

Abstract
We compared and determined the reasons for any differences in the review and approval times of tyrosine kinase inhibitors (TKIs) by the US Food and Drug Administration (FDA) and the European EMA/CHMP. Applications for these novel cancer drugs were submitted to them within a mean of 31.2 days of each other, providing a fair basis for comparison. The FDA had granted priority review to 12 TKIs but the EMA/CHMP did not grant the equivalent accelerated assessment to any. The FDA granted accelerated approvals to six (38%) and CHMP granted (the equivalent) conditional approvals to four (29%) of these agents. On average, the review and approval times were 205.3 days in the US compared with 409.6 days in the European Union (EU). The active review times, however, were comparable (225.4 days in the EU and 205.3 days in the US). Since oncology drug development lasts about 7 years, the 20 days difference in review times between the two agencies is inconsequential. Clock stops during review and the time required to issue an approval had added the extra 184.2 days to review time in the EU. We suggest possible solutions to expedite the EU review and approval processes. However, post-marketing emergence of adverse efficacy and safety data on gefitinib and lapatinib, respectively, indicate potential risks of expedited approvals. We challenge the widely prevalent myth that early approval translates into early access or beneficial impact on public health. Both the agencies collaborate closely but conduct independent assessments and make decisions based on distinct legislation, procedures, precedents and societal expectations.
Keywords: accelerated approval, conditional approval, EMA, FDA, priority review, tyrosine kinase inhibitors
Introduction
A number of recent studies have compared the drug review and approval performances of the US Food and Drug Administration (FDA) and the European Union's European Medicines Agency (EMA) and its expert advisory Committee for Medicinal Products for Human Use (CHMP) [1–5]. In general, these studies have focused on comparing the timelines of these two authorities without much regard to (i) the justifiable differences in regional legislations which in turn require different procedures and (ii) the impact, if any, of these differences on public health in the two regions. These comparisons are prompted by an underlying assumption that rapid review and approval of drugs, especially the novel ones, bring about better outcomes in terms of public health. Downing et al. [1] concluded that novel therapeutic agents approved between 2001 and 2010 were, on average, reviewed more quickly by the FDA than by the EMA or Health Canada, and that the vast majority of these agents were first approved for use in the US. These investigators later confirmed that their analysis had focused on regulatory review times and compared the time from submission of the application to market approval [2].
Specifically as it concerns oncology drugs, Trotta et al. [3] compared the approaches of the EMA and the FDA in the evaluation and approval of new anticancer indications and considered any possible clinical implications associated with the differences between the two agencies. They found that overall, 42 anticancer drugs were approved by the EMA between 1995 and 2008, corresponding to a total of 100 indications [3]. In 47 of these 100 indications, a difference was found. For 19 of these 47 indications, the difference was that one agency had approved an indication (16 by the FDA and three by the EMA), whereas the other agency had not. In the other 28 instances, the indication description had different wordings. The FDA was more restrictive in 13 of these 28 instances whereas the EMA was more restrictive in the remaining 15 cases. In 10 of these instances, these investigators considered the differences in therapeutic indications approved by the two agencies to be clinically relevant. Interestingly, they found an overall trend that suggested that the agency that was second in approving was usually more restrictive in the wording of the indication compared with the agency that provided approval first [3]. Roberts et al. [4] conducted a direct drug to drug comparison of the two regulatory agencies’ approvals of new oncology drugs and their subsequent market entry. They identified 35 new oncology drugs that were approved by either the FDA (n = 32) or the EMA (n = 26) in the period 2003–2010 and reported that the median time for approval for new cancer medicines in the US was just 6 months and that these new anticancer medicines were typically available in the US before they were in Europe. According to a study completed by The Tufts Centre for the Study of Drug Development, 40 oncology drugs received marketing approval in the US, compared with 30 in Europe, between 2000 and 2011 [5] and the approval times in the European Union (EU) were 27% shorter for non-oncology drugs, but 54% longer for oncology drugs, than similar approvals in the US [6]. The report also drew attention to an interesting finding that in both regions, there was little difference in approval times between products that had a special review designation, such as fast track, accelerated approval and orphan designation, and those that did not [6].
The introduction of small molecule tyrosine kinase inhibitors (TKIs) into clinical oncology over the last decade has transformed the treatment of certain forms of cancer. Since the approval of the first tyrosine kinase inhibitor, imatinib, in 2001, additional TKIs have been approved by both agencies, 15 by the FDA and 13 by the EMA as of 30 September 2012, and a large number of others are in development or under regulatory review (Shah RR, Morganroth J, Shah DR, unpublished data). The survey by Roberts et al. [4] had included only seven TKIs. For all TKIs approved by one or both agencies, we have analyzed the timelines of their review and approval with a focus on the reasons for any differences between them, which have not received sufficient consideration previously. We purposely restricted our comparison of the two agencies to the review and approval of TKIs for three reasons. Firstly, they are perceived as a major development in oncology therapeutics, secondly, they have all been reviewed over the last decade and therefore, provide a basis for the comparison of recent performances and finally, the majority of the applications were submitted to the two agencies within 2–3 months of each other, thus enabling a better comparison of the review of essentially the same data.
Both the agencies are active participants of and signatories to the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH), with the aim of setting common standards for data requirements. However, given the significant differences in the legislation that govern, and the consequential differences in review procedures of, the two agencies, we begin by describing the review procedures of the two agencies and explain why the system in the EU is more complex than that in the US. Although both agencies require a similar set of data and apply comparable and rigorous standards to document safety and efficacy, understanding the two systems of drug approval is the key to understanding the review timelines and any comparisons between them.
Approval procedures of the FDA and the EMA
Reasons for the complexity of the EU system
While the FDA is a federal agency that has a legally mandated authority to regulate drugs in the entire US, the EMA is a decentralized agency of the EU which, among other tasks, coordinates various expert committees that advise the European Commission (EC) on matters concerning medicinal products. It is the EC that is legally mandated to issue enforceable decisions, binding on all the 27 Member States (MS) of the EU. Each MS nominates an independent expert to the CHMP to provide an opinion and comments to the CHMP on the safety, efficacy and risk/benefit of the human medicinal product under review. The views and opinions of the CHMP members are exchanged and co-ordinated at the CHMP meetings with a view to building a consensus towards an all-inclusive CHMP opinion. Because this can only be done during the plenary meetings of the CHMP which meets monthly (except August), the review procedure is so planned that the various milestones of the centralized review procedure (described below) correspond to the dates of these monthly meetings.
Whether positive or negative, the CHMP opinion is communicated to the applicant and to the EC to issue a binding decision. The EC, before releasing the final decision, forwards a draft decision to a regulatory committee called the Standing Committee on Medicinal Products for Human Use where representatives of the MS have 30 days to raise objections. If important objections are raised, the CHMP is asked to formulate a reply, and a new Standing Committee procedure is started based on the CHMP answer. If the Standing Committee's opinion is favourable, the Commission proceeds with the decision-making process. For new EU authorizations, the overall duration from the CHMP opinion to the EC decision should not exceed 67 days, according to Article 10 of Regulation (EC) no. 726/2004 [7]. Inevitably, the system in the EU, compared with the system at the FDA, has to be more complex, given that EC is accountable to the citizens of the entire Union of sovereign MS. While it has the drawback of some potential delay, it also has the advantage of an application receiving a detailed and rigorous scrutiny through different perspectives of the 27 expert members of the CHMP.
Review of a new drug application by the FDA
The details of the review process are described elsewhere [8] and summarized below in brief. After a New Drug Application (NDA), or Biologics Licensing Application (BLA), is received by the agency, it is checked (validation) to ensure that sufficient data and information have been submitted to justify the initiation of its formal review. Incomplete applications are not filed for review and receive a formal ‘refusal to file’ notification.
Once an NDA is filed, an FDA review team is established to evaluate the drug for its safety and efficacy in the proposed indication and to show that the benefits of the drug appear to outweigh the risks. In 1992, Congress passed the Prescription Drug User Fee Act (PDFUA), which is re-authorized every 5 years. In compliance with the PDUFA, the FDA is expected to review and act on at least 90% of NDAs for standard drugs no later than 10 months after the applications are received [9, 10]. A regulatory project manager (RPM) is assigned to keep drug sponsors informed of the internal review timeline and to communicate any changes in this timeline. If the review team determines that a decision will not be made by the target date, the RPM will inform the sponsor and discuss relevant issues. If a sponsor submits a ‘major amendment’ (such as a major new study) within the last 3 months of the review, the official review clock may be extended for an additional 3 months.
Each reviewer prepares a written evaluation of the application and makes recommendations regarding whether or not the new drug should be approved and whether or not additional studies may be needed. If there are particular concerns with the choice of endpoint or trial design, troubling safety signals, unclear risk/benefit ratio, or the potential need for further studies, the FDA may call on advisory committees made up of outside experts [11, 12]. The FDA often takes the advice of advisory committees, but is not required to do so.
If it is decided that the benefits of a drug outweigh the risks for the proposed indication, the drug will receive approval for that indication and can be marketed in the US. If it is decided that the drug can probably be approved provided some issues are first resolved, it will received a designation of ‘approvable’, a decision that may necessitate the submission of a major amendment by the sponsor, and therefore an extension of the review time. Finally, if there are significant concerns or deficiencies with an NDA, the FDA will decide that the drug is ‘not approvable’ and provide the justification for its decision.
Provisions for special approvals
In order to expedite the development and availability of certain types of drugs that treat serious diseases, the FDA has developed three distinct approaches to making such drugs available as rapidly as possible. These are Priority review, Accelerated approval, and Fast track. Despite all these approaches implying speed, there are distinctions between them. An investigational agent can potentially be eligible for only one or a combination of these approaches
Fast track
Fast track designation is a process designed to facilitate the development, and expedite the review of drugs to treat serious diseases and fill an unmet medical need (providing a therapy where none exists or which may be potentially superior to existing therapy). Fast track designation can be granted at any time during the drug development process, and entails more frequent interactions between the FDA and the drug sponsor, and a rolling review of data as they accumulate, features intended to improve the efficiency of development by allowing the FDA to take a more active role in advising the drug sponsor.
Accelerated approval
The Accelerated approval pathway was created in 1992, allowing early approval on the basis of an improvement on a surrogate endpoint, such as decreased tumour burden, that is considered reasonably likely to predict a real clinical benefit, such as improved survival or quality of life. Because measuring true clinical benefits such as overall survival can take years, allowing earlier approval based on a surrogate endpoint can significantly expedite the time to approval. However, Accelerated approval is conditional in that post-marketing clinical trials are required to verify the anticipated clinical benefit. If these trials confirm the predicted clinical benefit, the Accelerated approval is converted into regular approval. If they do not, the drug may be removed from the market.
Priority review
Priority review shortens the regulatory review time from ten months to six months. This designation is given to drugs that are expected to offer major advances in treatment, or to provide a treatment where no adequate therapy exists. Unlike Fast track and Accelerated approval, Priority review is not restricted to drugs for serious diseases only. Priority review does not diminish or alter the quality of evidence necessary or the standards for approval.
Review of new drug application in the EU
Depending on the therapeutic class of the drug and the commercial strategies of the sponsor, three procedures (leaving aside national procedures for local authorizations only) are available for EU-wide approval of medicinal products. These are:
Centralized procedure.
Mutual recognition procedure.
Decentralized procedure.
These procedures are described in detail elsewhere [13–15].
Regulation (EC) no. 726/2004 [16] requires applications for all oncology drugs after 2005 to be submitted to the EMA and evaluated through the centralized procedure. A successful application under the EU (centralized) procedure delivers a single marketing authorization (a single decision from the EC) for a medicinal product, valid throughout the EU under a single trade name and a common Summary of Product Characteristics (SmPC, the EU equivalent of the US drug label), package leaflet and labelling. Legislation requires the procedure to be concluded within a maximum of 210 days by a scientific opinion from the CHMP, followed by a binding decision from the EC. At the conclusion of the procedure, the applicant ends up with either an approval to market the product in all MS or a refusal to market the product in any MS of the EU.
Following validation of a centralized application (within 20 days of receipt), it is presented to the CHMP for initiating a formal review. The CHMP appoints one of its members to act as a rapporteur for the coordination of the evaluation, and also a second member to act as a co-rapporteur. The CHMP is also assisted as required by a number of statutory Scientific Advisory Groups (SAGs), each composed of independent external academic experts in a designated therapeutic area, and EMA expert Working Parties which are made up of reviewers from national agencies. SAGs may be consulted by the CHMP on a whole range of general and specific issues and are therefore, loosely comparable to the FDA advisory committees.
Both the rapporteur and the co-rapporteur circulate their separate assessment reports to the CHMP members by day 80 from the start of the procedure. The CHMP members provide their comments and following plenary discussion, a final consolidated list of questions is agreed by the CHMP on day 120 and communicated to the applicant. The procedure clock is stopped, usually for up to 3 months but with a possibility to extend by a further 3 months maximum. This consolidated list of questions includes any major objections, points for clarification and changes to the SmPC/ Risk Management Plan.
On receipt of the responses from the applicant, the clock is re-started (day 121) and the rapporteur and co-rapporteur prepare a joint assessment report of the responses by day 150. Any issue(s) identified are discussed on day 180 during the plenary CHMP meeting and a decision may be made on whether to issue a positive CHMP opinion. If there still are any outstanding or unresolved issues, the clock is stopped again. Applicants normally respond within 1 month but in exceptional circumstances, a further extension of 1 month (or maximum 2 months) may be granted if justified by the applicant and agreed by the CHMP. Day 181 is the re-start of the clock when the response is assessed and an oral explanation may take place. On or before day 210, the CHMP adopts its opinion. The opinion requires a consensus or an absolute majority from its members. The deadline for adopting an opinion and finalization of the CHMP Assessment Report is day 210 of the procedure. A CHMP opinion, whether positive or negative, may be the subject of a re-examination (an appeal), a procedure that has its own time frame. In terms of the assessment phase and the clock stops during CHMP review, these periods averaging 168 days and 118 days, respectively, for all drugs given a positive opinion during 2009–2011 inclusive. The interval between a CHMP opinion to a binding decision by the EC averaged 58 days during the same 3-year period [17].
Provisions for special approvals
The EU pharmaceutical legislation also provides for special types of approvals of unique medicinal products, these being applications that qualify for accelerated assessment, approval under exceptional circumstances or conditional marketing authorization.
Accelerated assessment
For applications accepted for accelerated assessment, the time limit is reduced from 210 to 150 days. For a medicinal product that is of major public health interest and therapeutic innovation, the applicant may request an accelerated assessment, providing justifications for this request. Based on the justifications provided and the recommendations of the rapporteurs, the CHMP reaches a decision on the request for accelerated assessment. The CHMP itself may decide to conduct an accelerated assessment on its own volition. At any time during the accelerated assessment, the CHMP may also terminate conducting an accelerated assessment if no longer appropriate and continue the assessment under standard provisions.
Exceptional circumstances approval
In exceptional circumstances, and following consultation with the applicant, an authorization may be granted subject to specific sponsor obligations and a requirement for the applicant to introduce specific procedures concerning the safety of the product. Exceptional circumstances approval is granted when the applicant can show that he is unable to provide comprehensive data on the efficacy and safety under normal conditions of use in specific therapeutic indications. This may be because (i) the indications are encountered so rarely that the applicant cannot reasonably be expected to provide comprehensive evidence, (ii) in the present state of scientific knowledge, comprehensive information cannot be provided or (iii) it would be unethical to collect such information.
Conditional approval
Commission Regulation (EC) 507/2006 [18] provides the legal basis for conditional approval. In order to meet unmet medical needs of patients and in the interests of public health, medicines in certain categories may be approved on the basis of surrogate markers and/or other less complete data than is normally the case but subject to specific obligations. However, the benefit–risk balance of the product should have already been determined to be positive. A conditional marketing authorization is valid for 1 year and may be renewed annually as long as the benefit–risk is determined to be positive at each renewal. This requirement for annual renewal of conditional approvals provides an important safeguard against sponsors’ defaulting on fulfilling post-marketing requirements.
Comparing the FDA and the EMA expedited approval pathways
It is apparent that the priority review by the FDA (review time 6 months) is equivalent to accelerated assessment by the CHMP (review time 150 days) whereas the accelerated approval by the FDA is equivalent to the conditional approval by the CHMP (both approvals being based on a degree of uncertainty arising from use of surrogate markers and/or other less than the usually complete data). For appropriate medicines, both systems allow for an approval on a shorter review timeline combined with the use of surrogate endpoints. The two major differences between the two agencies are (a) the CHMP requirement for re-assessment of the conditional approvals for their benefit-risk on an annual basis and (b) the EU regulation providing for financial penalties in case of non-compliance with commitments agreed during conditional approval.
Approval of small molecule tyrosine kinase inhibitors
Since 2005, it has been mandatory in the EU to submit marketing authorization applications for oncology products through the centralized route. Notwithstanding, sponsors have submitted applications for all TKIs in the EU through the centralized route, including the application for the first TKI, imatinib, in 2001. The FDA had approved 16 TKIs, and of these, the CHMP/EMA had approved 14, as of 30 September 2012, with applications for bosutinib and regorafenib being still under review by the CHMP on that date. The regulatory exigency and the approval status of the TKIs approved by the FDA and the CHMP are summarized in Table 1. Both agencies have progressively approved additional indications following the initial approval. There are some differences in details of the wording of the indications for some of these TKIs. However, the broad indications approved by both the agencies appear to correspond well with each other. Although we have not examined the detailed wording of all indications of all the TKIs for minor clinically relevant differences, some differences in labelling were obviously significant. For example, only the EU label of erlotinib includes an indication for its use as first line treatment for patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) with EGFR activating mutations.
Table 1.
Regulatory aspects of approved small molecule tyrosine kinase inhibitors
| Drug | FDA review type | EU review type | Initial approval by FDA, EU | Summary of currently approved indications‡ |
|---|---|---|---|---|
| Axitinib | S | S | 27 January 2012* | Renal cell carcinoma |
| 03 September 2012† | ||||
| Bosutinib | O | O | 04 September 2012 | Chronic myeloid leukaemia |
| S | Under review | |||
| Crizotinib | O | C | 26 August 2011 | Non-small cell lung cancer |
| P | 23 October 2012 | |||
| A | ||||
| Dasatinib | O | O | 28 June 2006* | Chronic myeloid leukaemia |
| P | 20 November 2006 | Acute lymphoblastic leukaemia | ||
| A | ||||
| Erlotinib | P | S | 18 November 2004 | Non-small cell lung cancer |
| 19 September 2005† | Pancreatic cancer | |||
| Gefitinib | P | S | 05 May 2003* | Non-small cell lung cancer |
| A | 24 June 2009† | |||
| Imatinib | O | O | 10 May 2001 | Chronic myeloid leukaemia |
| P | 07 November 2001 | Acute lymphoblastic leukaemia | ||
| A | Hypereosinophilic syndrome | |||
| Myelodysplasia or proliferation | ||||
| Gastrointestinal stromal tumour | ||||
| Lapatinib | P ▪ | C | 13 March 2007 | HER2 positive breast cancer |
| 10 June 2008† | ||||
| Nilotinib | O | O | 29 October 2007 | Chronic myeloid leukaemia |
| A ▪ | 19 November 2007 | |||
| Pazopanib | S ▪ | C | 19 October 2009* | Renal cell carcinoma |
| 14 June 2010† | Soft tissue sarcoma | |||
| Regorafenib | P ▪ | ?? | 27 September 2012 | Colorectal cancer |
| Under review | ||||
| Ruxolitinib | O | O | 16 November 2011 | Myelofibrosis |
| P | 23 August 2012 | |||
| Sorafenib | O | O | 20 December 2005 | Renal cell carcinoma |
| P | 19 July 2006† | Hepatocellular carcinoma | ||
| Sunitinib | P | S | 26 January 2006 | Gastrointestinal stromal tumour |
| A ▪ | 19 July 2006‡ | Renal cell carcinoma | ||
| Pancreatic neuroendocrine tumours | ||||
| Vandetanib | O | C ▪ | 06 April 2011* | Medullary thyroid cancer |
| P ▪ | 17 February 2012† | |||
| Vemurafenib | O | S | 17 August 2011 | Melanoma with BRAFmutation(s) |
| P | 17 February 2012 |
Oncologic Drug Advisory Committee consulted by the FDA.
Scientific Advisory Group in Oncology consulted by the CHMP.
Indication(s) in italics and underlined: Indication(s) granted on first approval. The exact indications are highly qualified and the reader should check the prescribing information/label for details. A = Accelerated approval; C = Conditional approval; O = Orphan status; P = Priority review; S = Standard review; ▪ = Boxed warning.
The following summarizes the approaches of the two agencies when initially approving the drugs in this novel class of oncology agents. While the FDA granted priority review to 12 of these 16 TKIs, the CHMP did not grant accelerated assessment to any of the 14 TKIs approved as of 30 September 2012. Whereas the FDA had approved six (38%) TKIs under accelerated approval programme, the CHMP had granted (its equivalent of) conditional approval to four (29%). Interestingly, only one TKI (crizotinib) was granted accelerated or conditional approval by both agencies. The FDA afforded priority review as well as accelerated approval to five TKIs: crizotinib, dasatinib, gefitinib, imatinib and sunitinib. Of these five, only sunitinib carries a boxed warning (regarding its hepatotoxic potential). Orphan drug designation was granted to nine (56%) of the TKIs approved by the FDA and these included all the six (38%) TKIs also designated as orphan drugs by the EMA. This difference probably reflects the difference in the thresholds used by the two agencies for granting orphan designation. The FDA had consulted Oncologic Drugs Advisory Committee for five of the 16 TKIs approved by it whereas CHMP consulted (the equivalent) Oncology SAG for at least eight of the 14 TKIs approved by it.
Table 2 shows the dates when the applications were submitted to the two agencies. For 13 applications (all but bosutinib, gefitinib and regorafenib), submission to the FDA preceded the submission to the EMA by a mean of 31.2 days (range −2 to 120 days). Nine of these 13 applications were submitted to the two agencies within 4 weeks of each other. The successful application for gefitinib to the EMA, long-delayed after its submission to the FDA, is discussed later.
Table 2.
Dates applications submitted to the FDA and EMA
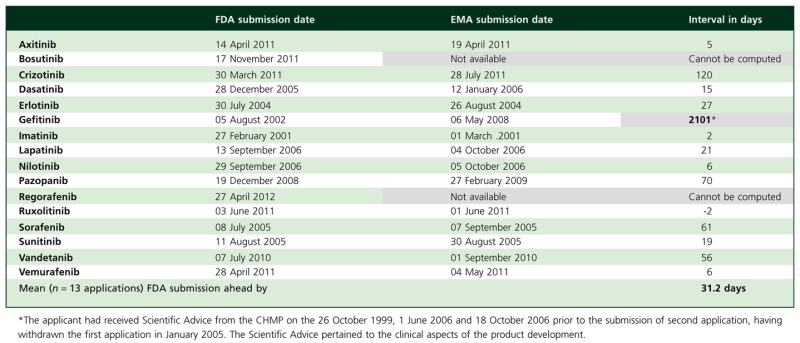 |
Not evident from the table is that the application for vandetanib was first submitted to the EMA on 30 June 2007, seeking approval for its use in combination with chemotherapy for the treatment of previously-treated patients with locally advanced or metastatic NSCLC. During its review, this application was withdrawn by the sponsor on 27 October 2009 after preliminary comments from the rapporteur and co-rapporteur indicated that the CHMP would be unlikely to conclude a favourable benefit–risk balance for the product in the indication requested [19, 20]. A fresh application for a different indication, treatment of aggressive and symptomatic medullary thyroid cancer in patients with unresectable locally advanced or metastatic disease, was submitted on 1 September 2010, 56 days after it was submitted to the FDA on 7 July 2010
Table 3 summarizes the FDA review and approval timelines in the US and Table 4 summarizes the EMA review and marketing authorization timelines in the EU. Marketing authorization in the EU took on average 409.6 days from the submission of the application compared with 205.3 days in the US. The extra time to market of 204.3 days in the EU, between the submission of the application and EC marketing authorization, was due to (i) an extra 20.1 days of active review time by the CHMP, (ii) clock stops during review which averaged 92.9 days and (iii) an interval which averaged 91.3 days between CHMP opinion and EC decision. When a homogeneous drug class such as TKIs is considered, the delay in the EU of about 184 days from a total time of 409 days (i.e. 45%), due to the above two non-review reasons, is substantially lower than that reported previously. Netzer [21] had previously reported a delay of 236 days (119 days for clock stop and 117 days from opinion to EC decision) from a mean total time of 429 days (i.e. 55%) for 20 oncology drugs between January 1995 and June 2004.
Table 3.
FDA approval times
| Submission date | Approval date | Interval in days | |
|---|---|---|---|
| Axitinib | 14 April 2011 | 27 January 2012 | 288 |
| Bosutinib | 17 November 2011 | 04 September 2012 | 292 |
| Crizotinib* | 30 March 2011 | 26 August 2011 | 149 |
| Dasatinib* | 28 December 2005 | 28 June 2006 | 182 |
| Erlotinib* | 30 July 2004 | 18 November 2004 | 111 |
| Gefitinib* | 05 August 2002 | 05 May 2003 | 273 |
| Imatinib* | 27 February 2001 | 10 May 2001 | 72 |
| Lapatinib* | 13 September 2006 | 13 March 2007 | 181 |
| Nilotinib | 29 September 2006 | 29 October 2007 | 396 |
| Pazopanib | 19 December 2008 | 19 October 2009 | 304 |
| Regorafenib* | 27 April 2012 | 27 September 2012 | 153 |
| Ruxolitinib* | 03 June 2011 | 16 November 2011 | 166 |
| Sorafenib* | 08 July 2005 | 20 December 2005 | 166 |
| Sunitinib* | 11 August 2005 | 26 January 2006 | 168 |
| Vandetanib* | 07 July 2010 | 06 April 2011 | 273 |
| Vemurafenib* | 28 April 2011 | 17 August 2011 | 111 |
| Mean (n = 16) | 205.3 | ||
| Range | 72–396 | ||
| Mean for Priority review drugs (n = 12) | 167.1 | ||
| Mean for Standard review drugs (n = 4) | 320.0 | ||
Designated for Priority review.
Table 4.
EMA approval times
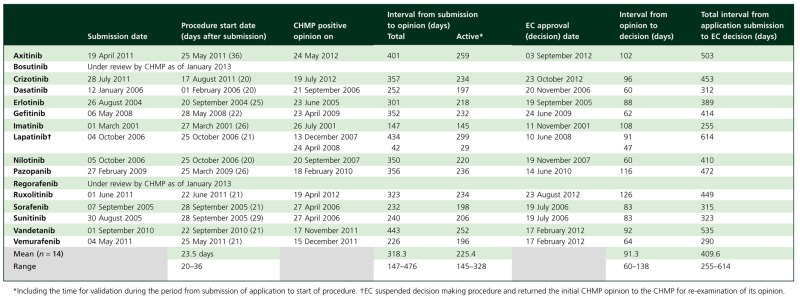 |
Both agencies have the authority to request, if necessary, additional information from sponsors during a review. For the EMA, requests for all new information are consolidated in a single list at day 120 when the clock is stopped. However, for the FDA, the review clock continues, and results in what appears as a longer review time, whilst the agency waits for the information. This was the case for nilotinib, pazopanib and vandetanib, resulting in longer than average FDA review times for these three drugs. We have no information on this waiting time but assuming that it was about 6 weeks for each, the adjusted FDA review time improves to 197.4 days. Therefore, in terms of active review time (including the time taken for validation of the application), the two agencies are comparable (197.4 or 205.3 days for the FDA and 225.4 days for the CHMP). This difference of 20–28 days in review times between the two agencies is minimal in the context of the long development times in oncology.
Drugs that receive FDA fast track designation are eligible for a rolling review of data as they accumulate during development, and the official ‘application submitted’ date is the date at which the final set of data is received. This is an issue not considered by other studies. For such applications, review of some of the data may have begun well before the ‘application submitted’ date. Thus, for these applications, the FDA review time is underestimated if the review time to approval is computed from ‘application submitted’ date to the approval date. We noticed that the FDA had accepted half of the 16 TKI applications on a rolling review basis. Although the ‘application submitted’ dates corresponded closely to the date of submission of initial data for four of the eight TKIs, the FDA had been provided the first set of data for review for the other four well ahead of the ‘application submitted’ dates as shown in Table 5. Assuming that the reviews of these four fast-tracked drugs had begun soon on receipt of the first set of data, the intervals to be added to the FDA review times (that are shown in Table 3) are shown in Table 5. If this is done, the mean review time of the 16 TKIs by the FDA computes at 244.3 days.
Table 5.
Dates of submission of data and applications to the FDA
| Drug | Dates of submission of | Interval in days | |
|---|---|---|---|
| First set of data for review | Complete ‘application’ | ||
| Erlotinib | 20 January 2004 | 30 July 2004 | 192 |
| Gefitinib | 30 July 2001 | 05 August 2002 | 371 |
| Nilotinib | 09 August 2006 | 29 September 2006 | 41 |
| Sorafenib | 17 June 2005 | 08 July 2005 | 19 |
Two atypical cases
There are two drugs that merit special comment when comparing the two agencies – gefitinib, for which the FDA submission was about 6 years ahead of the EMA submission, and lapatinib, for which the EU review time of 328 days was far longer than the FDA review time of 181 days. These two drugs provide valuable perspectives into the potential consequences of the differences in approach by the two agencies – gefitinib concerning efficacy and lapatinib concerning safety. Here, we briefly discuss these two drugs, exploring the reasons for the apparent wide disparity between the two agencies in terms of their approval and/or review timelines.
Gefitinib
The fast track application for gefitinib was submitted to the FDA on a rolling review basis with the first section of the NDA submitted on 30 July 2001. Following receipt of three supplementary submissions on the pulmonary toxicity data from Japan, the original PDUFA deadline for completion of review was reset to 5 May 2003 22. Ultimately, gefitinib was approved on 5 May 2003 for an unselected group of NSCLC patients. The approval was based on efficacy data from two trials (trials 39 and 16), both of which studied the effect of two doses (250 mg and 500 mg daily) of gefitinib on objective tumour response rate and disease-related symptom improvement rate and their safety. Secondary objectives included progression-free survival and overall survival. Trial 16 also evaluated potential differences between the Japanese and the non-Japanese patients. In study 39, dosing with 250 mg day−1 or 500 mg day−1 of gefitinib demonstrated objective tumour response rates of 11.8% and 8.8%, respectively and disease-related symptom improvement rates of 43.1% and 35.1%, respectively. The two doses were equivalent in terms of median progression-free survival and overall survival rates. Study 16 revealed significant differences between the Japanese and the non-Japanese patients with respect to tumour response, disease control, progression-free survival, and overall survival. The FDA concluded that the relevance of the symptom improvement data could not be adequately evaluated without a randomized, blinded study with an adequate control arm but nevertheless, granted an accelerated approval on 5 May 2003 and required the sponsor to conduct confirmatory trials as post-marketing commitments 23. One of three commitments for full approval was a randomized, placebo-controlled trial (Trial 709) in patients with locally advanced or metastatic NSCLC with the aim of evaluating the effect of gefitinib on overall survival.
In the EU, gefitinib was never approved for an unselected lung cancer population. In February 2003, the sponsor had submitted an application for gefitinib to the EMA for the treatment of locally advanced or metastatic NSCLC in patients who had failed prior chemotherapy [24]. In January 2005, however, the sponsor withdrew the application from the EMA since the survival data from IRESSA Survival Evaluation in Lung cancer (ISEL) did not meet the approval requirements of the CHMP [25]. Preliminary analysis of the ISEL trial had shown a statistically significant improvement in tumour shrinkage but this did not translate into a statistically significant survival benefit.
Following the FDA approval, Trial 709 was initiated and completed expediently and a survival analysis conducted in mid-December 2004 showed some increase in overall survival on gefitinib relative to placebo but it was not statistically significant in the overall population [26]. There were, however, subsets of patients where a survival benefit for gefitinib was seen, those of Asian descent and non-smokers. Results from Trial 709 led the sponsor to determine that physicians should consider other treatment options in the recurrent NSCLC patient population and, in consultation with the FDA, suspended promoting gefitinib.
In the EU, another application for gefitinib was submitted to the EMA on 6 May 2008 and the CHMP issued a positive opinion for its approval on 23 April 2009. Its indication was restricted to patients with NSCLC containing EGFR mutations since the benefit/risk balance was highly positive for this subgroup because almost all antineoplastic effects were confined to this subgroup 27. A further 62 days later, the Commission confirmed a pan-European approval of gefitinib on 24 June 2009.
The time taken from the receipt of the gefitinib application to its approval by the FDA in May 2003 was 273 days. By comparison, the CHMP had issued a positive opinion for approval on 23 April 2009, which was 232 days (excluding the clock stop time) from the receipt of the application by the EMA.
Lapatinib
The application for lapatinib was submitted to the FDA on 13 September 2006 and it was approved on 13 March 2007, with a review time of 181 days. The contents of the initially approved label did not signal any review concern regarding any hepatotoxic potential of lapatinib [28].
The application for lapatinib to the EMA was submitted on 4 October 2006 (21 days after its submission to the FDA) 29 and the CHMP issued a positive opinion on 13 December 2007 (434 days from start of procedure) about 275 days after its approval by the FDA. Three periods of clock stops, totalling 135 days, accounted for a significant proportion of this delay. On receipt of the responses to the second list of questions, the CHMP consulted SAG on Oncology on issues of efficacy and determined that additional efficacy data were needed. Ultimately, the indication initially approved by the CHMP was essentially the same as that approved by the FDA and the positive opinion of the CHMP was forwarded to the Commission for issuing a decision.
However, to complicate the matter, and reminiscent of the FDA experience with gefitinib and notification of unexpected pulmonary toxicity during its review process, the sponsor informed the EMA of a significant hepatotoxicity issue that emerged during the period of Commission deliberations. The Commission suspended its deliberations and required the CHMP to re-examine its opinion 29. The CHMP formulated a list of questions on 21 March 2008 to which the applicant responded on 2 April 2008 and the CHMP issued another positive opinion on 24 April 2008 when the sponsor provided an undertaking on the specific obligations and follow-up measures to be fulfilled post-authorization. The Commission issued a decision approving lapatinib on 10 June 2008.
The EU prescribing information for lapatinib included at the outset recommendations on liver function test monitoring and on discontinuation of treatment if changes were severe 30. By comparison, the FDA approved a revised label with a boxed warning on hepatotoxicity on 7 July 2008 [31]. Since its initial approval, the CHMP has regularly re-assessed the risk/benefit of lapatinib as required of all conditional approvals and concluded that this remains positive 32.
Expedited reviews vs. safety
There has been much debate in the literature as to whether expedited review compromises the assessment of drug safety. There is a regionally determined conflicting evidence in terms of the association between expedited reviews and subsequent safety problems in the EU and the US.
Boon et al. [33] reported that despite the fact that conditional approvals and approvals under exceptional circumstances in the EU are based on limited safety databases, there was no special safety issue associated with using these pathways.
Following a retrospective study of 289 new drugs (46 exceptional or conditional and 243 standard) approved in the EU and using the frequency and timing of a first ‘Dear Healthcare Professional’ communication as the outcome variable, Arnardottir et al. [34] also reported that exceptional circumstances or conditional approvals in the EU were not associated with more post-marketing safety alerts or safety-related withdrawals when used for drugs with unmet medical needs. However, compared with label changes, such communications are usually reserved for very significant changes and their numbers may not reflect new safety concerns that warranted only the label changes. Although the above evidence is encouraging, further studies are necessary to investigate any association between expedited reviews and subsequent safety problems in the EU.
The PDUFA has imposed deadlines for the completion of drug reviews by the FDA [9, 10]. With regard to the drugs approved in the US, Carpenter et al. [35] have reported that once medications are in clinical use, the discovery of safety problems is more likely for drugs approved immediately before a deadline than for those approved at other times. Berlin [36] has also examined the relationship between the frequency of oncology drug labelling revisions and the FDA review type. One hundred oncology drugs, designated by the FDA as accelerated approval, priority review, orphan drug, or traditional review, were identified from publicly available information. Drug information for each product was evaluated to assess the rate at which manufacturers revised product labelling. This study found that labelling for accelerated approval and priority review products is revised significantly more frequently than are labels for traditional products. However, it is unclear whether these were new safety issues or updating of safety issues noted pre-approval. Because these drugs are intended for the most serious of oncology settings with the highest unmet need, there may have been greater tolerance for any safety issues initially. The study by Richey et al. [37] suggests new safety issues. These investigators reported a higher likelihood of post-approval black box warnings to labels for oncology drugs that had received accelerated approvals compared with those that had received regular approval (17% vs. 9%). Of the four black box warnings added to the labels of oncology products with accelerated approval, three were added more than 2 years after approval, which led these investigators to suggest that safety signals could not have been recognized during clinical trials. In view of the small numbers, it is difficult to draw any firm conclusions but our analysis of boxed warnings on TKI labels tends to suggest otherwise. Labels of six of the 16 TKIs approved by the FDA carry boxed warnings. These are lapatinib, pazopanib, regorafenib and sunitinib with regard to hepatotoxicity and nilotinib and vandetanib with regard to their QT liability. For four of these, the boxed warnings were present at the outset of approval but two required inclusion much later. The FDA boxed warning on hepatotoxicity due to lapatinib (priority review) was inserted about 16 months after its approval and the one on sunitinib (accelerated approval) about 53 months later.
Nevertheless, the Oncologic Drugs Advisory Committee, at its February 2011 meeting, urged the FDA to raise its standards for granting experimental cancer drugs accelerated approval on the basis of surrogate markers [38]. The committee concluded that single arm trials, which were used to justify more than half the accelerated approvals since the programme's inception in 1992, should be accepted only for rare cancers or when the evidence of efficacy from a single arm trial was overwhelmingly positive. The committee also reached a consensus that the FDA should require at least two controlled trials as final proof of efficacy and recommended that those trials should be under way when accelerated approval is granted.
Approval, affordability and access
The emotive issue of access has often been used to support campaigns for an expedited review. A widely prevalent myth is that early approval guarantees, or is synonymous with, affordability and/or early access with significant beneficial impact on public health. Detailed discussion on affordability and access is beyond the scope of this review but we believe that a brief discussion is warranted to dispel this myth. Cohen et al. [39] have identified eight sub-dimensions of patient access to pharmaceuticals: marketing approvals, time of marketing approval, coverage, cost sharing, conditions of reimbursement, speed from marketing approval to reimbursement, extent to which beneficiaries control choice of their drug benefit and evenness of the availability of drugs to the population. Eichler et al. [40] have lucidly articulated on the myth equating review times with access time, ‘Over the past decade, the role of payers has become more prominent, and time-to-market no longer means time-to-licensing but time-to-reimbursement’. Non-adherence to prescription drugs due to lack of affordability is much wider than is generally believed [39, 41]. Even in countries such as Canada, which ranks highly in terms of societal values and affluence, there is evidence of cost-related non-adherence to prescription drugs [42–44]. In a small pilot study, Zheng et al. found 15% non-adherence related to cost [43], and a much larger study by Law et al. reported that about 1 in 10 Canadians who receive a prescription report cost-related non-adherence [44].
In an effort to control these costs and make medicines affordable, almost all the EU countries regulate prices of pharmaceutical products [45–47]. Drugs for cancer are no exception. Because of the way the provision of health care is structured, access is invariably delayed until the price is agreed upon. However, once agreed, the access is generally much wider. In contrast, access may be immediate in the US but not as widely as is generally believed or considered desirable. According to the 2007 Report from Office of Fair Trading [47], there appears to be a wide ranging misunderstanding of pricing and reimbursement policies in the US. There seems to be a view that access to the US market is ‘free’ in the sense of there being no requirement to agree on a price before a product can be reimbursed. However, reimbursement of new drugs in the US does not happen automatically but is subject to negotiations with several intermediaries representing private and public insurance organizations. Thus, in contrast to common belief, there is also an element of delay for reimbursement of new drugs in the US, although this will vary by plan and delays in US are in general brief by international standards [47]. In contrast to the US, several European countries also have health technology assessment programmes for drugs, many of which assess cost effectiveness.
According to the 2009 Comparator Report on Patient Access to Cancer Drugs in Europe [48], most of the countries in Europe have formal procedures for making national reimbursement decisions, although some countries such as the UK have no specific procedures before the drug may be prescribed under the reimbursement system. For countries with formal decision processes, the reimbursement decisions include price negotiations and estimates of the forecasts of sales. Although the UK and Germany lack overt restrictions on pricing, it does not mean that the authorities in these countries do not intervene with drug costs. In the UK, the Pharmaceutical Price Regulation Scheme of the Department of Health controls company profits and can ask for price cuts and paybacks from companies. In Belgium, Finland, the Netherlands, Norway, Portugal and Sweden, the formalized decision-making process requires an economic evaluation, and the issue of cost-effectiveness plays an important role. For Denmark and Switzerland the role of economic evaluation and cost-effectiveness is not a formalized part of the decision-making process, but the producer may submit supportive data of economic benefits, which may facilitate a positive decision.
Following their study of all anticancer drugs approved in the US from 2004 to 2008, Mason et al. [49] concluded that anticancer drug coverage decisions that consider cost effectiveness are associated with greater restrictions and slower time to coverage. However, Dr Cohen, research assistant professor at Tufts Centre for the Study of Drug Development who conducted the analysis referred to in the Introduction noted, ‘While greater access to more treatment options is definitely a positive for patients in the US, it is not clear if greater access leads to better health outcomes’ [5]. He also went on to say, ‘Although more oncology drugs are available in the US, and the costs for a higher share of them are reimbursed, the evidence-based approach adopted by European systems has improved the affordability of drugs in Europe that are considered to be cost-effective’ [5].
Finally, in the context of public health and the treatment of what are potentially fatal conditions, it is worth putting the risk/benefit of the TKIs in perspective. For the majority of them, their efficacy and the responder rates are modest at best. Following their review of the development and approval of cancer drugs in the EU between January 2001 and January 2012 (48 new drugs and 77 new indications for drugs already approved), Jonsson & Bergh reported that in most instances, the benefit–risk balance was considered to be only borderline favourable [50]. TKIs are known to be associated with a whole range of serious side effects. Because of their non-therapeutic on-target effects at sites remote from the cancer site, the efficacy and a variety of toxic effects of TKIs are often intricately linked to each other, so much so that these toxic effects are believed to have a potential role as biomarkers of effective pharmacological inhibition of the target pathway [51]. Consequently, targeted agents that lead to improvements in efficacy also increase treatment-related morbidity and mortality. Patients in pre-approval clinical trials are carefully selected but treatment of less selected patients in routine oncologic practice may increase the likelihood of toxicity and lower the probability of benefit [52], a clinical reality that is described as the efficacy-effectiveness gap [53]. This gap becomes all the more relevant because (i) during 1995–2008, phase III studies supported only 26% of accelerated approvals in contrast to 74% of regular approvals [37] and (ii) concerns have been expressed that sponsors were not completing the agreed phase III trials designed to verify improvements in clinical outcomes for oncology accelerated approvals in the US [54]. Therefore, while there is little doubt that the benefits of TKIs in life-threatening indications outweigh their risks, it is questionable if the delay in access, for whatever reasons, has as much a public health impact as is often claimed. We have not been able to locate any evidence-based data to support claims of adverse or improved public health impact as a result of 3–6 months differences in regulatory review times and access, taking into account the effect size of oncology drugs and the morbidity associated with their use.
Discussion and conclusions
Although our study sample of drugs is relatively small compared with that of some other studies, it has the advantage of studying a more homogeneous class of drugs that were submitted at about the same time to both the agencies for review. We believe this provides a basis for a more valid comparison. Our study supports the conclusion that the two agencies perform equally and comparably well with no one agency being better than the other, at least as far as the TKIs are concerned. The difference we have found of 20–28 days in review times between the two agencies is relatively small and should be seen in the context of the overall drug development time. According to the study by The Tufts Centre for the Study of Drug Development, the total development and approval time in the US for fast track drugs dropped by 20% – from 8.3 years in 2002–2006 to 6.6 years in 2007–2011 [6]. Richey et al. [37] reported median times from IND to approval of 6.7 years for accelerated and 7.0 years for regular oncology drugs during the period 2004–2008. For accelerated approvals, this was substantially lower than 9.3 years for the period 2001–2003. We believe that the concerns surrounding the availability of oncology drugs have inappropriately focused on review times rather than on the time for their development.
Trotta et al. [3] reported an interesting observation that the agency that was second in approving a drug was usually more restrictive in terms of wording of the indication compared with the agency that provided approval first. This would suggest that a small delay in approving may lead to a more refined assessment of safety, efficacy and risk/benefit. Gefitinib and lapatinib discussed above support such a suggestion. Experience with these two drugs demonstrates the potential risks of accelerated approvals since their adverse efficacy (gefitinib) and safety (lapatinib) data emerged during their post-marketing period.
In the final analysis, however, the average interval from submission of an application to the approval of a TKI was 409.6 days in the EU whereas it was 205.3 days in the US and it could be argued that the patient is less concerned with the reasons for this difference. Although this difference of 204 days is relatively small compared with the time required to develop and negotiate reimbursement of oncology drugs generally, a question inevitably arises whether the EU time frame is capable of abbreviation. Three possible solutions present themselves. First, novel oncology drugs may be considered for EU accelerated approval, a procedure hardly ever used, reducing the time frame for completing the review from 210 to 150 days. Second, the rapporteur and the co-rapporteur should be encouraged to interact frequently with each other and with the sponsor during primary review (before day 120) to enable earlier resolution with the sponsor of many issues (that currently form day 120 list of questions) as they arise during on-going review. Finally, the interval between a CHMP opinion and the EC decision may be capable of reduction to 30 days. Table 4 reveals that this interval exceeded the statutory 67 days for over 71% of TKIs approved in the EU. None of these three solutions, if implemented, is likely to compromise a pan-European consensus. Another innovative approach that may be worth exploring is a time-sensitive rolling review of a group of related studies (e.g., acute and chronic toxicity studies, clinical drug interactions and special populations studies and proof-of-concept and dose-ranging studies) as soon as they are completed during drug development.
In some cases, a product may be so effective (a breakthrough) that large beneficial effects are seen early in its development. In an effort to reduce the development times of such products, the recently enacted Food and Drug Administration Safety and Innovation Act (FDASIA) includes a provision that allows sponsors to request that their drug be designated as a Breakthrough Therapy. The FDA is in the process of developing guidance related to this designation. Breakthrough Therapy designation requires preliminary clinical evidence of exceptional activity in a serious or life-threatening disease with poor outcomes. The benefit is that this designation abbreviates or condenses the registration trials in a way that minimizes patient exposure to an ineffective placebo (if the effect size is really large, statistical significance can be achieved with a smaller trial). Similar initiatives are being explored in the EU and by other authorities (adaptive licensing) 55. Adaptive licensing seeks to maximize the positive impact of new drugs on public health by balancing timely access for patients with the need to provide adequate evolving information on benefits and harms.
The Office of Oncology Drug Products at the FDA and the EMA have undertaken to increase the dialogue between the two agencies to provide a deeper understanding of the basis for scientific advice, and to seize the opportunity to optimize product development and avoid unnecessary replication. Under a confidentiality arrangement finalized in September 2004 between the EC/EMA and the FDA, seven programmes and practices to increase the co-operation between the two agencies had been implemented by November 2005 in the Office of Oncology Drug Products at the FDA [54]. In his presentation of ‘New Drug Review: 2009 Update’, Dr John Jenkins of the FDA commented, ‘While comparisons are interesting, (the two agencies) do not consider (themselves) to be in a race. …’ [56]. With regard to the comparisons between the two agencies, he concluded that (i) there was concordance of action for ∼80% of new molecular entities (NMEs) submitted within 12 months to both agencies, (ii) there was little divergence on priority NMEs but greater divergence on standard NMEs which is probably not surprising given the lower public health priority of standard NMEs and many of these decisions are close judgment calls (i.e. marginal but statistically significant efficacy and safety concerns). He emphasized that the FDA and EMA communicate and share information on many applications, but both conduct independent assessments and make decisions based on distinct laws, regulations, precedents, and societal expectations.
Competing Interests
All three authors have completed the Unified Competing Interest Form, available on request from the corresponding author and declare no support from any organization for the submitted work, no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years and no other relationships or activities that could appear to have influenced the submitted work.
References
- 1.Downing NS, Aminawung JA, Shah ND, Braunstein JB, Krumholz HM, Ross JS. Regulatory review of novel therapeutics – comparison of three regulatory agencies. N Engl J Med. 2012;366:2284–2293. doi: 10.1056/NEJMsa1200223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Downing NS, Ross JS. Review of novel therapeutics by three regulatory agencies. N Engl J Med. 2012;367:1166–1167. doi: 10.1056/NEJMc1208803. [DOI] [PubMed] [Google Scholar]
- 3.Trotta F, Leufkens HG, Schellens JH, Laing R, Tafuri G. Evaluation of oncology drugs at the European Medicines Agency and US Food and Drug Administration: when differences have an impact on clinical practice. J Clin Oncol. 2011;29:2266–2272. doi: 10.1200/JCO.2010.34.1248. [DOI] [PubMed] [Google Scholar]
- 4.Roberts SA, Allen JD, Sigal EV. Despite criticism of the FDA review process, new cancer drugs reach patients sooner in the United States than in Europe. Health Aff (Millwood) 2011;30:1375–1381. doi: 10.1377/hlthaff.2011.0231. [DOI] [PubMed] [Google Scholar]
- 5.Tufts Center for the Study of Drug Development. U.S. offers patients faster, greater access to cancer drugs than Europe. July/August 2012, Vol. 14(4). Summary. Available at http://csdd.tufts.edu/files/uploads/08_-_july_10,_2012_-_cancer_drugs.pdf (last accessed 10 September 2012)
- 6.Tufts Center for the Study of Drug Development. Onocology drugs get faster approvals than non-oncology drugs in U.S September/October 2012, Vol. 14(5). Summary. Available at http://csdd.tufts.edu/files/uploads/09_-_sept_5,_2012_-_oncology_drugs.pdf (last accessed 10 September 2012)
- 7.European Commission. Procedures for marketing authorisation. EudraLex – Volume 2A – Chapter 6. Decision making procedure for the adoption of Commission Decisions (November 2005). Available at http://ec.europa.eu/health/files/eudralex/vol-2/a/vol2a_chap6_2005-11_en.pdf (last accessed 10 September 2012)
- 8.Food and Drug Administration. The CDER Handbook. 1998. Available at http://druganddevicelaw.net/CDER_handbook.pdf (last accessed 10 September 2012)
- 9.Food and Drug Administration. Performance report to the President and the Congress for the Prescription Drug User Fee Act. 2005. Available at http://www.fda.gov/downloads/AboutFDA/ReportsManualsForms/Reports/UserFeeReports/PerformanceReports/PDUFA/ucm095108.pdf (last accessed 10 September 2012)
- 10.Food and Drug Administration. PDUFA reauthorization performance goals and procedures. Available at http://www.fda.gov/downloads/ForIndustry/UserFees/PrescriptionDrugUserFee/UCM270412.pdf (last accessed 10 September 2012)
- 11.Food and Drug Administration. Guidance for the Public and FDA Staff on Convening Advisory Committee Meetings. Draft dated. August 2008. Available at http://www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM125651.pdf (last accessed 10 September 2012)
- 12.Roden DM, Temple R. The US Food and Drug Administration Cardiorenal Advisory Panel and the drug approval process. Circulation. 2005;111:1697–1702. doi: 10.1161/01.CIR.0000161370.77463.0F. [DOI] [PubMed] [Google Scholar]
- 13.European Commission. Pharmaceutical Legislation Notice to applicants and regulatory guidelines medicinal products for human use. EudraLex – Volume 2 – Chapter 1. Procedures for Marketing Authorisation (November 2005). Available at http://ec.europa.eu/health/files/eudralex/vol-2/a/vol2a_chap1_2005-11_en.pdf (last accessed 10 September 2012)
- 14.European Commission. Pharmaceutical Legislation Notice to applicants and regulatory guidelines medicinal products for human use. EudraLex – Volume 2 – Chapter 4. Centralised Procedure (April 2006). Available at http://ec.europa.eu/health/files/eudralex/vol-2/a/chap4rev200604_en.pdf (last accessed 10 September 2012)
- 15.European Commission. Pharmaceutical Legislation Notice to applicants and regulatory guidelines medicinal products for human use. EudraLex – Volume 2 – Chapter 2. Mutual recognition procedure and decentralised procedure (February 2007). Available at http://ec.europa.eu/health/files/eudralex/vol-2/a/vol2a_chap2_2007-02_en.pdf (last accessed 10 September 2012)
- 16.European Commission. REGULATION (EC) No 726/2004 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 31 March 2004 laying down Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency. Official Journal of the European Union. 2004;L136:1–33. Consolidated version. Available at http://ec.europa.eu/health/files/eudralex/vol-1/reg_2004_726/reg_2004_726_cons_en.pdf (last accessed 10 September 2012) [Google Scholar]
- 17.European Medicines Agency. Annual Report 2011. London: European Medicines Agency; 2012. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Annual_report/2012/06/WC500128162.pdf (last accessed 2 September 2012) [Google Scholar]
- 18.European Commission. COMMISSION REGULATION (EC) No 507/2006 of 29 March 2006 on the conditional marketing authorisation for medicinal products for human use falling within the scope of Regulation (EC) No 726/2004 of the European Parliament and of the Council. Official Journal of the European Union. 2006;L/92:6–9. Available at http://ec.europa.eu/health/files/eudralex/vol-1/reg_2006_507/reg_2006_507_en.pdf (last accessed 10 September 2012) [Google Scholar]
- 19.European Medicines Agency. PRESS RELEASE: AstraZeneca Withdraws Its Marketing Authorisation Application for Zactima (Vandetanib) (EMEA/698692/2009 Corr) London: European Medicines Agency; 2009. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Press_release/2009/11/WC500007024.pdf (last accessed 10 September 2012) [Google Scholar]
- 20.European Medicines Agency. Questions and Answers on the Withdrawal of the Marketing Authorisation Application for Zactima (Vandetanib) (EMEA/726915/2009) London: European Medicines Agency; 2009. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Medicine_QA/2010/01/WC500060209.pdf (last accessed 10 September 2012) [Google Scholar]
- 21.Netzer T. European Union centralised procedure for marketing authorisation of oncology drugs: an in-depth review of its efficiency. Eur J Cancer. 2006;42:446–455. doi: 10.1016/j.ejca.2005.04.045. [DOI] [PubMed] [Google Scholar]
- 22.Food and Drug Administration. Gefitinib: administrative documents – part 1. Available at http://www.accessdata.fda.gov/drugsatfda_docs/nda/2003/21-399_IRESSA_Admindocs_P1.pdf (last accessed 10 September 2012)
- 23.Food and Drug Administration. Gefitinib: approval letter. Available at http://www.accessdata.fda.gov/drugsatfda_docs/nda/2003/21-399_IRESSA_Approv.pdf (last accessed 10 September 2012)
- 24.AstraZeneca Global. Press release (11 February 2003): AstraZeneca submits IRESSA (gefitinib, ZD1839) for approval in Europe for advanced non-small cell lung cancer. Available at http://www.astrazeneca.com/Media/Press-releases/2003 (last accessed 10 September 2012)
- 25.AstraZeneca Global. Press release (4 January 2005): Gefitinib (IRESSA) marketing authorisation application withdrawn in EU. Available at http://www.astrazeneca.com/Media/Press-releases/2005 (last accessed 10 September 2012)
- 26.AstraZeneca Global. IRESSA (ZD1839, gefitinib) Tablets Oncologic Drugs Advisory Committee (ODAC) meeting briefing document. March 4, 2005. Available at http://www.fda.gov/ohrms/dockets/ac/05/briefing/2005-4095B2_01_01-AstraZeneca-Iressa.pdf (last accessed 10 September 2012)
- 27.European Medicines Agency. Assessment Report for IRESSA (Procedure No EMEA/H/C/001016). Available at http://www.emea.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/001016/WC500036361.pdf (last accessed 10 September 2012)
- 28.Food and Drug Administration. TYKERB (lapatinib) Label (13 March 2007). Available at http://www.accessdata.fda.gov/drugsatfda_docs/label/2007/022059lbl.pdf (last accessed 10 September 2012)
- 29.European Medicines Agency. Assessment Report for TYVERB (Procedure No EMEA/H/C/795). Available at http://www.emea.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/000795/WC500044960.pdf (last accessed 10 September 2012)
- 30.European Medicines Agency. Summary of product characteristics for TYVERB. Available at http://www.emea.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000795/WC500044957.pdf (last accessed 10 September 2012)
- 31.Food and Drug Administration. TYKERB (lapatinib) Label (7 July 2008). Available at http://www.accessdata.fda.gov/drugsatfda_docs/label/2008/022059s004lbl.pdf (last accessed 10 September 2012)
- 32.European Medicines Agency. Tyverb: procedural steps taken and scientific information after the authorisation. Available at http://www.emea.europa.eu/docs/en_GB/document_library/EPAR_-_Procedural_steps_taken_and_scientific_information_after_authorisation/human/000795/WC500044961.pdf (last accessed 10 September 2012)
- 33.Boon WP, Moors EH, Meijer A, Schellekens H. Conditional approval and approval under exceptional circumstances as regulatory instruments for stimulating responsible drug innovation in Europe. Clin Pharmacol Ther. 2010;88:848–853. doi: 10.1038/clpt.2010.207. [DOI] [PubMed] [Google Scholar]
- 34.Arnardottir AH, Haaijer-Ruskamp FM, Straus SM, Eichler HG, de Graeff PA, Mol PG. Additional safety risk to exceptionally approved drugs in Europe? Br J Clin Pharmacol. 2011;72:490–499. doi: 10.1111/j.1365-2125.2011.03995.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carpenter D, Zucker EJ, Avorn J. Drug-review deadlines and safety problems. N Engl J Med. 2008;358:1354–1361. doi: 10.1056/NEJMsa0706341. [DOI] [PubMed] [Google Scholar]
- 36.Berlin RJ. Examination of the relationship between oncology drug labeling revision frequency and FDA product categorization. Am J Public Health. 2009;99:1693–1698. doi: 10.2105/AJPH.2008.141010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Richey EA, Lyons EA, Nebeker JR, Shankaran V, McKoy JM, Luu TH, Nonzee N, Trifilio S, Sartor O, Benson AB, 3rd, Carson KR, Edwards BJ, Gilchrist-Scott D, Kuzel TM, Raisch DW, Tallman MS, West DP, Hirschfeld S, Grillo-Lopez AJ, Bennett CL. Accelerated approval of cancer drugs: improved access to therapeutic breakthroughs or early release of unsafe and ineffective drugs? J Clin Oncol. 2009;27:4398–4405. doi: 10.1200/JCO.2008.21.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Food and Drug Administration. Summary Minutes of the Oncologic Drugs Advisory Committee Meeting. February 8, 2011. Available at http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM250472.pdf (last accessed 10 September 2012)
- 39.Cohen J, Faden L, Predaris S, Young B. Patient access to pharmaceuticals: an international comparison. Eur J Health Econ. 2007;8:253–266. doi: 10.1007/s10198-006-0028-z. [DOI] [PubMed] [Google Scholar]
- 40.Eichler HG, Bloechl-Daum B, Abadie E, Barnett D, König F, Pearson S. Relative efficacy of drugs: an emerging issue between regulatory agencies and third-party payers. Nat Rev Drug Discov. 2010;9:277–291. doi: 10.1038/nrd3079. [DOI] [PubMed] [Google Scholar]
- 41.Boukus ER, Carrier ER. Americans’ access to prescription drugs stabilizes, 2007–2010. Track Rep. 2011;27:1–5. [PubMed] [Google Scholar]
- 42.Canadian Cancer Society. Cancer drug access for Canadians. September 2009. Available at http://www.cancer.ca/canada-wide/about%20us/media%20centre/cw-media%20releases/cw-2009/~/media/CCS/Canada%20wide/Files%20List/English%20files%20heading/pdf%20not%20in%20publications%20section/CANCER%20DRUG%20ACCESS%20FINAL%20-%20English.ashx (last accessed 10 September 2012)
- 43.Zheng B, Poulose A, Fulford M, Holbrook A. A pilot study on cost-related medication nonadherence in Ontario. J Popul Ther Clin Pharmacol. 2012;19:e239–247. [PubMed] [Google Scholar]
- 44.Law MR, Cheng L, Dhalla IA, Heard D, Morgan SG. The effect of cost on adherence to prescription medications in Canada. CMAJ. 2012;184:297–302. doi: 10.1503/cmaj.111270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vernon JA. Drug research and price controls. Regulation. 2002;25:22–25. Available at http://www.cato.org/pubs/regulation/regv25n4/v25n4.html (last accessed 10 September 2012) [Google Scholar]
- 46.Barros PP. Pharmaceutical policies in European countries. Adv Health Econ Health Serv Res. 2010;22:3–27. doi: 10.1108/s0731-2199(2010)0000022004. [DOI] [PubMed] [Google Scholar]
- 47.Office of Trade Trading. Annexe K: international survey of pharmaceutical pricing and reimbursement schemes. The Pharmaceutical Price Regulation Scheme’ (Product code: OFT885), February 2007 (Crown Copyright 2007). Available at http://www.oft.gov.uk/OFTwork/markets-work/pprs#named5 (last accessed 10 September 2012)
- 48.Wilking N, Jönsson B, Högberg D, Justo N. Comparator report on patient access to cancer drugs in Europe. Comparator Reports. 15 February 2009. Available at http://www.comparatorreports.se/ (last accessed 10 September 2012)
- 49.Mason A, Drummond M, Ramsey S, Campbell J, Raisch D. Comparison of anticancer drug coverage decisions in the United States and United Kingdom: does the evidence support the rhetoric? J Clin Oncol. 2010;28:3234–3238. doi: 10.1200/JCO.2009.26.2758. [DOI] [PubMed] [Google Scholar]
- 50.Jonsson B, Bergh J. Hurdles in anticancer drug development from a regulatory perspective. Nat Rev Clin Oncol. 2012;9:236–243. doi: 10.1038/nrclinonc.2012.14. [DOI] [PubMed] [Google Scholar]
- 51.Shah DR, Shah RR, Morganroth J. Tyrosine Kinase Inhibitors: their on-target toxicities as potential indicators of efficacy. Drug Saf. 2013 doi: 10.1007/s40264-013-0050-x. (in press) [DOI] [PubMed] [Google Scholar]
- 52.Niraula S, Seruga B, Ocana A, Shao T, Goldstein R, Tannock IF, Amir E. The price we pay for progress: a meta-analysis of harms of newly approved anticancer drugs. J Clin Oncol. 2012;30:3012–3019. doi: 10.1200/JCO.2011.40.3824. [DOI] [PubMed] [Google Scholar]
- 53.Eichler HG, Abadie E, Breckenridge A, Flamion B, Gustafsson LL, Leufkens H, Rowland M, Schneider CK, Bloechl-Daum B. Bridging the efficacy-effectiveness gap: a regulator's perspective on addressing variability of drug response. Nat Rev Drug Discov. 2011;10:495–506. doi: 10.1038/nrd3501. [DOI] [PubMed] [Google Scholar]
- 54.Food and Drug Administration. Transcript of the Oncologic Drugs Advisory Committee meeting of 8 November. 2005. Available at http://www.fda.gov/ohrms/dockets/ac/05/transcripts/2005-4191T1.pdf (last accessed 10 September 2012)
- 55.European Medicines Agency. Presentation by Eichler H-G. Adaptive Licensing: a useful approach for drug licensing in the EU? Available at http://www.ema.europa.eu/docs/en_GB/document_library/Presentation/2012/04/WC500124930.pdf (last accessed 16 January 2013)
- 56.Food and Drug Administration. New drug review 2009 updates. Presentation by Jenkins JK. FDA/CMS Summit, 3 December 2009. Available at http://www.fda.gov/downloads/AboutFDA/CentersOffices/CDER/UCM192786.pdf (last accessed 2 September 2012)