

Abstract
Many anticancer agents damage DNA and activate cell cycle checkpoints that permit time for the cells to repair their DNA and recover. These checkpoints have undergone intense investigation as potential therapeutic targets and Chk1 inhibitors have emerged as promising novel therapeutic agents. Chk1 was initially recognized as a regulator of the G2/M checkpoint, but has since been demonstrated to have additional roles in replication fork stability, replication origin firing and homologous recombination. Inhibition of these pathways can dramatically sensitize cells to some antimetabolites. Current clinical trials with Chk1 inhibitors are primarily focusing on their combination with gemcitabine. Here, we discuss the mechanisms of, and emerging uses for Chk1 inhibitors as single agents and in combination with antimetabolites. We also discuss the pharmacodynamic issues that need to be addressed in attaining maximum efficacy in vivo. Following administration of gemcitabine to mice and humans, tumour cells accumulate in S phase for at least 24 h before recovering. In addition, stalled replication forks evolve over time to become more Chk1 dependent. We emphasize the need to assess cell cycle perturbation and Chk1 dependence of tumours in patients administered gemcitabine. These assessments will define the optimum dose and schedule for administration of these drug combinations.
Keywords: cell cycle checkpoints, Chk1, gemcitabine, homologous recombination, hydroxyurea, pharmacodynamics
Introduction
The majority of cancer chemotherapeutic agents inhibit DNA synthesis, either by directly damaging the DNA or by inhibiting production of the necessary deoxyribonucleotide precursors. DNA damage induces cell cycle arrest through activation of a cell cycle checkpoint response whose goal is to prevent further DNA synthesis or mitosis until the damage is repaired (Figure 1). Chk1 is a critical kinase involved in halting the cell cycle in response to DNA damage. It has been known for over 40 years that caffeine and other methylxanthines can enhance the cytotoxicity of DNA damaging agents [1]. In 1982, caffeine was shown to abrogate the cell cycle arrest elicited by DNA damage, thereby limiting the time available for DNA repair [2]. Caffeine was eventually shown to inhibit ATM and ATR but the required concentrations could not be achieved in patients [3]. UCN-01 was later discovered to abrogate the DNA damage induced arrest [4, 5] via the inhibition of Chk1 [6, 7] and is 100 000 fold more potent than caffeine. Unfortunately, in phase I trials, UCN-01 was found to bind strongly to the α1-acid glycoprotein in plasma which resulted in a long half-life, very limited bioavailability and serious side effects when the binding capacity was exceeded [8]. Many Chk1 inhibitors have subsequently been synthesized with at least six entering clinical trials, although some have subsequently been terminated possibly because of inadequate selectivity (Table 1).
Figure 1.
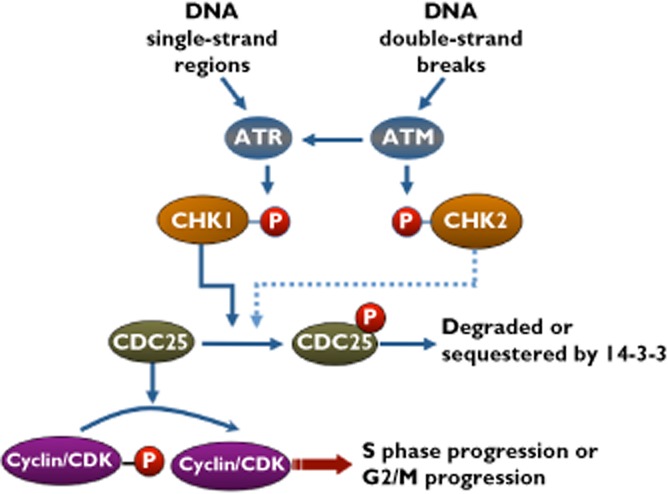
The cell cycle checkpoint pathway activated by DNA damage. ATR and ATM are activated by DNA single-strand regions or double-strand breaks, respectively. Chk1 and Chk2 are then activated, although Chk2 does not usually appear to elicit cell cycle arrest. Arrest is induced by phosphorylation and inhibition of CDC25 (either through degradation or sequestration). When Chk1 is inhibited, CDC25 is activated, which in turn activates cyclin/CDK complexes, driving the cell through the cell cycle, even when DNA damage persists
Table 1.
Selected Chk1 inhibitors in preclinical or clinical development
| Compound name | Company | Other targets | Phase of development | Reference |
|---|---|---|---|---|
| AZD7762 | AstraZeneca | CDK1, Chk2, CAMK, SRC-like kinase | Discontinued | [76–78] |
| SCH900776/ MK-8776 | Merck | Pim1 | Phase II | [72, 73] http://www.clinicaltrials.gov |
| IC83/ LY2603618 | Ely Lilly | Undisclosed | Phase I/II with pemetredex and cisplatin | http://www.clinicaltrials.gov |
| LY2606368 | Ely Lilly | Chk2 | Phase I | http://www.clinicaltrials.gov |
| GDC-0425 | Genentech | Undisclosed | Phase I | http://www.clinicaltrials.gov |
| PF-00477736 | Pfizer | Chk2, VEGFR2, Fms, Yes, Flt3, Ret | Discontinued | [79] |
| XL844 | Exelixis | Chk2 | Discontinued | http://www.clinicaltrials.gov |
| CEP-3891 | Cephalon | Undisclosed | Preclinical | [80] |
| SAR-020106 | Sareum | Undisclosed | Preclinical | [17] |
| CCT-244747 | Sareum | FLT3, Chk2, CDK1 | Preclinical | [81] |
| Arry-575 | Array | Undisclosed | Preclinical | http://www.arraybiopharma.com |
Despite the initial promise of Chk1 inhibitors in combination chemotherapy, there have been varying degrees of success reported with DNA damaging agents such as cisplatin and topoisomerase inhibitors in vitro. Early combination studies with UCN-01 demonstrated a 60-fold increase in cisplatin cytotoxicity in CHO cells [4] and potentiation of cisplatin cytotoxicity has also been seen with the more selective inhibitors Gö6976, PF-0477736 and SB218078 [9–11]. However, other studies have shown no sensitization of cells to cisplatin with the Chk1/Chk2 inhibitor AZD7762 [12] or with the more selective Chk1 inhibitor MK-8776 (previously known as SCH900776) [13]. It was also shown that MK-8776 did not sensitize cells to SN-38, the active metabolite of irinotecan, whereas AZD7762 and CHIR-124 were both found to enhance the antitumour effect of irinotecan in xenograft models [14–16]. The discrepancy between these observations may reflect the difference in assay used. Short term assays tend to show potentiation of cytotoxicity because inhibition of Chk1 may accelerate the rate of cell death, whereas long term assays reflect the overall level of cell death that eventually occurs.
The most dramatic sensitization has been observed when Chk1 inhibitors are combined with certain antimetabolites. We have demonstrated a 100-fold decrease in the IC50 (concentration that inhibits growth by 50%) for hydroxyurea upon addition of the Chk1 inhibitor MK-8776 [13]. Although the sensitization to gemcitabine was not as pronounced (∼10 fold), it has also been observed with other Chk1 inhibitors including PF-00477736 [10], AZD7762 [15], SAR-020106 [17] and XL-844 [18]. Potentiation of gemcitabine efficacy by these compounds has also been seen in xenograft models and clinical trials have focused primarily on combinations with gemcitabine [19].
Hydroxyurea and gemcitabine both inhibit ribonucleotide reductase thereby starving cells of deoxyribonucleotides and stalling replication fork progression. In addition, gemcitabine can be incorporated into the growing DNA strand, but induces chain termination after the addition of the next nucleotide. Potent sensitization by Chk1 inhibition has also been observed with cytarabine which stalls replication forks through chain termination [13]. However, Chk1 inhibitors do not appear to sensitize cells to all antimetabolites. 5-fluorouracil (5-FU) inhibits thymidylate synthase thus depleting cells of thymidine and stalling replication fork progression. We failed to observe sensitization to 5-FU when combined with MK-8776 [13]. Similar results have been reported in colon cancer cells in which 5-FU was shown to activate both ATM and ATR but inhibition of these kinases did not sensitive the cells [20]. In contrast, decreased clonogenic survival and increased DNA double-strand breaks were seen in the chicken B-lymphoma cell line DT40 when 5-FU was combined with UCN-01 [21]. Surprisingly, abrogation of 5-FU-induced cell cycle arrest has been reported upon combination with a Chk1 inhibitor [22, 23]. These results are unexpected as the lack of thymidine should cause arrest regardless of Chk1 status. However, the results can be explained by the methodology used whereby incorporation of BrdU was assessed. BrdU can substitute for thymidine and hence facilitate restart of replication. The discrepancy between these reports may reflect the different cell lines used, and in particular whether they are hypersensitive to Chk1 inhibition alone (see below). However, we are currently unable to explain why agents that appear to stall replication forks in a similar manner (i.e. by starving them of deoxyribonucleotides) should have such a difference in response when combined with Chk1 inhibitors.
How does inhibiting Chk1 sensitize cells to DNA damage?
It is now recognized that Chk1 has multiple roles in protecting cells from DNA damage and stalled replication. The most well recognized role of Chk1 is in the control of the cell cycle and in preventing premature entry into mitosis [24]. However, Chk1 also plays an important role in the stabilization of stalled replication forks [25], the control of replication origin firing and replication fork progression [26], and homologous recombination [27]. It is unclear whether it is the disruption of one or all of these pathways that results in the potentiation of antimetabolite cytotoxicity. The involvement of Chk1 in each of these pathways will be discussed here to provide a perspective of how the inhibition of these functions may lead to increased cytotoxicity and how this may impact the design of clinical trials.
The role of Chk1 in controlling entry into mitosis
Entry into mitosis is a tightly regulated process ultimately driven by the cyclin B/CDK1 complex [28]. In human cells, cyclin B is expressed in late S, G2 and early mitosis by regulated transcription and protein degradation. However, until the end of G2, cyclin B/CDK1 is kept inactive through inhibitory phosphorylation at threonine 14 and tyrosine 15 of CDK1. To enter mitosis, these sites must be dephosphorylated by CDC25 phosphatase. The most well known role of Chk1 is to prevent passage of a damaged cell through S and G2 and this is achieved by phosphorylation and inhibition of CDC25 [29]. While there are three CDC25 phosphatases (A, B and C) that function at different phases of the cell cycle [30], it will suffice for this review to consider them as a single entity.
Treatment with a DNA damaging agent, such as SN38 or cisplatin, results in phosphorylation and activation of Chk1, inhibition of CDC25 and cell cycle arrest (Figure 1). Inhibition of Chk1 results in activation of CDC25, inappropriate cell cycle progression and cell death [9, 13]. This enhanced killing only occurs in cells that are damaged and hence have activated Chk1 and arrested. Arrest and cell death are not discriminated in most cytotoxicity assays and may explain why different conclusions have been reported as to whether cells are sensitized to DNA damaging agents by Chk1 inhibitors [4, 5, 9–15].
In contrast, cell cycle arrest induced by antimetabolites does not require checkpoint activation as cells cannot complete DNA replication without deoxyribonucleotides. Indeed, we have observed S phase arrest of hydroxyurea- and gemcitabine-treated cells with little to no activation of Chk1 ([13] and unpublished observations). Because of the lack of deoxyribonucleotides, Chk1 inhibitors do not cause cell cycle progression when combined with hydroxyurea or gemcitabine. However, inhibition of Chk1 in gemcitabine-treated cells can induce premature entry of S phase cells into mitosis [24, 31]. Premature mitosis from the inhibition of Chk1 in mammalian cells leads to the phenomenon of mitotic catastrophe whereby chromosomes segregate aberrantly resulting in multiple micronuclei [32]. This is generally considered a lethal event that progresses to apoptosis.
The role of Chk1 in the stabilization of stalled replication forks
As a human cell prepares to enter S phase, it loads pre-replication complexes at many sites in the genome. Replication is initiated at 10–50 000 origins firing temporally during S phase [33], but many more remain dormant and never fire unless the DNA is damaged [34]. When damage is detected, the activated checkpoint rapidly suppresses firing of late origins of replication to protect the DNA integrity. Later, the dormant origins may fire to resolve problems that arise at irreversibly inhibited replication forks [35].
As each replication fork progresses, the leading helicase unwinds the DNA allowing the accompanying polymerase to synthesize new strands of DNA [36]. Upon incubation with hydroxyurea or gemcitabine the replication forks stall rapidly, and the helicase may separate from the polymerase creating a bubble in the DNA that is covered with the single-strand DNA (ssDNA) binding protein, RPA (Figure 2). Eventually, the fork will regress primarily because of torsion created by the supercoiled DNA ahead of the helicase [37]. The regressed fork generates a fourth branch in which the two daughter strands are annealed (often termed a ‘chicken foot’). This branch may contain ssDNA but recision by various nucleases including Mre11 and Exo1, create further ssDNA and RPA binding [38, 39] that provide the precursor for homologous recombination. RPA is replaced by RAD51 which then invades the homologous parent strand and is extended. However, because of the continued presence of the antimetabolite, the invading strand cannot be extended leading to stalled recombination. This recombination intermediate represents a four-way ‘Holliday junction’ which is normally resolved by double-strand cleavage (e.g. by Mus81 endonuclease) [40]. This cleavage is meant to facilitate recombination, yet in the case of stalled recombination, it is likely to result in additional breaks that cannot be repaired. In summary, the participation of many nucleases is intended to enhance survival of a cell. Chk1 plays many critical roles in the regulation of these nucleases to protect the replication forks and, as discussed in the next section, facilitate homologous recombination. The inhibition of Chk1 can exacerbate these nucleases leading to the formation of new DNA breaks and increased cytotoxicity.
Figure 2.
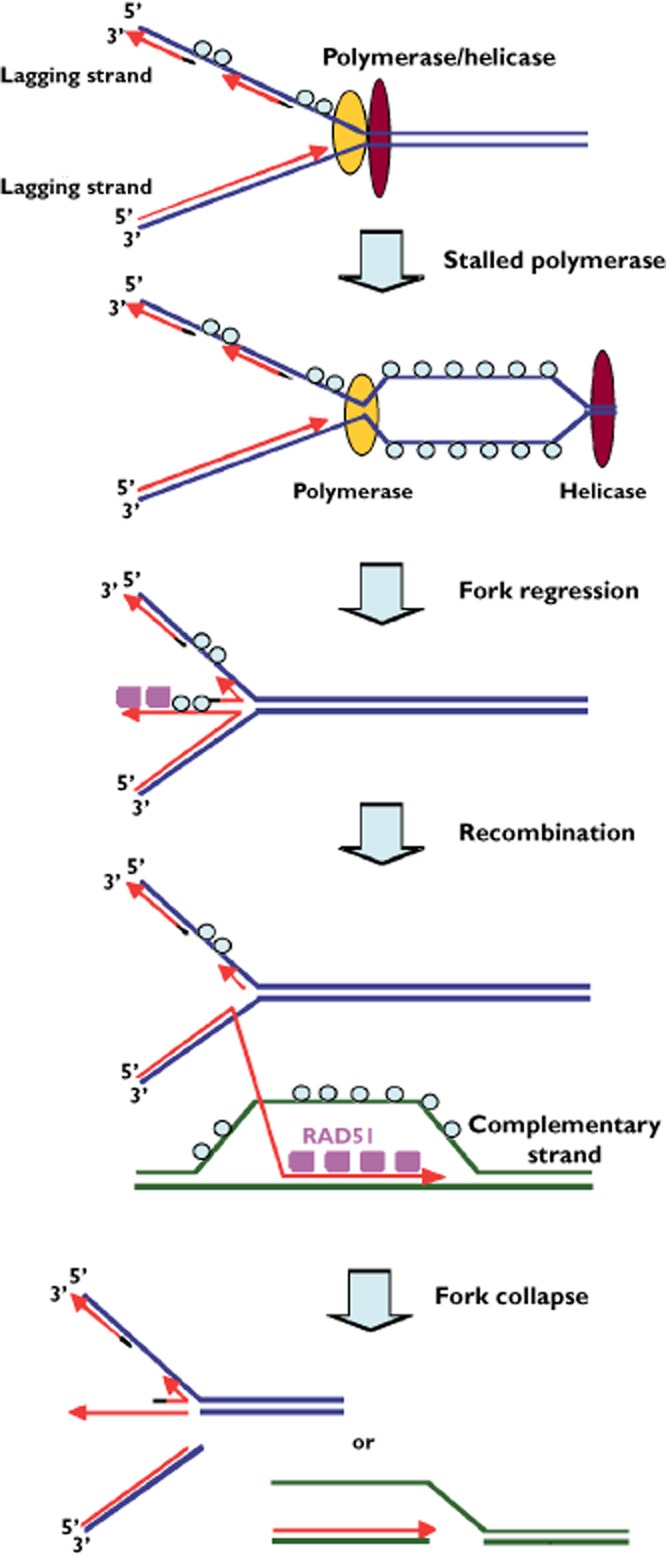
The events that occur at a stalled replication fork. See text for details. RPA (○); RAD51 ( )
)
Much of the research into the mechanism of fork stalling and replication restart has been performed in the fission yeast Saccharomyces cerevisiae. The fission yeast Rad53 has greatest sequence homology with the metazoan Chk2, although functionally it is more equivalent to the metazoan Chk1 [41]. When DNA replication forks are stalled by hydroxyurea, they remain competent to resume replication following the removal of hydroxyurea. Mutants of Rad53 lack this ability and as a consequence, stalled replication forks regress (this event is sometimes misleadingly called fork collapse, although this may occur subsequently; see below) [42–44].
In the budding yeast S. pombe, fork regression has also been documented in checkpoint-deficient cells following treatment with hydroxyurea [45]. Dna2 is an endonuclease that cleaves ssDNA. In S. pombe, Dna2 is phosphorylated by Cds1Chk2. This phosphorylation and subsequent nuclease activity are required to prevent fork regression by cleaving the first newly-synthesized strand that dissociates from the template DNA (either leading or lagging strand). Consequently, if Chk1 is inhibited and Dna2 is not activated, the number of regressed forks increases.
Whether Chk1 mediated activation of Dna2 prevents fork regression in human cells arrested with hydroxyurea remains to be determined. However, there is contrasting evidence that fork regression is enhanced rather than inhibited by an activated checkpoint, and this occurs through phosphorylation of SMARCAL1 (albeit by ATM, ATR and DNA-PK rather than Chk1) [46, 47]. The activation of both Dna2 and SMARCAL1 have been reported to protect from the eventual collapse of the replication fork. These conflicting observations need to be resolved.
As noted above, the term ‘collapsed fork’ has been used ambiguously in the literature. Most investigators agree that a collapsed fork involves the formation of a double-strand break and requires homologous recombination for restart [35, 39], while others refer to a regressed fork as a collapsed fork [44, 45]. Importantly, a regressed fork gives rise to a Holliday junction that is a potential substrate for the Mus81 endonuclease (Figure 2). Mus81 is responsible for cleaving the Holliday junctions that occur at the conclusion of homologous recombination. However, the Holliday junctions at regressed forks are structurally identical and can also be cleaved by Mus81 in the absence of an active replication checkpoint in fission yeast [48]. In budding yeast in response to hydroxyurea, Mus81 is phosphorylated in a Cds1Chk2 dependent manner and dissociates from chromatin, thus preventing it from cleaving stalled forks [49]. It has also been suggested that in mammalian cells, both active Chk1 and SMARCAL1 protect replication forks from Mus81 in a similar manner [47, 50].
The different definitions of fork collapse could be due to the fact that double-strand breaks are commonly inferred from the appearance of γH2AX (the phosphorylated form of the novel histone H2AX). However, γH2AX has been shown to appear well in advance of DNA fragmentation as visualized by other techniques (pulsed field gel electrophoresis or comet assay) in cells treated with hydroxyurea [35, 40]. It has been hypothesized that γH2AX is recruited to stalled forks and spreads out from the stalled fork [39]. We postulate an alternate explanation whereby phosphorylation of H2AX results from regression of the replication fork, localizing to the double-strand end of the regressed arm. Accordingly, γH2AX may detect double-strand ends, yet only after Mus81 cleavage are there true double-strand breaks.
These data demonstrate a clear rationale for the inhibition of Chk1 in combination with antimetabolites. Antimetabolites alone result in the accumulation of stalled replication forks. These forks would normally remain stalled until the removal of the antimetabolite upon which they would retain the capacity for restart. In the absence of Chk1, however, the stalled forks are not protected by Dna2 or SMARCAL1 and would become susceptible to Mus81 cleavage and increased cytotoxicity.
The role of Chk1 in homologous recombination
The mechanism of homologous recombination was primarily realized from studies into the repair of DNA double-strand breaks as induced by γ-radiation. The first step in homologous recombination is the resection of the DNA ends by one or several nucleases including Mre11, Exo1 and Dna2 to give regions of ssDNA for strand invasion [51]. This role for Dna2 has been reported both in S. cerevisiae and human cells [52, 53], yet appears to contrast with its role in preventing fork regression in S. pombe as discussed above. Interestingly, Dna2 activity in S. cerevisiae is regulated by CDK1-mediated phosphorylation [52], yet the phosphorylation sites are not conserved in S. pombe suggesting differential regulation and perhaps even different functions of Dna2 between these species.
As ssDNA is generated in the sequences flanking the double-strand break, replication protein A binds. It is subsequently displaced by RAD51, a process that is facilitated by the human breast cancer susceptibility protein BRCA2 [54–56]. The binding of RAD51 to BRCA2 is dependent on the phosphorylation of the BRCA2 C-terminal domain by Chk1. In addition, Chk1 can directly phosphorylate RAD51 and this is also required for recruitment of RAD51 to ssDNA [27, 54]. The loading of BRCA2 and RAD51 on to DNA also prevents further nuclease digestion by Mre11 [57, 58]. RAD51 is then responsible for catalyzing invasion of the ssDNA into the complementary parent strand, creating a primed double-stranded DNA that can be extended. The Holliday junction created by this recombination event is eventually resolved by Mus-81-mediated cleavage and ligation. Depletion of Chk1 results in the loss of RAD51 localization to nuclear foci in response to DNA damage demonstrating the involvement of Chk1 in homologous recombination [27].
Many of the same steps occur when a replication fork stalls due to depletion of deoxyribonucleotides. However, the initial substrate appears to be a regressed replication fork that may already contain ssDNA (Figure 2). For example, if both leading and lagging strands of the replication fork stall simultaneously, the leading strand will always be longer such that a regressed fork could have an extended 3′ end which is required for homologous recombination. In this case, Mre11 nuclease may be dispensable for the creation of ssDNA. However, the nuclease activity may also be required to remove any blocking lesions from the 3′ terminus such as occurs when gemcitabine is incorporated into DNA. During incubation with hydroxyurea, RAD51 foci do not appear immediately after fork arrest, but are detected by 24 h which suggests a delay in the fork regression or the generation of ssDNA [35]. Completion of recombination then appears critical for fork recovery, yet in the continued absence of deoxyribonucleotides the stalled recombination cannot be resolved. As discussed above, inhibition of Chk1 prevents RAD51 loading, and thereby prevents the generation of a recombination intermediate, yet the fork still collapses in a process that has been attributed to Mus81-mediated cleavage [40]. This cleavage likely results from digestion of the Holliday junction that persists at the regressed fork (Figure 2). One question that has not been addressed is the impact of Chk1 inhibition on the stalled recombination after RAD51 has mediated strand invasion. Replication forks still collapse under this circumstance and given the presence of the Holliday junction, it seems likely that Mus81 is still involved.
These data provide further mechanistic evidence to support the concept of combining antimetabolites with Chk1 inhibitors. The inability of cells lacking Chk1 activity to load RAD51 effectively and thus repair regressed replication forks means that cells would be unable to resume replication following the removal of the antimetabolite.
The above discussion highlights many substrates of Chk1. It is likely that a combination of some or all of these substrates is involved in the overall potentiation of gemcitabine and hydroxyurea cytotoxicity upon Chk1 inhibition. Following replication fork stalling, the lack of active Chk1 leads to fork regression through dysregulation of proteins such as Dna2. Homologous recombination is required to rescue the regressed fork. However, Chk1 is also required for loading of BRCA2 and RAD51 meaning that attempts at recombination will fail if Chk1 is inhibited. At this point, the safest thing for the cell to do would be to remain in S phase until the damage can be repaired but the lack of functional Chk1 results in the cells being forced into mitosis with incompletely replicated DNA. All these steps can lead to further DNA damage and cell death.
This discussion also provides an explanation for the greater impact of Chk1 inhibitors when combined with hydroxyurea and gemcitabine than cisplatin or topoisomerase I inhibitors. The latter agents directly damage DNA leading to the activation of Chk1 thereby inducing arrest. In contrast, hydroxyurea and gemcitabine can arrest cells in a manner that induces no direct DNA damage and may not even activate Chk1. The stalled replication can recover rapidly upon removal of the drug. However, while the forks are stalled, inhibition of Chk1 results in the collapse of stalled replication forks and the induction of lethal double-strand breaks.
Chk1 inhibitors kill cells as single agents
Due to the well-established role for Chk1 in cell cycle regulation following DNA damage, Chk1 inhibitors were originally developed and tested as chemo-sensitizing agents. In most cell lines, inhibition of Chk1 alone is well tolerated during short incubations, so despite the fact that it has been known for over a decade that Chk1 is an essential gene in mouse embryonic development [59], it was not generally suspected that cells would be sensitive to Chk1 inhibitors as single agents. It is only recently that the value of Chk1 inhibitors as single agents has been recognized. UCN-01 alone has been shown to induce apoptosis in cortical neurons [60] and the small molecule inhibitor Chk1-A has demonstrated anti-proliferative activity in human cancer cells both in vitro and in xenograft models [61]. We have also shown significant differences in sensitivity to the Chk1 inhibitor MK-8776 across multiple human cancer cell lines [13].
The essential function of Chk1 is poorly understood. Chk1 localizes to the chromatin in normal cycling cells where it is phosphorylated at low levels on ser345 (but not ser317). This phosphorylation is not required for chromatin association of Chk1 but rather prevents nuclear export [62]. Furthermore, phosphorylation of Chk1 has been shown to induce dissociation from chromatin [63–65], questioning whether it functions while un-phosphorylated on DNA or when phosphorylated and dissociated from DNA (or both).
Inhibition of Chk1 results in increased levels of CDC25A, increased CDK activation and a transient increase in the rate of DNA synthesis [66]. However, individual replication forks progress at a much slower rate in Chk1-depleted or inhibited cells [67]. This apparent discrepancy is explained by Chk1-mediated suppression of origin firing in normal S phase. When Chk1 is inhibited, more origins fire but each progress at a slower rate [26]. UCN-01 also reduced fork speed in homologous recombination-deficient cells, demonstrating that the involvement of Chk1 in replication fork progression is unrelated to any role in homologous recombination [67]. By generating site-specific phosphorylation mutations, it has been established that the essential and non-essential functions of Chk1 are regulated through different phosphorylation sites [68]. An S317A mutant was viable but failed to degrade CDC25A and failed to arrest irradiated cells in G2. This mutant also exhibited slower replication fork progression similar to that observed upon inhibition of Chk1. In contrast, attempts to create an S345A-mutant cell line failed suggesting this site is essential for viability. The essential function was associated with mitotic control and appeared to be related to phosphorylation of Chk1 on S345 at the centrosome during an unperturbed mitosis.
We have screened >70 cell lines for sensitivity to the Chk1 inhibitor MK-8776 and have found that about 15% are hypersensitive, dying rapidly when incubated with <1 μm, while the majority of cell lines continued to grow in the presence of 10 μm MK-8776 ([13] and unpublished observations). The hypersensitive cells die rapidly while in S phase suggesting that this phenomenon is not related to the essential function of Chk1 [69]. This cytotoxicity is associated with the rapid induction of both single-strand DNA regions and double-strand breaks and can be prevented by inhibiting CDK1 and CDK2. This suggests the critical action of the Chk1 inhibitor is through activation of CDC25 leading to inappropriate activation of CDK1/2 [66]. The DNA double-strand breaks that subsequently appear are a consequence of cleavage by the Mus81 endonuclease [50]. It was suggested that Chk1 protects replication forks from Mus81 cleavage under normal conditions. We have since demonstrated the involvement of the nuclease activity of Mre11 in generating single-stranded DNA prior to Mus81 cleavage [69]. The link between CDK1/2 and Mre11 activation may be attributed to CDK2-mediated phosphorylation of the Mre11 partner protein CtIP [70]. The reason why Mre11 creates single-stranded DNA in an otherwise unperturbed S phase remains to be determined. Cell lines resistant to Chk1 inhibitors fail to activate CDK1/2 or recruit and activate Mre11 (for example, Mre11-defective cells are resistant) [69]. We propose that the cells hypersensitive to Chk1 inhibitors have a defect in the normal regulation of this pathway and that this may provide a means to target and kill these cells selectively.
Pharmacokinetics and pharmacodynamics inform clinical development
Both the preclinical data and mechanistic studies advocate the use for Chk1 inhibitors in combination with selected antimetabolites (e.g. hydroxyurea or gemcitabine) as potential anticancer therapy. The discussion above addressed phenomena in cell culture. How do these issues play out in a patient? When considering a combination of two drugs, it is necessary to address the pharmacokinetics and pharmacodynamics of both drugs and to define the schedule by which each drug will have the greatest therapeutic impact.
As previously discussed, following replication block, cells continue to accumulate in S phase, but if the block is reversible as it is following administration of hydroxyurea, replication can recover rapidly. Replication forks also need to arrest for more than 12 h before they appear to become fully Chk1 dependent. However, this is not achievable with the routinely used oral dosing of hydroxyurea. Hydroxyurea diffuses freely through the body and plasma concentrations of 1 mm can be achieved. However, its half-life in plasma is 4 h, so concentrations rapidly decrease to below those that cause arrest. One study reported that hydroxyurea administered every 4 h for 72 h was tolerated by patients and the plasma concentration was maintained above 0.5 mm [71]. Whether this administration schedule does cause prolonged S phase arrest in tumours needs to be established as a prelude to combining with a Chk1 inhibitor.
The pharmacokinetics of gemcitabine are very different. Gemcitabine is a pro-drug that must first be metabolized to the activated metabolites dFdCDP and dFdCTP, the former of which inhibits ribonucleotide reductase, while the latter can be incorporated into DNA and induce chain termination. As a consequence, plasma concentrations provide little information, rather one must consider the intracellular concentrations of these metabolites. In a human study with administered doses from 22.5 to 1000 mg m−2, peak dFdCTP concentration (20 μm) in peripheral mononuclear cells was achieved at 350 mg m−2. At the clinically administered dose of 1000 mg m−2, the half-life of dFdCTP was almost 20 h. Furthermore, gemcitabine irreversibly inhibits ribonucleotide reductase meaning that the effects within cells may persist even longer.
In our ongoing research, we have briefly incubated cells with gemcitabine to reflect the bolus treatment that occurs in patients more closely. This permits us to follow the recovery from stalled replication as would occur clinically. Following treatment, the rate of recovery depends on the concentration of gemcitabine, with S arrest lasting for 24 h at low concentrations and 48–72 h at higher concentrations. We have also assessed cell cycle perturbation in human tumour xenografts in mice. Tumours were stained for Ki67 which measures cells at all phases of the cell cycle except Go, and geminin which stains cells in S and G2 phase. Results are expressed as the ratio of geminin : Ki67, that is the ratio of cells in cycle that are in S/G2. In untreated U251 brain tumour xenografts growing in the mouse flank, about 25% of the cells were in S/G2, while administration of 30–150 mg kg−1 gemcitabine (equivalent to approximately 90–450 mg m−2 in humans) showed 80–90% of the cycling cells were arrested in S/G2 24 h after drug administration. We are currently conducting a clinical trial in human bladder cancer to assess the cell cycle perturbation that occurs following administration of a standard dose of 1000 mg m−2 gemcitabine. Initial results show a significant S/G2 arrest 24 h after drug administration. Further experiments are required to assess the persistence of this arrest in both xenograft models and human tumours.
Having established conditions that induce persistent S phase arrest, we next have to consider the pharmacokinetics of the Chk1 inhibitors. Their peak plasma concentration and half-life likely varies considerably, so we will use MK-8776 as the example. A phase I dose escalation study of MK-8776 in patients with solid tumours revealed it to have a half-life of 6.29–9.38 h, but concentrations required to inhibit Chk1 (i.e. ∼1 μm) were attained for only 6 h [72, 73]. Given the continued accumulation of cells in S phase after treatment with gemcitabine or hydroxyurea, and that stalled replication forks are more susceptible to Chk1 inhibition 18 h after the initial replication block, concurrent treatment with MK-8776 would result in inadequate drug remaining at the time it would be most effective (Figure 3). The ideal scheduling for these drug combinations would involve MK-8776 administered at the time that the maximum number of stalled replication forks have become Chk1 dependent but before the cells begin to recover. Our in vitro results have shown that addition of MK-8776 18 h after administration of hydroxyurea [13] and gemcitabine (unpublished observations) is far more effective than concurrent treatment and this time frame is consistent with the gemcitabine-mediated S phase accumulation observed in xenograft models and human bladder cancer. However, even later administration of the Chk1 inhibitor might be indicated if the replication arrest is more persistent in human tumours, or perhaps even multiple dosing of the Chk1 inhibitor as has recently been used in a xenograft model [16]. Clearly, there is a need for further pharmacodynamic assessments to establish the optimum schedule for these drug administrations.
Figure 3.
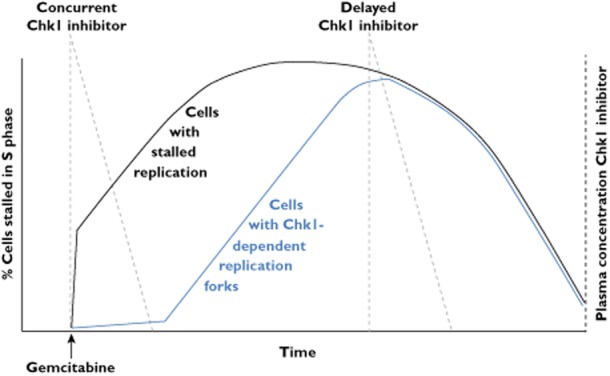
The impact of schedule for the administration of gemcitabine and a Chk1 inhibitor. S phase cells incubated with gemcitabine arrest rapidly. As more cells continue to enter S phase, they also stall leading to a further increase in arrested cells. After an undefined time (at least 24 h in mouse and human experiments), the cells recover and the tumour continues to grow. Stalled replication forks also evolve to become more Chk1 dependent after 12 h, which correlates with the onset of homologous recombination. Maximum cell killing would then occur when the Chk1 inhibitor is administered about 12 h after the time of peak S arrest. This model is based on MK-8776 where an effective plasma concentration is only maintained for about 6 h
The schedule of drug administration that is predicted to have the maximum tumour cell killing may, unfortunately, also elicit increased toxicity to normal tissues, and whether there is any selectivity for the tumour remains to be established. Toxicity would most likely occur in the highly proliferative normal tissues (e.g. bone marrow, gastrointestinal tract), but whether these normal cells also continue to accumulate in S phase following administration of an antimetabolite needs to be determined. If unacceptable toxicity is observed, we have previously shown that activation of the p53 pathway by drugs such as nutlin can protect cells from hydroxyurea plus MK-8776 [74], and this approach has been shown to elicit a systemic and non-toxic induction of the p53 pathway in mice [75]. In addition, we have demonstrated that some cell lines require much lower concentrations of MK-8776 to enhance hydroxyurea-mediated cell killing [13]. These sensitive cell lines are the same ones which are hypersensitive to MK-8776 alone and we are optimistic that some patient tumours will also be hypersensitive to these drug combinations.
Conclusions
Chk1 inhibitors have been explored as chemopotentiating agents since the discovery of UCN-01. Although the efficacy of combinations with DNA damaging agents have been inconsistent, treatment with Chk1 inhibitors has recently emerged as an effective way of sensitizing cancer cells to treatment with the antimetabolites, gemcitabine and hydroxyurea. There is a strong mechanistic basis for this combination and at least four distinct means by which Chk1 protects from replication stress; stabilization of stalled replication forks, control of replication origin firing, mediation of homologous recombination and control of cell cycle progression. It is likely that all of these processes play a role in the sensitization of cells to gemcitabine and hydroxyurea.
It is also critical to consider the impact of drug schedule when combining these agents. Cells continue to accumulate in S phase during replication stress, and stalled replication forks become more Chk1 dependent with time. Chk1 inhibitors will be most effective if administered when the maximum number of cells have arrested in S phase. Our ongoing research is making such determinations in patients receiving gemcitabine.
Finally, and perhaps most exciting, is the observation that some tumours are hypersensitive to Chk1 inhibitors alone. As a consequence, these tumours are likely more responsive to the combination of antimetabolite plus Chk1 inhibitor. It is important to understand the mechanism of this hypersensitivity so that appropriate patients can be selected for study. These patients may have a much greater therapeutic window and much greater benefit from administration of a Chk1 inhibitor.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare no support from any organization for the submitted work, no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years and no other relationships or activities that could appear to have influenced the submitted work.
References
- 1.Rauth AM. Evidence for dark-reactivation of ultraviolet light damage in mouse L cells. Radiat Res. 1967;31:121–138. [PubMed] [Google Scholar]
- 2.Lau CC, Pardee AB. Mechanism by which caffeine potentiates lethality of nitrogen mustard. Proc Natl Acad Sci U S A. 1982;79:2942–2946. doi: 10.1073/pnas.79.9.2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eastman A. Cell cycle checkpoints and their impact on anticancer therapeutic strategies. J Cell Biochem. 2004;91:223–231. doi: 10.1002/jcb.10699. [DOI] [PubMed] [Google Scholar]
- 4.Bunch RT, Eastman A. Enhancement of cisplatin-induced cytotoxicity by 7-hydroxystaurosporine (UCN-01), a new G2-checkpoint inhibitor. Clin Cancer Res. 1996;2:791–797. [PubMed] [Google Scholar]
- 5.Wang Q, Fan S, Eastman A, Worland PJ, Sausville EA, O'Connor PM. UCN-01: a potent abrogator of G2 checkpoint function in cancer cells with disrupted p53. J Natl Cancer Inst. 1996;88:956–965. doi: 10.1093/jnci/88.14.956. [DOI] [PubMed] [Google Scholar]
- 6.Graves PR, Yu L, Schwarz JK, Gales J, Sausville EA, O'Connor PM, Piwnica-Worms H. The Chk1 protein kinase and the Cdc25C regulatory pathways are targets of the anticancer agent UCN-01. J Biol Chem. 2000;275:5600–5605. doi: 10.1074/jbc.275.8.5600. [DOI] [PubMed] [Google Scholar]
- 7.Busby EC, Leistritz DF, Abraham RT, Karnitz LM, Sarkaria JN. The radiosensitizing agent 7-hydroxystaurosporine (UCN-01) inhibits the DNA damage checkpoint kinase hChk1. Cancer Res. 2000;60:2108–2112. [PubMed] [Google Scholar]
- 8.Fuse E, Tanii H, Kurata N, Kobayashi H, Shimada Y, Tamura T, Sasaki Y, Tanigawara Y, Lush RD, Headlee D, Figg WD, Arbuck SG, Senderowicz AM, Sausville EA, Akinaga S, Kuwabara T, Kobayashi S. Unpredicted clinical pharmacology of UCN-01 caused by specific binding to human alpha1-acid glycoprotein. Cancer Res. 1998;58:3248–3253. [PubMed] [Google Scholar]
- 9.Thompson R, Meuth M, Woll P, Zhu Y, Danson S. Treatment with the Chk1 inhibitor Go6976 enhances cisplatin cytotoxicity in SCLC cells. Int J Oncol. 2012;40:194–202. doi: 10.3892/ijo.2011.1187. [DOI] [PubMed] [Google Scholar]
- 10.Blasina A, Hallin J, Chen E, Arango ME, Kraynov E, Register J, Grant S, Ninkovic S, Chen P, Nichols T, O'Connor P, Anderes K. Breaching the DNA damage checkpoint via PF-00477736, a novel small-molecule inhibitor of checkpoint kinase 1. Mol Cancer Ther. 2008;7:2394–2404. doi: 10.1158/1535-7163.MCT-07-2391. [DOI] [PubMed] [Google Scholar]
- 11.Zenvirt S, Kravchenko-Balasha N, Levitzki A. Status of p53 in human cancer cells does not predict efficacy of CHK1 kinase inhibitors combined with chemotherapeutic agents. Oncogene. 2010;29:6149–6159. doi: 10.1038/onc.2010.343. [DOI] [PubMed] [Google Scholar]
- 12.Wagner JM, Karnitz LM. Cisplatin-induced DNA damage activates replication checkpoint signaling components that differentially affect tumor cell survival. Mol Pharmacol. 2009;76:208–214. doi: 10.1124/mol.109.055178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Montano R, Chung I, Garner KM, Parry D, Eastman A. Preclinical development of the novel Chk1 inhibitor SCH900776 in combination with DNA-damaging agents and antimetabolites. Mol Cancer Ther. 2012;11:427–438. doi: 10.1158/1535-7163.MCT-11-0406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tse AN, Rendahl KG, Sheikh T, Cheema H, Aardalen K, Embry M, Ma S, Moler EJ, Ni ZJ, Lopes de Menezes DE, Hibner B, Gesner TG, Schwartz GK. CHIR-124, a novel potent inhibitor of Chk1, potentiates the cytotoxicity of topoisomerase I poisons in vitro and in vivo. Clin Cancer Res. 2007;13:591–602. doi: 10.1158/1078-0432.CCR-06-1424. [DOI] [PubMed] [Google Scholar]
- 15.Zabludoff SD, Deng C, Grondine MR, Sheehy AM, Ashwell S, Caleb BL, Green S, Haye HR, Horn CL, Janetka JW, Liu D, Mouchet E, Ready S, Rosenthal JL, Queva C, Schwartz GK, Taylor KJ, Tse AN, Walker GE, White AM. AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol Cancer Ther. 2008;7:2955–2966. doi: 10.1158/1535-7163.MCT-08-0492. [DOI] [PubMed] [Google Scholar]
- 16.Ma CX, Cai S, Li S, Ryan CE, Guo Z, Schaiff WT, Lin L, Hoog J, Goiffon RJ, Prat A, Aft RL, Ellis MJ, Piwnica-Worms H. Targeting Chk1 in p53-deficient triple-negative breast cancer is therapeutically beneficial in human-in-mouse tumor models. J Clin Invest. 2012;122:1541–1552. doi: 10.1172/JCI58765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walton MI, Eve PD, Hayes A, Valenti M, De Haven Brandon A, Box G, Boxall KJ, Aherne GW, Eccles SA, Raynaud FI, Williams DH, Reader JC, Collins I, Garrett MD. The preclinical pharmacology and therapeutic activity of the novel CHK1 inhibitor SAR-020106. Mol Cancer Ther. 2010;9:89–100. doi: 10.1158/1535-7163.MCT-09-0938. [DOI] [PubMed] [Google Scholar]
- 18.Matthews DJ, Yakes FM, Chen J, Tadano M, Bornheim L, Clary DO, Tai A, Wagner JM, Miller N, Kim YD, Robertson S, Murray L, Karnitz LM. Pharmacological abrogation of S-phase checkpoint enhances the anti-tumor activity of gemcitabine in vivo. Cell Cycle. 2007;6:104–110. doi: 10.4161/cc.6.1.3699. [DOI] [PubMed] [Google Scholar]
- 19.Garrett MD, Collins I. Anticancer therapy with checkpoint inhibitors: what, where and when? Trends Pharmacol Sci. 2011;32:308–316. doi: 10.1016/j.tips.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 20.Geng L, Huehls AM, Wagner JM, Huntoon CJ, Karnitz LM. Checkpoint signaling, base excision repair, and PARP promote survival of colon cancer cells treated with 5-fluorodeoxyuridine but not 5-fluorouracil. PLoS ONE. 2011;6:e28862. doi: 10.1371/journal.pone.0028862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fujinaka Y, Matsuoka K, Iimori M, Tuul M, Sakasai R, Yoshinaga K, Saeki H, Morita M, Kakeji Y, Gillespie DA, Yamamoto K, Takata M, Kitao H, Maehara Y. ATR-Chk1 signaling pathway and homologous recombinational repair protect cells from 5-fluorouracil cytotoxicity. DNA Repair (Amst) 2012;11:247–258. doi: 10.1016/j.dnarep.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 22.Robinson HM, Jones R, Walker M, Zachos G, Brown R, Cassidy J, Gillespie DA. Chk1-dependent slowing of S-phase progression protects DT40 B-lymphoma cells against killing by the nucleoside analogue 5-fluorouracil. Oncogene. 2006;25:5359–5369. doi: 10.1038/sj.onc.1209532. [DOI] [PubMed] [Google Scholar]
- 23.Xiao Z, Xue J, Sowin TJ, Rosenberg SH, Zhang H. A novel mechanism of checkpoint abrogation conferred by Chk1 downregulation. Oncogene. 2005;24:1403–1411. doi: 10.1038/sj.onc.1208309. [DOI] [PubMed] [Google Scholar]
- 24.McNeely S, Conti C, Sheikh T, Patel H, Zabludoff S, Pommier Y, Schwartz G, Tse A. Chk1 inhibition after replicative stress activates a double strand break response mediated by ATM and DNA-dependent protein kinase. Cell Cycle. 2010;9:995–1004. doi: 10.4161/cc.9.5.10935. [DOI] [PubMed] [Google Scholar]
- 25.Scorah J, McGowan CH. Claspin and Chk1 regulate replication fork stability by different mechanisms. Cell Cycle. 2009;8:1036–1043. doi: 10.4161/cc.8.7.8040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petermann E, Woodcock M, Helleday T. Chk1 promotes replication fork progression by controlling replication initiation. Proc Natl Acad Sci U S A. 2010;107:16090–16095. doi: 10.1073/pnas.1005031107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bahassi EM, Ovesen JL, Riesenberg AL, Bernstein WZ, Hasty PE, Stambrook PJ. The checkpoint kinases Chk1 and Chk2 regulate the functional associations between hBRCA2 and Rad51 in response to DNA damage. Oncogene. 2008;27:3977–3985. doi: 10.1038/onc.2008.17. [DOI] [PubMed] [Google Scholar]
- 28.Lindqvist A, Rodriguez-Bravo V, Medema RH. The decision to enter mitosis: feedback and redundancy in the mitotic entry network. J Cell Biol. 2009;185:193–202. doi: 10.1083/jcb.200812045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O'Connell MJ, Raleigh JM, Verkade HM, Nurse P. Chk1 is a wee1 kinase in the G2 DNA damage checkpoint inhibiting cdc2 by Y15 phosphorylation. EMBO J. 1997;16:545–554. doi: 10.1093/emboj/16.3.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boutros R, Lobjois V, Ducommun B. CDC25 phosphatases in cancer cells: key players? Good targets? Nat Rev Cancer. 2007;7:495–507. doi: 10.1038/nrc2169. [DOI] [PubMed] [Google Scholar]
- 31.Aarts M, Sharpe R, Garcia-Murillas I, Gevensleben H, Hurd MS, Shumway SD, Toniatti C, Ashworth A, Turner NC. Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov. 2012;2:524–539. doi: 10.1158/2159-8290.CD-11-0320. [DOI] [PubMed] [Google Scholar]
- 32.Niida H, Tsuge S, Katsuno Y, Konishi A, Takeda N, Nakanishi M. Depletion of Chk1 leads to premature activation of Cdc2-cyclin B and mitotic catastrophe. J Biol Chem. 2005;280:39246–39252. doi: 10.1074/jbc.M505009200. [DOI] [PubMed] [Google Scholar]
- 33.Berezney R, Dubey DD, Huberman JA. Heterogeneity of eukaryotic replicons, replicon clusters, and replication foci. Chromosoma. 2000;108:471–484. doi: 10.1007/s004120050399. [DOI] [PubMed] [Google Scholar]
- 34.Blow JJ, Ge XQ. Replication forks, chromatin loops and dormant replication origins. Genome Biol. 2008;9:244. doi: 10.1186/gb-2008-9-12-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell. 2010;37:492–502. doi: 10.1016/j.molcel.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lou H, Komata M, Katou Y, Guan Z, Reis CC, Budd M, Shirahige K, Campbell JL. Mrc1 and DNA polymerase epsilon function together in linking DNA replication and the S phase checkpoint. Mol Cell. 2008;32:106–117. doi: 10.1016/j.molcel.2008.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Atkinson J, McGlynn P. Replication fork reversal and the maintenance of genome stability. Nucleic Acids Res. 2009;37:3475–3492. doi: 10.1093/nar/gkp244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cotta-Ramusino C, Fachinetti D, Lucca C, Doksani Y, Lopes M, Sogo J, Foiani M. Exo1 processes stalled replication forks and counteracts fork reversal in checkpoint-defective cells. Mol Cell. 2005;17:153–159. doi: 10.1016/j.molcel.2004.11.032. [DOI] [PubMed] [Google Scholar]
- 39.Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, Cortez D. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 2011;25:1320–1327. doi: 10.1101/gad.2053211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hanada K, Budzowska M, Davies SL, van Drunen E, Onizawa H, Beverloo HB, Maas A, Essers J, Hickson ID, Kanaar R. The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat Struct Mol Biol. 2007;14:1096–1104. doi: 10.1038/nsmb1313. [DOI] [PubMed] [Google Scholar]
- 41.Khanna KK, Shiloh Y. The DNA Damage Response: Implications on Cancer Formation and Treatment. Dordrecht, New York: Springer; 2009. [Google Scholar]
- 42.Lopes M, Cotta-Ramusino C, Pellicioli A, Liberi G, Plevani P, Muzi-Falconi M, Newlon CS, Foiani M. The DNA replication checkpoint response stabilizes stalled replication forks. Nature. 2001;412:557–561. doi: 10.1038/35087613. [DOI] [PubMed] [Google Scholar]
- 43.Desany BA, Alcasabas AA, Bachant JB, Elledge SJ. Recovery from DNA replicational stress is the essential function of the S-phase checkpoint pathway. Genes Dev. 1998;12:2956–2970. doi: 10.1101/gad.12.18.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sogo JM, Lopes M, Foiani M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science. 2002;297:599–602. doi: 10.1126/science.1074023. [DOI] [PubMed] [Google Scholar]
- 45.Hu J, Sun L, Shen F, Chen Y, Hua Y, Liu Y, Zhang M, Hu Y, Wang Q, Xu W, Sun F, Ji J, Murray JM, Carr AM, Kong D. The intra-S phase checkpoint targets Dna2 to prevent stalled replication forks from reversing. Cell. 2012;149:1221–1232. doi: 10.1016/j.cell.2012.04.030. [DOI] [PubMed] [Google Scholar]
- 46.Bansbach CE, Betous R, Lovejoy CA, Glick GG, Cortez D. The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev. 2009;23:2405–2414. doi: 10.1101/gad.1839909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Betous R, Mason AC, Rambo RP, Bansbach CE, Badu-Nkansah A, Sirbu BM, Eichman BF, Cortez D. SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 2012;26:151–162. doi: 10.1101/gad.178459.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Froget B, Blaisonneau J, Lambert S, Baldacci G. Cleavage of stalled forks by fission yeast Mus81/Eme1 in absence of DNA replication checkpoint. Mol Biol Cell. 2008;19:445–456. doi: 10.1091/mbc.E07-07-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kai M, Boddy MN, Russell P, Wang TS. Replication checkpoint kinase Cds1 regulates Mus81 to preserve genome integrity during replication stress. Genes Dev. 2005;19:919–932. doi: 10.1101/gad.1304305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Forment JV, Blasius M, Guerini I, Jackson SP. Structure-specific DNA endonuclease Mus81/Eme1 generates DNA damage caused by Chk1 inactivation. PLoS ONE. 2011;6:e23517. doi: 10.1371/journal.pone.0023517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mimitou EP, Symington LS. DNA end resection – unraveling the tail. DNA Repair (Amst) 2011;10:344–348. doi: 10.1016/j.dnarep.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen X, Niu H, Chung WH, Zhu Z, Papusha A, Shim EY, Lee SE, Sung P, Ira G. Cell cycle regulation of DNA double-strand break end resection by Cdk1-dependent Dna2 phosphorylation. Nat Struct Mol Biol. 2011;18:1015–1019. doi: 10.1038/nsmb.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peng G, Dai H, Zhang W, Hsieh HJ, Pan MR, Park YY, Tsai RY, Bedrosian I, Lee JS, Ira G, Lin SY. Human nuclease/helicase DNA2 alleviates replication stress by promoting DNA end resection. Cancer Res. 2012;72:2802–2813. doi: 10.1158/0008-5472.CAN-11-3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sorensen CS, Hansen LT, Dziegielewski J, Syljuasen RG, Lundin C, Bartek J, Helleday T. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol. 2005;7:195–201. doi: 10.1038/ncb1212. [DOI] [PubMed] [Google Scholar]
- 55.Thompson LH. Recognition, signaling, and repair of DNA double-strand breaks produced by ionizing radiation in mammalian cells: the molecular choreography. Mutat Res. 2012;751:158–246. doi: 10.1016/j.mrrev.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 56.Errico A, Costanzo V. Differences in the DNA replication of unicellular eukaryotes and metazoans: known unknowns. EMBO Rep. 2010;11:270–278. doi: 10.1038/embor.2010.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hashimoto Y, Ray Chaudhuri A, Lopes M, Costanzo V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol. 2010;17:1305–1311. doi: 10.1038/nsmb.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ying S, Hamdy FC, Helleday T. Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res. 2012;72:2814–2821. doi: 10.1158/0008-5472.CAN-11-3417. [DOI] [PubMed] [Google Scholar]
- 59.Takai H, Tominaga K, Motoyama N, Minamishima YA, Nagahama H, Tsukiyama T, Ikeda K, Nakayama K, Nakanishi M. Aberrant cell cycle checkpoint function and early embryonic death in Chk1(-/-) mice. Genes Dev. 2000;14:1439–1447. [PMC free article] [PubMed] [Google Scholar]
- 60.Ye W, Blain SW. Chk1 has an essential role in the survival of differentiated cortical neurons in the absence of DNA damage. Apoptosis. 2011;16:449–459. doi: 10.1007/s10495-011-0579-z. [DOI] [PubMed] [Google Scholar]
- 61.Davies KD, Humphries MJ, Sullivan FX, von Carlowitz I, Le Huerou Y, Mohr PJ, Wang B, Blake JF, Lyon MA, Gunawardana I, Chicarelli M, Wallace E, Gross S. Single-agent inhibition of Chk1 is antiproliferative in human cancer cell lines in vitro and inhibits tumor xenograft growth in vivo. Oncol Res. 2011;19:349–363. doi: 10.3727/096504011x13079697132961. [DOI] [PubMed] [Google Scholar]
- 62.Jiang K, Pereira E, Maxfield M, Russell B, Goudelock DM, Sanchez Y. Regulation of Chk1 includes chromatin association and 14-3-3 binding following phosphorylation on Ser-345. J Biol Chem. 2003;278:25207–25217. doi: 10.1074/jbc.M300070200. [DOI] [PubMed] [Google Scholar]
- 63.Smits VA, Reaper PM, Jackson SP. Rapid PIKK-dependent release of Chk1 from chromatin promotes the DNA-damage checkpoint response. Curr Biol. 2006;16:150–159. doi: 10.1016/j.cub.2005.11.066. [DOI] [PubMed] [Google Scholar]
- 64.Zhang YW, Otterness DM, Chiang GG, Xie W, Liu YC, Mercurio F, Abraham RT. Genotoxic stress targets human Chk1 for degradation by the ubiquitin-proteasome pathway. Mol Cell. 2005;19:607–618. doi: 10.1016/j.molcel.2005.07.019. [DOI] [PubMed] [Google Scholar]
- 65.Smits VA. Spreading the signal: dissociation of Chk1 from chromatin. Cell Cycle. 2006;5:1039–1043. doi: 10.4161/cc.5.10.2761. [DOI] [PubMed] [Google Scholar]
- 66.Syljuasen RG, Sorensen CS, Hansen LT, Fugger K, Lundin C, Johansson F, Helleday T, Sehested M, Lukas J, Bartek J. Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol Cell Biol. 2005;25:3553–3562. doi: 10.1128/MCB.25.9.3553-3562.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Petermann E, Maya-Mendoza A, Zachos G, Gillespie DA, Jackson DA, Caldecott KW. Chk1 requirement for high global rates of replication fork progression during normal vertebrate S phase. Mol Cell Biol. 2006;26:3319–3326. doi: 10.1128/MCB.26.8.3319-3326.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wilsker D, Petermann E, Helleday T, Bunz F. Essential function of Chk1 can be uncoupled from DNA damage checkpoint and replication control. Proc Natl Acad Sci U S A. 2008;105:20752–20757. doi: 10.1073/pnas.0806917106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thompson R, Montano R, Eastman A. The mre11 nuclease is critical for the sensitivity of cells to chk1 inhibition. PLoS ONE. 2012;7:e44021. doi: 10.1371/journal.pone.0044021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huertas P, Jackson SP. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J Biol Chem. 2009;284:9558–9565. doi: 10.1074/jbc.M808906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Belt RJ, Haas CD, Kennedy J, Taylor S. Studies of hydroxyurea administered by continuous infusion: toxicity, pharmacokinetics, and cell synchronization. Cancer. 1980;46:455–462. doi: 10.1002/1097-0142(19800801)46:3<455::aid-cncr2820460306>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 72.Daud A, Springett GM, Mendelson DS, Munster PN, Goldman JR, Strosberg JR, Kato G, Nesheiwat T, Isaacs R, Rosen LS. A Phase I dose-escalation study of SCH 900776, a selective inhibitor of checkpoint kinase 1 (CHK1), in combination with gemcitabine (Gem) in subjects with advanced solid tumors. J Clin Oncol. 2010;28(Suppl):abstract 3064. [Google Scholar]
- 73.Karp JE, Thomas BM, Greer JM, Sorge C, Gore SD, Pratz KW, Smith BD, Flatten KS, Peterson K, Schneider P, Mackey K, Freshwater T, Levis MJ, McDevitt MA, Carraway HE, Gladstone DE, Showel MM, Loechner S, Parry DA, Horowitz JA, Isaacs R, Kaufmann SH. Phase I and pharmacologic trial of cytosine arabinoside with the selective checkpoint 1 inhibitor SCH 900776 in refractory acute leukemias. Clin Cancer Res. 2012;18:6723–6731. doi: 10.1158/1078-0432.CCR-12-2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chang LJ, Eastman A. Differential regulation of p21 (waf1) protein half-life by DNA damage and Nutlin-3 in p53 wild-type tumors and its therapeutic implications. Cancer Biol Ther. 2012;13:1047–1057. doi: 10.4161/cbt.21047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shangary S, Qin D, McEachern D, Liu M, Miller RS, Qiu S, Nikolovska-Coleska Z, Ding K, Wang G, Chen J, Bernard D, Zhang J, Lu Y, Gu Q, Shah RB, Pienta KJ, Ling X, Kang S, Guo M, Sun Y, Yang D, Wang S. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Natl Acad Sci U S A. 2008;105:3933–3938. doi: 10.1073/pnas.0708917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sausville EA, LoRusso P, Carducci MA, Barker PN, Agbo F, Oakes P, Senderowicz AM. Phase I dose-escalation study of AZD7762 in combination with gemcitabine (gem) in patients (pts) with advanced solid tumors. J Clin Oncol. 2011;29(Suppl):abstr 3058. [Google Scholar]
- 77.Ho AL, Bendell JC, Cleary JM, Schwartz GK, Burris HA, Oakes P, Agbo F, Barker PN, Senderowicz AM, Shapiro G. Phase I, open-label, dose-escalation study of AZD7762 in combination with irinotecan (irino) in patients (pts) with advanced solid tumors. J Clin Oncol. 2011;29(Suppl):abstr 3033. [Google Scholar]
- 78.Seto T, Esaki T, Hirai F, Arita S, Nosaki K, Makiyama A, Kometani T, Fujimoto C, Hamatake M, Takeoka H, Agbo F, Shi X. Phase I dose-escalation study of AZD7762 alone and in combination with gemcitabine in Japanese patients with advanced solid tumors. J Clin Oncol. 2012;30 doi: 10.1007/s00280-013-2234-6. (Suppl.): abst 3045. [DOI] [PubMed] [Google Scholar]
- 79.Brega N, McArthur GA, Britten C, Wong SG, Wang E, Wilner KD, Blasina A, Schwartz GK, Gallo J, Tse AN. Phase I clinical trial of gemcitabine (GEM) in combination with PF-00477736 (PF-736), a selective inhibitor of CHK1 kinase. J Clin Oncol. 2010;28(Suppl):abstract 3062. [Google Scholar]
- 80.Syljuasen RG, Sorensen CS, Nylandsted J, Lukas C, Lukas J, Bartek J. Inhibition of Chk1 by CEP-3891 accelerates mitotic nuclear fragmentation in response to ionizing radiation. Cancer Res. 2004;64:9035–9040. doi: 10.1158/0008-5472.CAN-04-2434. [DOI] [PubMed] [Google Scholar]
- 81.Walton MI, Eve PD, Hayes A, Valenti MR, De Haven Brandon AK, Box G, Hallsworth A, Smith EL, Boxall KJ, Lainchbury M, Matthews TP, Jamin Y, Robinson SP, Aherne GW, Reader JC, Chesler L, Raynaud FI, Eccles SA, Collins I, Garrett MD. CCT244747 is a novel potent and selective CHK1 inhibitor with oral efficacy alone and in combination with genotoxic anticancer drugs. Clin Cancer Res. 2012;18:5650–5661. doi: 10.1158/1078-0432.CCR-12-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]