

Abstract
Imaging single epidermal growth factor receptors (EGFR) in intact cells is presently limited by the available microscopy methods. Environmental scanning electron microscopy (ESEM) of whole cells in hydrated state in combination with specific labeling with gold nanoparticles was used to localize activated EGFRs in the plasma membranes of COS7 and A549 cells. The use of a scanning transmission electron microscopy (STEM) detector yielded a spatial resolution of 3 nm, sufficient to identify the locations of individual EGFR dimer subunits. The sizes and distribution of dimers and higher order clusters of EGFRs were determined. The distance between labels bound to dimers amounted to 19 nm, consistent with a molecular model. A fraction of the EGFRs was found in higher order clusters with sizes ranging from 32–56 nm. ESEM can be used for quantitative whole cell screening studies of membrane receptors, and for the study of nanoparticle-cell interactions in general.
The EGFR plays a critical role in the pathogenesis and progression of many different types of cancer and is one of the best-studied transmembrane receptors1,2. Studying EGFR dimerization is demanding partly because methods are lacking to image the subunits within the context of intact cells. Cellular macromolecules are mostly imaged with transmission electron microscopy (TEM) yielding nanometer resolution on conventional thin sections, cryo sections, membrane sheets, or freeze-fracture samples, or via serial sectioning3,4,5. In particular sectioning remains challenging regarding artifacts destroying the cellular context of membrane proteins5. Intact cells in liquid are traditionally imaged with light microscopy. But even recently developed super resolution fluorescence techniques6,7 lack about an order of magnitude in their spatial resolution as required for resolving the subunits of macromolecular complexes. A variety of other techniques exists but all with their specific limitations. Förster Resonance Energy Transfer (FRET) analysis, for instance, is sensitive to intermolecular distances but is an ensemble-averaging technique only8.
Here, we describe the localization and analysis of the EGFR dimer and higher order clusters of EGFRs in intact hydrated cells. Live cells were grown on supporting membrane windows. The cells were then incubated with the epidermal growth factor (EGF) coupled to gold nanoparticles, fixed, and subsequently imaged with ESEM in a saturated water vapor environment adjusted in such way that a thin liquid layer resided over the cells (Fig. 1). The sample preparation was minimal and similar to that of light microscopy. Usage of the STEM detector9 resulted in high contrast on the labels10 such that their positions could be determined with nanometer resolution. A series of whole cells was studied and the EGFR distribution analyzed.
Figure 1. Schematic representation of environmental scanning electron microscopy (ESEM) of a whole eukaryotic cell in hydrated state.
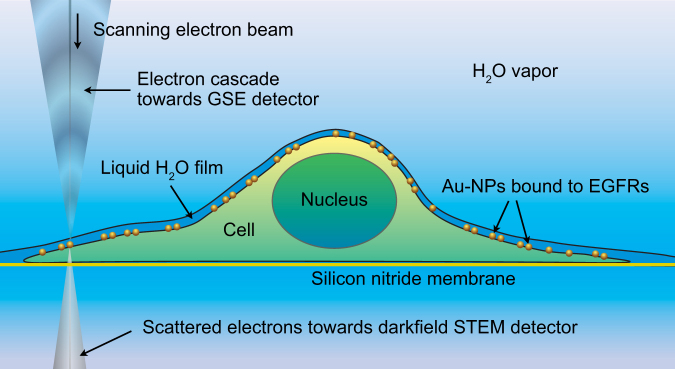
A cell on a supporting silicon nitride membrane is maintained in a saturated water vapor atmosphere; the hydrated cell is covered with a thin layer of pure water. Epidermal growth factor receptor (EGFR) proteins labeled with gold nanoparticles (Au-NPs) reside in the plasma membrane. Imaging is done by scanning a focused electron beam over the cell, mostly at its thinner regions. Two different types of signals are used for image formation, 1) secondary electrons from the sample surface cause a cascade of electrons (and ions) which are recorded with a gaseous secondary electron (GSE) detector located above the sample, 2) transmitted electrons are recorded with the scanning transmission electron microscopy (STEM) detector located beneath the sample.
Results
Preparation of the cellular samples
Live cells were grown on silicon microchips with thin electron transparent windows of silicon nitride10,11 (SiN). Two different cell lines were studied, COS7 fibroblast cells and A549 lung cancer cells. Gold nanoparticles (AuNPs) of an average diameter of 12 nm containing one streptavidin each were conjugated to the ligand epidermal growth factor (EGF) linked to biotin, resulting in the compound EGF-AuNP10. Compared to the commonly used immuno-labeling, relying on primary and secondary antibodies, the bound EGF-AuNP label exhibits a reduced distance between the electron dense AuNP and the targeted protein12. The cells were incubated for 5 minutes with EGF-AuNPs, sufficiently long for activation and subsequent dimerization to occur for a fraction of the EGFRs13. The cells were then fixed with glutaraldehyde, and washed with deionized water as needed for ESEM imaging. The chemical fixation was needed to provide the network of cellular protein structures sufficient resistance against the low osmotic pressure during the rinse with water prior to ESEM imaging, and to provide sample stability against electron beam irradiation. The preservation of cellular fine structures by glutaraldehyde is recognized at least down to the level of macromolecular resolution14. The sample preparation was minimal and similar to that of light microscopy.
ESEM imaging of hydrated A549 and COS7 cells
A series of STEM dark field images from A549 cells were recorded with increasing magnification. Fig. 2a served as an overview image at a magnification of 1,500×. Three different cells light up on a dark background. Thicker and electron denser areas appear brighter, for instance the oval white spot of the left cell representing the nucleus. At the thinner and peripheral regions of the cells the structured intracellular space is visible. Cell borders and protrusions like the broad lamellipodia, and fine filopodia, stretching from the upper right cell to the adjacent left cell are clearly discernable. The existence of the water layer was verified by comparing the signals from different detectors (Supplementary Fig. S1). Fig. 2b is an image recorded with 5,336× magnification at the location indicated by the rectangle in Fig. 2a showing details of the lamellipodia, and some clusters of AuNPs appeared as tiny bright dots. Fig. 2c was recorded at a magnification of 50,000× sufficient to resolve the individual labels. The spatial resolution was measured to amount to 3 nm from line-scans in the images at the locations of AuNPs (Supplementary Fig. S2). Several pairs of labels (Fig. 2d, e) were visible, and were attributed to EGFR dimers, which were absent in control experiments (Supplementary Fig. S3). Several larger clusters were also present (Fig. 2f, g).
Figure 2. ESEM of a whole fixed A549 cell in hydrated state.

(a) Overview dark field ESEM-STEM image showing the flat regions of the cells in grey, and the thicker cellular areas in white. The pixel size s = 87 nm and the magnification M = 1,500×. (b) Image recorded at the location of the rectangle in a using s = 25 nm and M = 5,336×. (c) Image showing individual Au-NPs as white spots for the region shown as rectangle in b, s = 2.7 nm and M = 50,000×. (d)–(g) Magnified regions from c showing individual AuNPs, dimers, and larger clusters indicated with numbers 1–4, respectively, in c.
The total dose for this series was 1.9 e−/nm2, only a factor of two larger than that used for cryo TEM studies of cells5. The effect of radiation damage was evaluated in a control experiment (Supplementary Fig. S4). Minor sample shrinkage (<1%) was observed but the AuNP distances in the relevant range of up to 300 nm were not influenced by electron beam irradiation for a dose of up to 7.5 e−/nm2.
A total of 22 regions of 8 different A549 cells was imaged and used for analysis of the spatial distribution of EGFRs. Similar images were recorded for COS7 cells for a total of 23 regions of 7 different cells (data not shown). Also in these images similar dimers and larger clusters of the labeled EGFR were visible.
Analysis of the locations of the EGF-AuNPs
The positions of a total of 1411 EGF-AuNPs from 20 micrographs recorded of both cell types were automatically detected and studied with particle analysis algorithms (Supplementary Fig. S5). Firstly, a density-based spatial clustering algorithm was applied to describe the development of the particle numbers with increasing distance from the particle neighborhood. The resulting cluster size distribution histogram (Fig. 3a) shows that most AuNPs were found as single objects, whereas about 18% existed in pairs, and 14% were found in higher order clusters for COS7 cells, and similarly for A549 cells.
Figure 3. Analysis of the spatial distribution patterns of labeled EGFRs on COS7 and A549 cells.
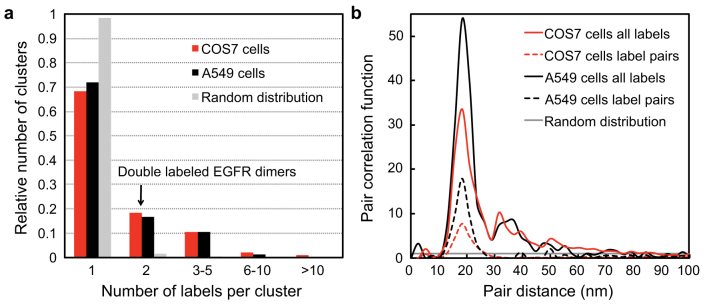
(a) Size distribution histogram of EGFR clusters in COS7 (red) and A549 cells (black). A total of 20 images (M = 50,000×) for both cell types was first processed to determine the positions of 1411 EGFR labels followed by the application of a density-based spatial clustering algorithm. A random distribution generated by Monte Carlo simulation (grey) is included for comparison. (b) Normalized pair correlation function for both cell types. The peak maxima indicating the likeliest average distance between the two labels are found at 19 nm for both curves. The subsets of labels assigned to a cluster size of two are included as dashed lines. The grey line indicates the level for a random distribution.
Secondly, the cluster size was analyzed using the pair correlation function15 measuring the probability of finding a particle at a certain radius from another particle. The curves for both cell types (Fig. 3b) show a main peak at 19 nm and several smaller peaks. Beyond 80 nm, the correlation function became smaller than unity, the value for a random distribution. The main peak reflects double-labeled dimers. Indeed, applying the pair correlation function to dimers only by filtering the data using the clustering algorithm of the above resulted in single peaks for both cell types with equal locations of the maxima (Fig. 3b). The ranges of the dimer curves above unity reached from 13 to 28 nm around the maximum. The peak value of 19 nm indicates the most probable distance between the centers of the two AuNP labels of a double-labeled dimer, while the range values specify the limits of the upper and lower confidence distances for two labels bound to the same dimer, and also include the precision of the pair distance measurement of 3 nm. The same AuNP dimer distance was found from an established but much more elaborative method using membrane sheets (Supplementary Fig. S6). Note that the membrane sheet does not provide the location of the analyzed membrane patch within the cellular context, and would miss internalized nanoparticles.
Additional peaks of the correlation function were found around 32, 40, and 50 nm for COS7 cells, and around 36, 48, and 56 nm for A549 cells. These values are interpreted as the sizes of higher order clusters.
Discussion
The incubation of live cells with small AuNP labels allowed the localization of individual EGFR-bound labels with a resolution of 3 nm within the context of intact and hydrated cells (Fig. 2). Previous electron microscopic studies of the spatial distribution of EFGRs relied on membrane sheet or freeze-fracture membrane preparations in combination with immunolabeling by primary and secondary, gold-conjugated, antibodies16,17,18,19. However, such a detection system of primary and secondary antibodies is too large20 to detect the subunits of EGFR dimers on account of steric hindrance. Using the information that both ligand binding sites in the dimer conformation reside at the outer periphery at opposite sides21, a molecular model was constructed from the EGFR dimer, streptavidin, and biotin protein database structures, and an assumed model for the Au-NP including its coating (Fig. 4). The experimentally determined most probable label distance of 19 nm matches a realistic configuration of the molecular model, while the range of 13–28 nm can be explained by the flexibility of the streptavidin-biotin linker. These findings indicate that the measured label distances in double labeled EGFR dimers originate from the native configuration of the EGFR dimer.
Figure 4. Molecular model of the EGFR dimer with Au-NPs conjugated via a streptavidin-biotin bond to the epidermal growth factor (EGF).

The distance between the centers of the two AuNPs was estimated to amount to ~19 nm for a tightly packed structure. A larger distance of up 29 nm can be accomplished by rotating the AuNP on the streptavidin. The scaled drawings of streptavidin, EGF and the EGFR were derived from CPK models of the 1stp (streptavidin), 1EGF (EGF), 1NQL, 2JWA, 1M17, 1IVO and 2GS6 (EGFR) structures as found in the RCSB Protein Protein Databank, created by Jmol Version 12.2.15. Biotin size and structure was derived from the biotin model as drawn in RCSB Ligand Explorer Version 1.0.
The results shown in Fig. 4b demonstrate that in addition to single EGFR, and EGFR dimers, a small fraction of the EGFR resides in larger clusters, after 5 minutes of incubation at room temperature for the A549 and COS7 cells. EGFR clustering is possibly related to lipid rafts in the cell membrane, which are believed to exert a modulatory function on receptor ligand binding and activation22. Other electron microscopic studies have already shown the occurrence of EGFR clusters16,23,24 but did not detect dimers. Obtaining simultaneous information about the label distance in EGFR dimer subunits, cluster sizes, and the nanoscale cellular location of individual EGFRs is not possible with other electron microscopy methods or with fluorescence microscopy25,26,27.
A key advance reported here is the detection and analysis of nanoparticles attached to the EGFR subunits in intact and hydrated COS7 and A549 cells using a method essentially not more difficult than fluorescence microscopy. In contrast to most methods available in electron microcopy, the sample preparation was minimal, and as a result, information of 1411 labels from 15 different cells was readily obtained. Avoiding the limitations of sectioning, whole cell quantitative analyses are feasible as was demonstrated here, opening new routes for quantitative screening studies of EGF binding under various conditions. Such approach could potentially aid the development of new therapeutics for cancer, for example, via testing the influence of drug compounds on receptor distributions.
The aggregation state of other oligomers and macromolecular complexes than EGFR could readily be studied in future. Nanoparticle labels of different sizes, shapes, and materials could be used to label macromolecular complexes assembled from different types of subunits. Intracellular proteins are accessible via application of, e.g., genetic labeling protocols28. In addition to the study of macromolecular complexes, ESEM could also be used for the imaging of electron dense nanoparticles in cells, with useful applications in areas such as nano-toxicology29,30,31, for the testing of new labels for molecular imaging32 or for therapeutics33. Enhanced capabilities to study the molecular machinery of cells are also of importance for bio-nanotechnology aiming to design future technology based on principles governing the intracellular world34.
Methods
Materials
Epidermal growth factor biotin-XX conjugate (biotin-EGF), Dulbecco's Phosphate Buffered Saline (PBS), (DPBS), Dulbecco's Modified Eagle's Medium (DMEM) and Fetal Bovine Serum (FBS), were from Invitrogen, Carlsbad, CA, USA. Size exclusion Micro Bio-Spin 30 Columns in Tris buffer were from Bio-Rad, Hercules, CA, USA. Regular electron mciroscopy grade glutaraldehyde 25%, was from Electron Microscopy Sciences, Hatfield, PA, USA. CellStripper was from Mediatech, Herndon, VA, USA. L-glutamine solution, Tyrode's salts with sodium bicarbonate (liquid), PBS (10×), albumin from bovine serum (BSA), D-glucose, glycine, Poly-L-lysine (PLL) solution (0.01%, mol wt 70,000–150,000), sodium cacodylate trihydrate, acetone, ethanol, and deionized water were all of HPLC grade from Sigma-Aldrich, St. Louis, MO, USA. Gold-labeled streptavidin (Strept-AuNP), was from KPL, Gaithersburg, MD, USA. The diameter indicated by the manufacturer was 40 nm, however transmission electron microscopy measurements showed an average diameter of 12 ± 1 nm. Sample support microchips10,11 with a central SiN membrane window of a dimension of 50 × 400 μm and a thickness of 50 nm were custom made by Protochips, Raleigh, NC, USA.
Generation of EGF-labeled AuNPs
Strept-AuNP was diluted 1:5 in PBS containing 0.5% BSA (PBS-BSA) and centrifuged for 20 min at 7,000 × g. The pellet was dissolved in 250 μL PBS-BSA and centrifuged for 13 min at 7,000 × g. The Strept-AuNP pellet was again dissolved in 30 μL of PBS-BSA and conjugated with EGF by adding 0.7 μL of biotin-EGF (6 μM, dissolved in PBS-BSA). The solution was incubated at 35°C for 1 h in a water bath, with repeated mixing every 10 min. The buffer of a size exclusion centrifugation column was exchanged four times with 500 μL PBS-BSA each, and the EGF-AuNP solution was cleaned from unbound biotin-EGF by centrifugation for 4 minutes at 1,000 × g. The filtrate was diluted with PBS-BSA to yield a total volume of 44 μL corresponding to approx. 4.5 nM of EGF-AuNP.
SiN membrane microchip preparation for cell settlement
A batch of a dozen SiN membrane microchips11 were prepared for COS7 or A549 cells by rinsing the microchips for 2 min in 100 mL of acetone first, followed by a 2 min rinse in 100 mL of ethanol. The dried microchips were cleaned with ambient air plasma for 5 min. Thereafter, microchips were directly placed at room temperature in 0.01% PLL for 5 min, rinsed twice in water, and kept in water until the cell suspension was ready (within 10–15 min).
Cell culture seeding
COS7 cells (African Green Monkey SV40-transfected kidney fibroblast cell line) were cultured in flasks (25 cm2) with DMEM, supplemented with 10% FBS, 2 mM L-glutamine, in a 5% CO2 atmosphere, at 37°C. A549 cells (human lung carcinoma cell line) were cultured under the same conditions except that the medium was not supplied with L-glutamine and the CO2 concentration was 9%. Confluent cells were harvested by rinsing the attached cell layer in DPBS, subsequent dissociation with CellStripper (3 min at 37°C), followed by a quench in the respective media. Volumes of 20 μL of harvested cells in suspension were added to each previously prepared SiN membrane microchip, lying each at the bottom of a well in a 96 well plate, which was filled with 200 μL of supplemented media. After 5–10 min, the microchips, with usually 3–6 cells adhering on the SiN window, were transferred into new wells, filled with 200 μL of media, and incubated for another 4 h in a 5% CO2 atmosphere for the COS7 cells, and 9% CO2 for the A549 cells, at 37°C.
Labeling with EGF-AuNPs and fixation
The day before labeling, the microchips with the adherent cells were transferred into new wells, filled with serum free DMEM. After 18 h of serum starvation the cells were rinsed once and then incubated for 2 min with PBS-BSA. For the EGFR labeling, lids of small plastic tubes (200 μL volume, sold for polymerase chain reactions) were cut off and fixed upside down on a microscope slide11. Per microchip, one 11 μL droplet of EGF-Au solution was used11. The microchips were placed inclined (such that the cells were oriented slightly upside down) against a droplet and incubated for 5 min at room temperature. The microchips were then rinsed three times with PBS, and once with 0.1 M cacodylate buffer, pH 7.4 (CB). Subsequently, the samples were incubated for 10 min in 3% glutaraldehyde in CB. Following fixation, all microchips were rinsed once with CB, three times with PBS and incubated for 3 minutes in 100 mM glycine in PBS. Microchips were again rinsed three times with PBS and stored in PBS at 4°C until imaging. Control experiments were performed under the same experimental conditions, but with Strept-AuNPs, thus without EGF bound to the label (see Fig. S3).
ESEM
The labeled cells on the SiN support membranes were imaged in hydrated state with ESEM (Quanta 400 FEG, FEI, USA). The electron beam energy was 30 kV, the spot size was 3, and the working distance was set between 6.8 and 7.4 mm. These settings resulted in a theoretical spot size of ~1.0 nm and a probe current of 0.6 nA. The image size was always 1024 × 884 pixels but selected regions are shown in Fig. 2. The pixel-dwell time was 30 μs for Fig. 2. Other images used for analysis of the EGFR were recorded with pixel-dwell times of 10, 30, and 100 μs. A detailed description of the setup for STEM in ESEM can be found elsewhere9. In short, the stage was equipped with a special sample holder, including a Peltier cooling element, and a two-segment solid-state detector, mounted underneath the sample serving as STEM detector. The two detector segments could be used independently, enabling bright field or dark field contrast mode. Note that it was not possible to translate the detector with respect to the sample, and, as a consequence, certain sample regions were imaged in dark field mode, while other regions were imaged in bright field. It was not possible to determine the detection collection angle for this detector. A GSE detector mounted at the pole piece (above the sample) was used to generate a synchronous set of images, serving mainly as control for the thin water film on the cell. The stage temperature was kept at 3°C, and a pressure of 740 Pa was chosen in most cases in order to create a saturated water vapor atmosphere (= 100% relative humidity) in the ESEM chamber as needed to ensure the constant coverage of the cells with a thin film of water.
Loading a sample into the ESEM and preparation for imaging
For the loading of a sample into the ESEM stage, a microchip was taken out of the cooled storage buffer (PBS), rinsed twice for a few seconds in cooled ultrapure water. The backside of the microchip was blotted on a lint-free clean room wipe, and placed on the precooled Peltier stage (3°C), with a working distance previously set to a value between 6.5 and 7.5 mm. Visual control of the upper side of the sample after the blotting and during the sample transfer confirmed that the cells remained wet (a wet sample had a shiny/reflective surface that would loose its shininess upon drying). As soon as the sample was mounted in the stage (1–2 min after the blotting), 3 μL cooled ultrapure water was pipetted onto the sample surface. Three additional 3 μL water droplets were placed close to the sample on the Peltier stage as water source.
Immediately afterwards the ESEM chamber was closed, and the pump procedure was started. An optimized pumping sequence was used in order to control evaporation from, and condensation on the sample during the filling of the specimen chamber with saturated water vapor9,35. A sequence was applied consisting of a fivefold cycling of the pressure in the specimen chamber between 800 Pa and 1,300 Pa. At the end of the sequence the pressure was held at 800 Pa, a value above the water vapor pressure at 3°C. The beam valve was then opened and ESEM at low magnification was used to locate the SiN window. At this time, usually no signals could reach the STEM detector because the water film on the sample was too thick, and therefore, the signal from the GSED detector was used to locate the sample. After the SiN window was found and positioned, thinning of the water film was initiated by stepwise reduction of the pressure to a final value of 740 Pa, taking about 3 minutes. As soon as the water film was thin enough an image in the STEM became visible (see Fig. S1).
Electron dose calculation
The electron dose q per unit area of the sample surface was calculated from q = I × t/d2 × e, with probe current I, pixel size d, pixel dwell time t, and the elementary charge e. On account of interactions of the electron beam with the water layer over the sample, it was assumed that the probe size was broadened from its value in vacuum to a value approximately equal to the resolution of 3 nm, and that the pixel size of 2.7 nm could be used as measure for the dose calculation. A fraction of 50% of the images used to study the spatial distribution of EGFRs were recorded at a magnification of 50,000×, a pixel size of 2.7 nm, and 100 μs pixel dwell time, leading to the maximal electron dose used in this study of q = 4.9 e−/nm2, while all other images were recorded at a smaller pixel-dwell time and thus received lower electron doses.
Image analysis
The positions of the nanoparticles in the images were determined with an automated procedure (see Fig. S5). The particle distribution was then analyzed using a density-based spatial clustering algorithm15. The algorithm considered particles to belong to a cluster if their inter-particle distance did not exceed 29 nm as was estimated from the model of Fig. 4 and was verified using the correlation function (see below). The algorithm sorted the nanoparticles on their existence in a cluster of a certain size, i.e., single particles, pairs, clusters of three nanoparticle, etc. A random distribution was simulated of 10 nm-diameter nanoparticles of a similar particle density per image as the experimental data using a Monte Carlo method.
Distributions of the particle pair distances were studied by applying the pair correlation function15,36 that can be calculated as the difference quotient of Ripley's K-function37 by:
with r as the radius around the tested particles. This function describes the probability that a particle can be found at a radius r from another particle, normalized to the case of a random distribution. Here, g(r) = 1 represents randomness and g(r) > 1 and <1 describe clustering and dispersion, respectively. For error reduction, in case the particle detection ring was partly exceeding the boundary of the micrograph, the toroidal edge correction was used38. The pair correlation function was first calculated for all particles, and then for the dimers only. The pair correlation function filtered for dimers was also used to verify the maximum inter-particle distance for deciding if a particle belonged to a cluster or not. This limit was set to the spatial value at which the correlation function was <1 (thus below the g(r) for randomness), being 27 and 29 nm for A549 and COS7 cells, respectively.
Author Contributions
D.B.P. designed and performed experiments, analyzed data, and wrote the paper. J.P.B. and M.E. performed experiments. U.W. analyzed data. N.J. designed experiments, and wrote the paper.
Supplementary Material
Supplementary Results and Figures
Acknowledgments
This paper is dedicated to Agnès Bogner whom we also thank for the introduction into the WetSTEM technique. We thank M. Koch for help with the ESEM, A. Kraegeloh for support of the experiments, D.W. Piston for discussions, and Protochips Inc, NC, USA for providing the microchips. We thank E. Arzt for his support through INM. Research in part supported by NIH grant 2R44-EB008589 (to J. Damiano).
References
- Normanno N. et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 366, 2–16 (2006). [DOI] [PubMed] [Google Scholar]
- Arkhipov A. et al. Architecture and membrane interactions of the EGF receptor. Cell 152, 557–569 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson C. V., Sali A. & Baumeister W. The molecular sociology of the cell. Nature 450, 973–982 (2007). [DOI] [PubMed] [Google Scholar]
- Pierson J., Sani M., Tomova C., Godsave S. & Peters P. J. Toward visualization of nanomachines in their native cellular environment. Histochem. Cell Biol. 132, 253–262 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoenger A. & Bouchet-Marquis C. Cellular tomography. Adv. Protein Chem. Struct. Biol. 82, 67–90 (2011). [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J. & Manley S. Putting super-resolution fluorescence microscopy to work. Nat. Meth. 6, 21–23 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell S. W. Far-field optical nanoscopy. Science 316, 1153–1158 (2007). [DOI] [PubMed] [Google Scholar]
- Bader A. N., Hofman E. G., Voortman J., en Henegouwen P. M. & Gerritsen H. C. Homo-FRET imaging enables quantification of protein cluster sizes with subcellular resolution. Biophys. J. 97, 2613–2622 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogner A., Thollet G., Basset D., Jouneau P. H. & Gauthier C. Wet STEM: A new development in environmental SEM for imaging nano-objects included in a liquid phase. Ultramicroscopy 104, 290–301 (2005). [DOI] [PubMed] [Google Scholar]
- de Jonge N., Peckys D. B., Kremers G. J. & Piston D. W. Electron microscopy of whole cells in liquid with nanometer resolution. Proc. Natl. Acad. Sci. 106, 2159–2164 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring E. A., Peckys D. B., Dukes M. J., Baudoin J. P. & de Jonge N. Silicon nitride windows for electron microscopy of whole cells. J. Microsc. 243, 273–283 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann R., Walther P. & Muller M. Immunogold labeling in scanning electron microscopy. Histochem. Cell Biol. 106, 31–39 (1996). [DOI] [PubMed] [Google Scholar]
- Lidke D. S. et al. Quantum dot ligands provide new insights into erB/HER receptor-mediated signal transduction. Nat. Biotechnol. 22, 198–203 (2004). [DOI] [PubMed] [Google Scholar]
- Hayat M. A. Principles and Techniques of Electron Microscopy: Biological Applications. 4th edn, 564 (Cambridge University Press, 2000). [Google Scholar]
- Ester M., Kriegel H.-P., Sander J. & Xu X. in Second International Conference on Knowledge Discovery and Data Mining. 226–231 (AAAI Press). [Google Scholar]
- Ariotti N. et al. Epidermal growth factor receptor activation remodels the plasma membrane lipid environment to induce nanocluster formation. Mol. Cell Biol. 30, 3795–3804 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S. et al. Mapping ErbB receptors on breast cancer cell membranes during signal transduction. J. Cell Sci. 120, 2763–2773 (2007). [DOI] [PubMed] [Google Scholar]
- van Belzen N. et al. Direct visualization and quantitative analysis of epidermal growth factor-induced receptor clustering. J. Cell Physiol. 134, 413–420 (1988). [DOI] [PubMed] [Google Scholar]
- Wiegant F. A. et al. Epidermal growth factor receptors associated to cytoskeletal elements of epidermoid carcinoma (A431) cells. J. Cell Biol. 103, 87–94 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann R., Walther P. & Muller M. Immunogold labeling in scanning electron microscopy. Histochem. Cell Biol. 106, 31–39 (1996). [DOI] [PubMed] [Google Scholar]
- Ogiso H. et al. Crystal Structure of the Complex of Human Epidermal Growth Factor and Receptor Extracellular Domains. Cell 110, 775–787 (2002). [DOI] [PubMed] [Google Scholar]
- Lillemeier B. F., Pfeiffer J. R., Surviladze Z., Wilson B. S. & Davis M. M. Plasma membrane-associated proteins are clustered into islands attached to the cytoskeleton. Proc. Natl. Acad. Sci. 103, 18992–18997 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Belzen N. et al. Direct visualization and quantitative analysis of epidermal growth factor-induced receptor clustering. J. Cell. Physiol. 134, 413–420 (1988). [DOI] [PubMed] [Google Scholar]
- Yang S. et al. Mapping ErbB receptors on breast cancer cell membranes during signal transduction. J. Cell Sci. 120, 2763–2773 (2007). [DOI] [PubMed] [Google Scholar]
- Cambi A. & Lidke D. S. Nanoscale membrane organization: where biochemistry meets advanced microscopy. ACS Chem. Biol. 7, 139–149 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endres N. F. et al. Conformational Coupling across the Plasma Membrane in Activation of the EGF Receptor. Cell 152, 543–556 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating E., Nohe A. & Petersen N. O. Studies of distribution, location and dynamic properties of EGFR on the cell surface measured by image correlation spectroscopy. Eur. Biophys. J. 37, 469–481 (2008). [DOI] [PubMed] [Google Scholar]
- Shu X. et al. A genetically encoded tag for correlated light and electron microscopy of intact cells, tissues, and organisms. PLoS Biol. 9, e1001041 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsaesser A. et al. Quantification of nanoparticle uptake by cells using microscopical and analytical techniques. Nanomedicine 5, 1447–1457 (2010). [DOI] [PubMed] [Google Scholar]
- Tantra R. & Shard A. We need answers. Nat. Nanotechnol. 8, 71 (2013). [DOI] [PubMed] [Google Scholar]
- Tantra R. & Knight A. Cellular uptake and intracellular fate of engineered nanoparticles: a review on the application of imaging techniques. Nanotoxicol. 5, 381–392 (2011). [DOI] [PubMed] [Google Scholar]
- Hahn M. A., Singh A. K., Sharma P., Brown S. C. & Moudgil B. M. Nanoparticles as contrast agents for in-vivo bioimaging: current status and future perspectives. Anal. Bioanal. Chem. 399, 3–27 (2011). [DOI] [PubMed] [Google Scholar]
- Davis M. E., Chen Z. G. & Shin D. M. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat. Rev. Drug. Discov. 7, 771–782 (2008). [DOI] [PubMed] [Google Scholar]
- Simons K. Engineering the future with cell biology. Mol. Biol. Cell. 21, 3822 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk S., Skepper J. & Donald A. M. Application of environmental scanning electron microscopy to determine biological surface structure. J. Microsc. 233, 205–224 (2009). [DOI] [PubMed] [Google Scholar]
- Stoyan D. & Stoyan H. Estimating pair correlation functions of planar cluster processes. Biom. J. 38, 259–271 (1996). [Google Scholar]
- Ripley B. D. The Second-Order Analysis of Stationary Point Processes. J. Appl. Probability 13, 255–266 (1976). [Google Scholar]
- Yamada I. & Rogerson P. A. An Empirical Comparison of Edge Effect Correction Methods Applied to K-function Analysis. Geographical Analysis 35, 97–109 (2003). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Results and Figures