SHP2 can be targeted by inhibitory drugs for the control of PTPN11 mutation-associated malignancies.
While #220–324 is a drug-like small molecule that is bioavailable in cells, its affinity is not high, with an IC50 in the range of 10–20 μM. We tested the activity of #220–324 in the presence of the detergent Triton X-100. We found that the inhibitory function of #220–324 was decreased by ~30% in the presence of Triton X-100 at the concentration of 0.001% (Supplementary Fig. 4), raising the possibility that the interaction between #220–324 and SHP2 is weak and that this binding may be partially mediated by a promiscuous effect. Accordingly, further studies are required to optimize this compound to improve the binding to SHP2 in order to develop new SHP2 inhibitors with increased activities and selectivity suitable for pre-clinical and clinical studies. Such efforts would be greatly facilitated by co-crystallization of this compound with SHP2 that would reveal structural details of the binding interaction that would be used to guide optimization of the lead compound.
Supplementary Material
Acknowledgments
Grant Support: This work was supported by the National Institutes of Health grants HD070716 (C.K. Qu), HL068212 (C.K. Qu), and CA082103 (F.P. McCormick).
This work was supported by the National Institutes of Health grants HD070716 and HL068212 (to C.K. Qu). We would also like to thank the University of California, San Francisco Tissue Cancer Cell Bank funded by the National Institutes of Health grant CA082103 (F.P. McCormick) for providing JMML cells.
Footnotes
Competing financial interests: The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
B.Y., W.L., W.M.Y., S.A., O.G. performed the experiments and summarized the data. A.D.M, L.D.T., and C.K.Q. designed the experiments. M.L. provided critical reagents. W.L., O.G., A.D.M., and C.K.Q. wrote the manuscript.
References
- 1.Xu D, Qu CK. Protein tyrosine phosphatases in the JAK/STAT pathway. Front Biosci. 2008;13:4925–32. doi: 10.2741/3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chan G, Kalaitzidis D, Neel BG. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev. 2008;27:179–92. doi: 10.1007/s10555-008-9126-y. [DOI] [PubMed] [Google Scholar]
- 3.Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol. 2006;7:833–46. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 4.Zhao R, Fu X, Teng L, Li Q, Zhao ZJ. Blocking the function of tyrosine phosphatase SHP-2 by targeting its Src homology 2 domains. J Biol Chem. 2003;278:42893–8. doi: 10.1074/jbc.M306136200. [DOI] [PubMed] [Google Scholar]
- 5.Pawson T. Specificity in signal transduction: from phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell. 2004;116:191–203. doi: 10.1016/s0092-8674(03)01077-8. [DOI] [PubMed] [Google Scholar]
- 6.Barford D, Neel BG. Revealing mechanisms for SH2 domain mediated regulation of the protein tyrosine phosphatase SHP-2. Structure. 1998;6:249–54. doi: 10.1016/s0969-2126(98)00027-6. [DOI] [PubMed] [Google Scholar]
- 7.Eck MJ, Pluskey S, Trub T, Harrison SC, Shoelson SE. Spatial constraints on the recognition of phosphoproteins by the tandem SH2 domains of the phosphatase SH-PTP2. Nature. 1996;379:277–80. doi: 10.1038/379277a0. [DOI] [PubMed] [Google Scholar]
- 8.Hof P, Pluskey S, Dhe-Paganon S, Eck MJ, Shoelson SE. Crystal structure of the tyrosine phosphatase SHP-2. Cell. 1998;92:441–50. doi: 10.1016/s0092-8674(00)80938-1. [DOI] [PubMed] [Google Scholar]
- 9.Loh ML, Vattikuti S, Schubbert S, Reynolds MG, Carlson E, Lieuw KH, et al. Mutations in PTPN11 implicate the SHP-2 phosphatase in leukemogenesis. Blood. 2004;103:2325–31. doi: 10.1182/blood-2003-09-3287. [DOI] [PubMed] [Google Scholar]
- 10.Loh ML, Reynolds MG, Vattikuti S, Gerbing RB, Alonzo TA, Carlson E, et al. PTPN11 mutations in pediatric patients with acute myeloid leukemia: results from the Children’s Cancer Group. Leukemia. 2004;18:1831–4. doi: 10.1038/sj.leu.2403492. [DOI] [PubMed] [Google Scholar]
- 11.Tartaglia M, Martinelli S, Cazzaniga G, Cordeddu V, Iavarone I, Spinelli M, et al. Genetic evidence for lineage-related and differentiation stage-related contribution of somatic PTPN11 mutations to leukemogenesis in childhood acute leukemia. Blood. 2004;104:307–13. doi: 10.1182/blood-2003-11-3876. [DOI] [PubMed] [Google Scholar]
- 12.Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. 2001;29:465–8. doi: 10.1038/ng772. [DOI] [PubMed] [Google Scholar]
- 13.Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J, Jung A, et al. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. 2003;34:148–50. doi: 10.1038/ng1156. [DOI] [PubMed] [Google Scholar]
- 14.Bentires-Alj M, Paez JG, David FS, Keilhack H, Halmos B, Naoki K, et al. Activating mutations of the noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res. 2004;64:8816–20. doi: 10.1158/0008-5472.CAN-04-1923. [DOI] [PubMed] [Google Scholar]
- 15.Keilhack H, David FS, McGregor M, Cantley LC, Neel BG. Diverse biochemical properties of Shp2 mutants. Implications for disease phenotypes. J Biol Chem. 2005;280:30984–93. doi: 10.1074/jbc.M504699200. [DOI] [PubMed] [Google Scholar]
- 16.Loh ML, Sakai DS, Flotho C, Kang M, Fliegauf M, Archambeault S, et al. Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood. 2009;114:1859–63. doi: 10.1182/blood-2009-01-198416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muramatsu H, Makishima H, Jankowska AM, Cazzolli H, O’Keefe C, Yoshida N, et al. Mutations of an E3 ubiquitin ligase c-Cbl but not TET2 mutations are pathogenic in juvenile myelomonocytic leukemia. Blood. 2010;115:1969–75. doi: 10.1182/blood-2009-06-226340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Araki T, Mohi MG, Ismat FA, Bronson RT, Williams IR, Kutok JL, et al. Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutation. Nat Med. 2004;10:849–57. doi: 10.1038/nm1084. [DOI] [PubMed] [Google Scholar]
- 19.Chan G, Kalaitzidis D, Usenko T, Kutok JL, Yang W, Mohi MG, et al. Leukemogenic Ptpn11 causes fatal myeloproliferative disorder via cell-autonomous effects on multiple stages of hematopoiesis. Blood. 2009;113:4414–24. doi: 10.1182/blood-2008-10-182626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chan RJ, Leedy MB, Munugalavadla V, Voorhorst CS, Li Y, Yu M, et al. Human somatic PTPN11 mutations induce hematopoietic-cell hypersensitivity to granulocyte-macrophage colony-stimulating factor. Blood. 2005;105:3737–42. doi: 10.1182/blood-2004-10-4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schubbert S, Lieuw K, Rowe SL, Lee CM, Li X, Loh ML, et al. Functional analysis of leukemia-associated PTPN11 mutations in primary hematopoietic cells. Blood. 2005;106:311–7. doi: 10.1182/blood-2004-11-4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu D, Liu X, Yu WM, Meyerson HJ, Guo C, Gerson SL, et al. Non-lineage/stage-restricted effects of a gain-of-function mutation in tyrosine phosphatase Ptpn11 (Shp2) on malignant transformation of hematopoietic cells. J Exp Med. 2011;208:1977–88. doi: 10.1084/jem.20110450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu WM, Daino H, Chen J, Bunting KD, Qu CK. Effects of a leukemia-associated gain-of-function mutation of SHP-2 phosphatase on interleukin-3 signaling. J Biol Chem. 2006;281:5426–34. doi: 10.1074/jbc.M507622200. [DOI] [PubMed] [Google Scholar]
- 24.Yu WM, Guvench O, Mackerell AD, Qu CK. Identification of small molecular weight inhibitors of Src homology 2 domain-containing tyrosine phosphatase 2 (SHP-2) via in silico database screening combined with experimental assay. J Med Chem. 2008;51:7396–404. doi: 10.1021/jm800229d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burch JD, Belley M, Fortin R, Deschenes D, Girard M, Colucci J, et al. Structure-activity relationships and pharmacokinetic parameters of quinoline acylsulfonamides as potent and selective antagonists of the EP(4) receptor. Bioorg Med Chem Lett. 2008;18:2048–54. doi: 10.1016/j.bmcl.2008.01.103. [DOI] [PubMed] [Google Scholar]
- 26.Best RB, Zhu X, Shim J, Lopes PE, Mittal J, Feig M, et al. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J Chem Theory Comput. 2012;8:3257–73. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem. 2010;31:671–90. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee MS, Feig M, Salsbury FR, Jr, Brooks CL., 3rd New analytic approximation to the standard molecular volume definition and its application to generalized Born calculations. J Comput Chem. 2003;24:1348–56. doi: 10.1002/jcc.10272. [DOI] [PubMed] [Google Scholar]
- 29.Lee MS, Salsbury FR, Brooks CL., III Novel generalized Born methods. J Chem Phys. 2002;116(24):10606–14. [Google Scholar]
- 30.Vanommeslaeghe K, MacKerell AD., Jr Automation of the CHARMM General Force Field (CGenFF) I: bond perception and atom typing. J Chem Inf Model. 2012;52:3144–54. doi: 10.1021/ci300363c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vanommeslaeghe K, Raman EP, MacKerell AD., Jr Automation of the CHARMM General Force Field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J Chem Inf Model. 2012;52:3155–68. doi: 10.1021/ci3003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23(3):327–41. [Google Scholar]
- 33.Steinbach PJ, Brooks BR. New spherical-cutoff methods for long-range forces in macromolecular simulation. J Comput Chem. 1994;15(7):667–83. [Google Scholar]
- 34.Pastor RW. Techniques and Applications of Langevin Dynamics Simulations. In: Luckhurst GR, Veracini CA, editors. The Molecular Dynamics of Liquid Crystals. The Netherlands: Kluwer Academic Publishers; 1994. pp. 85–138. [Google Scholar]
- 35.Brooks BR, Brooks CL, 3rd, Mackerell AD, Jr, Nilsson L, Petrella RJ, Roux B, et al. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30:1545–614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–8. 27–8. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 37.Macias AT, Mia MY, Xia G, Hayashi J, MacKerell AD., Jr Lead validation and SAR development via chemical similarity searching; application to compounds targeting the pY+3 site of the SH2 domain of p56lck. J Chem Inf Model. 2005;45:1759–66. doi: 10.1021/ci050225z. [DOI] [PubMed] [Google Scholar]
- 38.Tartaglia M, Cotter PD, Zampino G, Gelb BD, Rauen KA. Exclusion of PTPN11 mutations in Costello syndrome: further evidence for distinct genetic etiologies for Noonan, cardio-facio-cutaneous and Costello syndromes. Clin Genet. 2003;63:423–6. doi: 10.1034/j.1399-0004.2003.00076.x. [DOI] [PubMed] [Google Scholar]
- 39.Tartaglia M, Martinelli S, Stella L, Bocchinfuso G, Flex E, Cordeddu V, et al. Diversity and functional consequences of germline and somatic PTPN11 mutations in human disease. Am J Hum Genet. 2006;78:279–90. doi: 10.1086/499925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu WM, Hawley TS, Hawley RG, Qu CK. Catalytic-dependent and -independent roles of SHP-2 tyrosine phosphatase in interleukin-3 signaling. Oncogene. 2003;22:5995–6004. doi: 10.1038/sj.onc.1206846. [DOI] [PubMed] [Google Scholar]
- 41.Bennett AM, Hausdorff SF, O’Reilly AM, Freeman RM, Neel BG. Multiple requirements for SHPTP2 in epidermal growth factor-mediated cell cycle progression. Mol Cell Biol. 1996;16:1189–202. doi: 10.1128/mcb.16.3.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nishida K, Hirano T. The role of Gab family scaffolding adapter proteins in the signal transduction of cytokine and growth factor receptors. Cancer Sci. 2003;94:1029–33. doi: 10.1111/j.1349-7006.2003.tb01396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gu H, Neel BG. The “Gab” in signal transduction. Trends Cell Biol. 2003;13:122–30. doi: 10.1016/s0962-8924(03)00002-3. [DOI] [PubMed] [Google Scholar]
- 44.Emanuel PD, Bates LJ, Castleberry RP, Gualtieri RJ, Zuckerman KS. Selective hypersensitivity to granulocyte-macrophage colony-stimulating factor by juvenile chronic myeloid leukemia hematopoietic progenitors. Blood. 1991;77:925–9. [PubMed] [Google Scholar]
- 45.Lyubynska N, Gorman MF, Lauchle JO, Hong WX, Akutagawa JK, Shannon K, et al. A MEK inhibitor abrogates myeloproliferative disease in Kras mutant mice. Sci Transl Med. 2011;3:76ra27. doi: 10.1126/scitranslmed.3001069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang X, He Y, Liu S, Yu Z, Jiang ZX, Yang Z, et al. Salicylic acid based small molecule inhibitor for the oncogenic Src homology-2 domain containing protein tyrosine phosphatase-2 (SHP2) J Med Chem. 2010;53:2482–93. doi: 10.1021/jm901645u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hellmuth K, Grosskopf S, Lum CT, Wurtele M, Roder N, von Kries JP, et al. Specific inhibitors of the protein tyrosine phosphatase Shp2 identified by high-throughput docking. Proc Natl Acad Sci U S A. 2008;105:7275–80. doi: 10.1073/pnas.0710468105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dawson MI, Xia Z, Jiang T, Ye M, Fontana JA, Farhana L, et al. Adamantyl-substituted retinoid-derived molecules that interact with the orphan nuclear receptor small heterodimer partner: effects of replacing the 1-adamantyl or hydroxyl group on inhibition of cancer cell growth, induction of cancer cell apoptosis, and inhibition of SRC homology 2 domain-containing protein tyrosine phosphatase-2 activity. J Med Chem. 2008;51:5650–62. doi: 10.1021/jm800456k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen L, Sung SS, Yip ML, Lawrence HR, Ren Y, Guida WC, et al. Discovery of a novel shp2 protein tyrosine phosphatase inhibitor. Mol Pharmacol. 2006;70:562–70. doi: 10.1124/mol.106.025536. [DOI] [PubMed] [Google Scholar]
- 50.Chan G, Cheung LS, Yang W, Milyavsky M, Sanders AD, Gu S, et al. Essential role for Ptpn11 in survival of hematopoietic stem and progenitor cells. Blood. 2011;117:4253–61. doi: 10.1182/blood-2010-11-319517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang W, Klaman LD, Chen B, Araki T, Harada H, Thomas SM, et al. An Shp2/SFK/Ras/Erk signaling pathway controls trophoblast stem cell survival. Dev Cell. 2006;10:317–27. doi: 10.1016/j.devcel.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 52.Zhu HH, Ji K, Alderson N, He Z, Li S, Liu W, et al. Kit-Shp2-Kit signaling acts to maintain a functional hematopoietic stem and progenitor cell pool. Blood. 2011;117:5350–61. doi: 10.1182/blood-2011-01-333476. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.