

Abstract
Aims
Eribulin mesilate is an inhibitor of microtubule dynamics that is approved for the treatment of late-stage metastatic breast cancer. Neutropenia is one of the major dose-limiting adverse effects of eribulin. The objective of this analysis was to develop a population pharmacokinetic–pharmacodynamic model for eribulin-associated neutropenia.
Methods
A combined data set of 12 phase I, II and III studies for eribulin mesilate was analysed. The population pharmacokinetics of eribulin was described using a previously developed model. The relationship between eribulin pharmacokinetic and neutropenia was described using a semi-physiological lifespan model for haematological toxicity. Patient characteristics predictive of increased sensitivity to develop neutropenia were evaluated using a simulation framework.
Results
Absolute neutrophil counts were available from 1579 patients. In the final covariate model, the baseline neutrophil count (ANC0) was estimated to be 4.03 × 109 neutrophils l−1 [relative standard error (RSE) 1.2%], with interindividual variability (IIV, 37.3 coefficient of variation % [CV%]). The mean transition time was estimated to be 109 h (RSE 1.8%, IIV 13.9CV%), the feedback constant (γ) was estimated to be 0.216 (RSE 1.4%, IIV 12.2CV%), and the linear drug effect coefficient (SLOPE) was estimated to be 0.0451 μg l−1 (RSE 3.2%, IIV 54CV%). Albumin, aspartate transaminase and receival of granulocyte colony-stimulating factor (G-CSF) were identified as significant covariates on SLOPE, and albumin, bilirubin, G-CSF, alkaline phosphatase and lactate dehydrogenase were identified as significant covariates on mean transition time.
Conclusions
The developed model can be applied to investigate optimal treatment strategies quantitatively across different patient groups with respect to neutropenia. Albumin was identified as the most clinically important covariate predictive of interindividual variability in the neutropenia time course.
Keywords: eribulin mesilate, haematological toxicity, modelling, neutropenia, pharmacodynamics, NONMEM
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Eribulin mesilate is an inhibitor of microtubule dynamics that is approved for the treatment of late-stage metastatic breast cancer.
One of the major dose-limiting effects of eribulin mesilate is neutropenia.
WHAT THIS STUDY ADDS
Eribulin-associated neutropenia was described using a semi-physiological population pharmacokinetic–pharmacodynamic model.
Patient characteristics predictive for interindividual variability in pharmacodynamic parameters were identified, with albumin being a clinically important covariate.
Eribulin dosing guidelines to optimize dose for neutropenia based on expected changes in model parameters were generated.
Introduction
Treatment options for metastatic breast cancer that increase overall survival are limited, and include anthracyclines and taxanes. However, patients may not always show adequate response to these agents or may develop resistance [1]. The anticancer drug eribulin mesilate (E7389) is an inhibitor of microtubule dynamics and is the first in a new class of anticancer drugs referred to as the halichondrins.
The pivotal phase III trial (EMBRACE) of eribulin mesilate showed promising results for women with heavily pretreated metastatic breast cancer, demonstrating a significant benefit in the improvement of median overall survival of 2.5 months compared with the treatment of physician's choice [2]. Based on this trial, eribulin mesilate has been approved by the Food and Drug Administration, European Medicines Agency and Japanese regulatory authorities. In the USA, eribulin mesilate is approved for the treatment of patients with metastatic breast cancer who have previously received at least two chemotherapeutic regimens for the treatment of metastatic disease. Prior therapy should have included an anthracycline and a taxane in either the adjuvant or metastatic setting.
One of the major dose-limiting adverse effects of eribulin mesilate is occurrence of neutropenia. It is therefore important that the incidence and dynamics of this clinically important adverse effect are well understood across the full population of patients who are being treated with eribulin mesilate.
Population pharmacokinetic–pharmacodynamic (PK-PD) modelling may be used to describe the time course of neutropenia. Friberg et al. [3] have described a semi-physiological PK-PD model for haematological toxicity, consisting of system-specific and drug-specific parameters [3]. Parameter consistency has been shown across drugs, and interspecies scaling has also been demonstrated [4]. The model has also been demonstrated to have predictive value during early clinical drug development [5], and has successfully been applied for a range of different drugs [6]. Moreover, a population PK-PD modelling approach allows identification of patient characteristics that are predictive of interpatient variability in PK or PD [7, 8]. This concept may also be applied to identify patient characteristics predictive for increased sensitivity to develop haematological toxicity, in order ultimately to determine optimized dosing schedules in different patient groups [9].
The objectives of this analysis were as follows: (i) the development of a semi-physiological population PK-PD model for eribulin-associated neutropenia; (ii) univariate identification of covariates predictive for interindividual variability in pharmacodynamic model parameters; and (iii) development of a multivariate covariate model that is predictive of interindividual variability in model parameters describing the dynamics of the absolute neutrophil count time course. Ultimately, this model may be applied to optimize drug treatment further in patients treated with eribulin mesilate and to support further clinical development.
Methods
Clinical studies
This analysis was conducted using a pooled data set of phase I, II and III studies of eribulin mesilate. An overview of the included studies is given in Table 1. The data set included 1579 individuals with 23 427 observed absolute neutrophil counts (ANC). Pharmacokinetic data were available for 428 patients (27%).
Table 1.
Overview of clinical studies included in the analysis
| Study | Phase | Indication | Subjects | Objective | Dose | Dose times | PK data | PD data | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | I | Solid tumours | 33 | MTD | 0.25–1.4 mg m−2 IV | Days 1, 8,15 q 21 | Yes | Yes | Goel et al. 2009 |
| 2 | I | Solid tumours | 21 | MTD | 0.25–4.0 mg m−2 IV | Day 1 q 21 | Yes | Yes | Tan et al. 2009 |
| 3 | II | Breast cancer | 104 | ORR | 1.4 mg m−2 IV | Days 1,8,15 q 28 | No | Yes | Vahdat et al. 2009 |
| 4 | II | NSCLC | 106 | ORR | 1.4 mg m−2 IV | Days 1,8,15 q 28 or 1,8 q 21 | No | Yes | Spira et al. 2012 |
| 5 | I | Solid tumours | 6 | ADME | 1.4 mg m−2 IV* | Days 1,8 q 21* | Yes | No | Dubbelman et al. 2012 |
| 6 | I | Solid tumours | 17 | Liver function | 0.7–1.4 mg m−2 IV | – | Yes | Yes | Devriese et al. 2012 |
| 7 | I | Solid tumours | 12 | DDI | 0.7–1.4 mg m−2 IV | – | Yes | Yes | Devriese, Witteveen et al. 2013; Devriese, Mergui-Roelvink et al. 2013 |
| 8 | I | Solid tumours | 26 | QTc prolongation | 1.4 mg m−2 IV | Days 1, 8 q 21 | Yes | Yes | Lesimple et al. 2012 |
| 9 | II | Prostate cancer | 108 | ORR | 1.4 mg m−2 IV | Days 1, 8 q 21 | No | Yes | De Bono et al. 2012 |
| 10 | II | Breast cancer | 298 | ORR | 1.4 mg m−2 IV | Days 1, 8 q 21 | Yes | Yes | Cortes et al. 2010 |
| 11 | III | Breast cancer | 761 | OS | 1.4 mg m−2 IV | Days 1,8 q 21 | Yes | Cortes et al. 2011 | |
| 12 | I | Solid tumours | 15 | MTD | 0.7–2.0 mg m−2 IV | Days 1, 8 q 21 | Yes | Yes | Mukohara et al. 2012 |
| 13 | II | Breast cancer | 81 | ORR | 1.4 mg m−2 IV | Days 1, 8 q 21 | No | Yes | Aogi et al. 2012 |
First cycle a flat 2 mg dose of 14C-eribulin mesylate on day 1 cycle 1 only; thereafter, 1.4 mg m−2 on days 1 and 8. Abbreviations are as follows: ADME, absorption, distribution, metabolism, excretion study; DDI, drug–drug interaction study; IV, intravenous; MTD, maximal tolerated dose; NSCLC, nonsmall cell lung cancer; ORR, objective response rate; OS, overall survival; q, every; QTc, corrected QT interval.
All studies were approved by an Institutional Review Board or Independent Ethics Committee and conducted in accordance with International Conference on Harmonisation guidelines, the Declaration of Helsinki and good clinical practice.
Estimation method and software
R (version 2.10) [10] was used for database handling and generation of diagnostic plots. Pirana (version 2.3.0) [11] was used for the model-building process. Parameter estimation and simulation of the nonlinear mixed effect models were performed using NONMEM (version 7.1.0) [12]. The use of the first-order conditional estimation with interaction method was preferred. However, if it proved not to be computationally feasible, the first-order estimation method was used.
Pharmacokinetic data integration
A previously developed population PK model [unpublished data on file, Dr Z. Hussein (ziad_hussein@eisai.net), Clinical Pharmacology & Translational Medicine, Eisai Limited, Hatfield, UK] including covariates was used to generate predicted PK profiles.
The PK model was systematically developed based on statistical significance of all included parameters, adequate goodness-of-fit plots, bootstrapping and visual predictive checks using both internal and external data sets. In summary, the PK model was a three-compartmental model with linear elimination. The covariates albumin, alkaline phosphatase (ALP) and bilirubin were related to clearance, the covariate dose was related to intercompartmental clearance, and fixed allometric scaling using bodyweight was included on all model parameters using exponents of 0.75 on clearances (CL, Q1 and Q2), and an exponent of 1 on volumes.
Using the previously developed PK model, either typical covariate-adjusted population parameters were used when no PK information was available or, alternatively, when PK observations were available, empirical Bayes estimates were generated which described the individual PK profiles.
Subsequently, the individual or typical covariate-adjusted PK parameters were used as input for the sequential semi-physiological model for neutropenia.
The PK and sequential PK/PD modelling were based on the dose amount of the eribulin free base.
Pharmacodynamic structural model development
The relationship between eribulin exposure and decrease in ANC was described using a semi-physiological lifespan model for haematological toxicity as described by Friberg et al. [3]. This model consists of drug-specific (e.g. linear drug effect coefficient [SLOPE] or concentration of 50% of the maximum effect [EC50] and maximum drug effect [Emax]) and system-specific parameters [e.g. feedback parameter (γ) and the mean transition time (MTT)], and is schematically depicted in Figure 1. Observed absolute neutrophil counts were log-transformed prior to analysis to improve the symmetry of the residual error distribution. The structural model can be described using the following differential equations:
Figure 1.
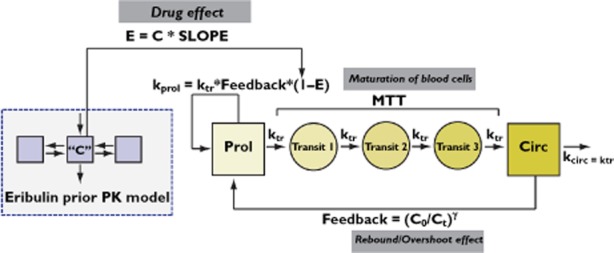
Schematic representation of the semi-physiological model for haematological toxicity. C, drug plasma concentration; C0, circulating neutrophils at time = 0; Ct, circulating neutrophils at time = t; E, drug effect; gamma, feedback constant; kcirc, decay rate of circulating neutrophils; kprol, proliferation rate of neutrophils; ktr, maturation rate of neutrophils; MTT, mean transition time; PK, pharmacokinetics; SLOPE, linear drug effect coefficient.
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
Here, the Prol compartment represents proliferative cells, the compartments Transit1–3 represent transit compartments mimicking the cell maturation occurring with rate ktr, and the Circ compartment represents the observed number of circulating neutrophils. The generation of new cells in the proliferating cells compartment is dependent on the following factors: (i) the number of cells in the compartment; (ii) a proliferation rate constant, kprol, determining the rate of cell division; and (iii) a feedback mechanism from circulating cells (γ). The drug concentration C in the central PK compartment is assumed to reduce the proliferation rate or induce cell loss. This is most commonly modelled using either a linear relationship (Eq. 6) or an Emax relationship (Eq. 7):
| (6) |
| (7) |
Model discrimination was guided by the change in objective function value (OFV) between models, changes in interindividual variability (IIV) and residual unexplained variability (RUV), the magnitude of asymptotic relative standard errors (RSE), goodness of fit and residual error diagnostics. For hierarchical models, a difference in OFV of >7.88 (P < 0.005, d.f. = 1) was used to select the best model. The base model and final model were also evaluated using a visual predictive check. Parameter precision of the final model was evaluated using a nonparametric bootstrap analysis (n = 200).
Statistical model development
The IIV on structural population parameters was described using a log-normal distribution, as follows (Eq. 8):
| (8) |
where Pi represents the individual parameter value, PP,COV represents the covariate adjusted typical parameter value, and ηi is an independent random variable with a distribution of N(0, ω2), where ω represents the population variance for interindividual variability distribution estimated.
Residual error on ANCs was included as a proportional relationship, as follows:
| (9) |
where ANCij,obs represents the observed ANC for the ith individual and the jth observations, ANCij,pred represents the individual predicted ANC value (i.e. based on equations 1–7) for the ith individual and the jth observations, and εij represents the residual error distributed N(0, Σ2), where Σ2 represents the population variance for residual unexplained variability.
Covariate model development
A covariate-screening step was performed for available covariates (Table 2) using plots of empirical Bayes estimates vs. covariates, also taking into account the clinical relevance and biological plausibility of parameter–covariate relationships, as identified in previous implementations of this model for other compounds. Covariates with an expected impact on model parameters were evaluated also when parameter–covariate plots were ambiguous, for instance, due to increased magnitudes of η-shrinkage. We then performed a univariate covariate analysis identifying potential covariates predictive of interindividual variability in model parameters. The clinical relevance of the parameters identified in the univariate analysis was supported by a simulation study, in which the incidences in grade 3 and grade 4 neutropenia were computed.
Table 2.
Patient demographics of the pooled analysis data set
| Description | Unit | Value |
|---|---|---|
| Sex (male/female) | n | 229/1359 |
| Ethnicity | ||
| Caucasian | n | 1253 |
| Black/African American | n | 83 |
| Asian/Pacific Islander | n | 23 |
| Japanese | n | 96 |
| Other/unknown | n | 133 |
| Previous chemotherapy (yes/no) | n | 1527/61 |
| Previous Pt-containing chemotherapy (yes/no) | n | 446/1142 |
| Previous radiotherapy (yes/no) | n | 1212/375 |
| Previous hormonal therapy (yes/no) | n | 923/761 |
| Blood transfusions (yes/no) | n | 161/1523 |
| Received G-CSF (yes/no) | n | 382/1302 |
| Age [median (IQR)] | years | 58.0 (49.0–66.0) |
| Bodyweight [median (IQR)] | kg | 67.7* (59.0–77.6) |
| Body surface area [median (IQR)] | m2 | 1.73 (1.61–1.86) |
| Height [median (IQR)] | cm | 162 (157–168) |
| Albumin [median (IQR)] | g dl−1 | 3.90† (3.6–4.27) |
| Alkaline phosphatase [median (IQR)] | IU l−1 | 118‡ (82.0–206) |
| Alanine transaminase [median (IQR)] | IU l−1 | 25.0 (17.0–39.6) |
| Aspartate transaminase [median (IQR)] | IU l−1 | 30.0 (22.0–46.0) |
| Bilirubin [median (IQR)] | mg dl−1 | 0.50§ (0.40–0.70) |
| Serum creatinine [median (IQR)] | mg dl−1 | 0.80 (0.68–0.91) |
| Lactate dehydrogenase [median (IQR)] | IU l−1 | 328¶ (211–486) |
| Platelets [median (IQR)] | 109 l−1 | 260 (209–326) |
| Protein [median (IQR)] | g dl−1 | 7.1 (6.7–7.5) |
Abbreviations are as follows: G-CSF, granulocyte colony-stimulating factor; IQR, interquartile range; Pt, platinum-containing. Covariates were missing for <6% of subjects for the first or last observation over time per patient, and <13% for intermediate missing observations over time per patient. For <6% of subjects, all covariates were missing, when excluding lactate dehydrogenase (7.82%) and platelets (8.98%). The scaling value used in the covariate model deviated from the median and was set at *70 kg, †4, ‡100, §2 and ¶238.
The effect of continuous covariates covi→n and dichotomous covariates covi→m on the population parameter PP were included as follows (Eq. 10):
| (10) |
where PP,COV represents the covariates adjusted population parameter value, PP represents the typical population parameter estimate, and θi→n and θm→j represent covariate effect parameters. Multilevel categorical covariates, such as race, were evaluated by estimating separate covariate effects for each category.
Covariates that showed a drop in OFV larger than 10.8 (P < 0.001) when tested univariately, were added to the full model. Subsequently, covariates were deleted from the full model in a stepwise backward elimination procedure (again using an OFV difference of 10.8, P < 0.001). A conservative P value of P < 0.001 was used in order to take into account potential deviations from the nominal P value under the first-order estimation method.
Evaluation of the impact of covariates on risk for neutropenia
In order to evaluate the clinical relevance of covariates identified as significant in the univariate analysis on ANC0, and the covariates identified as significant in the final covariate model, simulations were performed by computing the incidences of grade 3 and 4 neutropenia for each of the parameter–covariate relationships separately. Simulations were conducted using the parameter estimates from the univariate runs. For each of these simulation scenarios, the parameter estimates obtained for each of the associated models were used.
Grade 3 toxicity was defined as a ANC < 1 × 109 cells l−1 for >7 days. Grade 4 toxicity was defined as ANC < 0.5 × 109 cells l−1 for >7 days. Patient cohorts were simulated based on the approved dosing regimen of 1.4 mg m−2 at day 1 and 8 for a 21 day treatment cycle. Body surface area (BSA) was simulated from the observed BSA distribution in the full data set, with a mean of 1.57 m2 and standard deviation of 0.22. The BSA was truncated for values within 1–3 mg m−2. A patient cohort of 2000 patients was simulated for each scenario in order to obtain reliable 95% prediction intervals.
Evaluation of the impact of covariates on dosing guidelines
In order to evaluate the impact of covariates identified on model parameters, we evaluated the dose adjustment necessary to match the nadir as predicted for a typical patient receiving a dose of 1.4 mg m−2, for all relevant combinations of model parameters. This was done in the following steps: (i) definition of the final PK-PD model differential equations in the R-package deSolve; (ii) simulation of a single ANC–time profile for a dose of 1.4 mg m−2 using the final covariate model PD parameters and a typical BSA of 1.73 m2; (iii) computation of typical ANC nadir value; and (iv) repeated simulations for different possible deviations in PD parameter values, optimizing dose to match the nadir value obtained in the typical patient, using the optimization function in R.
The resulting matrix of dose adjustments was then depicted graphically, and can be used for easy derivation of recommended dose adjustments, based on specific combinations of patient covariates.
In addition, we simulated ANC–time profiles using the recommended dose of 1.4 mg m−2 for the typical individual, for different combinations of deviations in parameter values. This was done to assess the expected impact of various covariate-induced changes in PD parameter values on ANC–time profiles.
Evaluation of the relationship between exposure and risk for neutropenia
The simulation approach used to compute incidence of grade 3 and 4 neutropenia for separate univariate covariate models described above was also utilized to evaluate the relationship between eribulin exposure (in terms of area under the concentration–time curve, AUC), and the incidence of grade 3 and 4 neutropenia.
Simulations were performed using the base model for three dose levels (0.6–4 mg m−2), with 200 patients per dose level in each data set. Different dose levels were used to obtain a wide range of AUC values. BSA values to calculate absolute dose amounts were simulated according to the same algorithm as described for the clinical evaluation of covariate relationships. For each simulated individual, it was determined whether grade 3 or 4 toxicity had occurred. Then the incidence of grade 3 and 4 neutropenia at different AUC levels was calculated for each data set.
Results
Base model development
Model development
An overview of patient demographics in the final pooled data set is summarized in Table 2. The first-order estimation method was used because first-order conditional estimation with interaction was not computationally feasible. Both SLOPE and Emax models were evaluated. Parameter estimates for the SLOPE model had superior precision compared with the Emax model. Also, we identified a high correlation between the fixed effects of EC50 and Emax. Therefore, the SLOPE model was selected for further model development. Interindividual variability was estimated for all fixed effects. Estimation of interoccasion variability (IOV) and off-diagonal covariances in IIV were not computationally feasible. Residual unexplained variability was best described by a proportional relationship.
Parameter estimates and model evaluation
The parameter estimates of the base model are given in Table 3. Relatively high interindividual variability (56.8 coefficient of variation % [CV%]) was found on the linear drug effect (SLOPE). All parameters were estimated with adequate precision (RSE < 25%). Shrinkage was minimal for baseline neutrophil count (ANC0) and SLOPE (<27.6 CV%), but was high for MTT and γ (37.3 and 51.0%, respectively). The magnitude of residual unexplained variability (49.6 CV%) was high. No relevant trend in observed vs. predicted values was observed.
Table 3.
Parameter estimates of the base model and final covariate model
| Base model | ||||
|---|---|---|---|---|
| Parameter | Units | Estimate (RSE%) | η-shrinkage (%) | |
| Baseline neutrophils (ANC0) | 109 l−1 | 3.97 (1.2) | – | |
| Mean transition time (MTT) | h | 96.7 (5.9) | – | |
| Feedback (γ) | – | 0.206 (2.8) | – | |
| Linear drug effect (SLOPE) | μg l−1 | 0.0414 (4.4) | – | |
| Proportional residual error | CV% | 49.7 (3.2) | – | |
| Between-subject variability | ||||
| Baseline neutrophils (ANC0) | CV% | 37 (2.7) | 12.3 | |
| Mean transition time (MTT) | CV% | 23.2 (20.1) | 37.3 | |
| Feedback (γ) | CV% | 19.9 (25) | 51 | |
| Linear drug effect (SLOPE) | CV% | 56.8 (3.7) | 27.6 | |
| Residual variability | ||||
| Proportional residual error | CV% | 49.7 (3.2) | – |
| Full covariate model | ||||
|---|---|---|---|---|
| Parameter | Units | Estimate (RSE%) | Bootstrap median (IQR)‡ | η-shrinkage (%) |
| Baseline neutrophils (ANC0) | 109 l−1 | 4.03 (1.2) | 4.02 (3.99–4.06) | – |
| Mean transition time (MTT) | h | 109 (1.8) | 109 (107–110) | – |
| Albumin* | 0.374 (23.6) | 0.375 (0.342–0.412) | – | |
| Bilirubin* | −0.046 (25.7) | −0.046 (−0.054 to −0.037) | – | |
| Alkaline phosphatase* | −0.0337 (30.3) | −0.034 (−0.041 to −0.026) | – | |
| Lactate dehydrogenase* | −0.0561 (20.5) | −0.056 (−0.063 to −0.048) | – | |
| Received G-CSF† | 0.883 (2.2) | 0.884 (0.870–0.896) | – | |
| Feedback (γ) | – | 0.216 (1.4) | 0.216 (0.214–0.218) | – |
| Linear drug effect (SLOPE) | μg l−1 | 0.0451 (3.2) | 0.045 (0.044–0.046) | – |
| Albumin* | 0.763 (18.6) | 0.771 (0.682–0.831) | – | |
| Aspartate transaminase* | 0.119 (24.4) | 0.121 (0.099–0.139) | – | |
| Received G-CSF† | 1.3 (8.2) | 1.310 (1.270–1.366) | – | |
| Between-subject variability | ||||
| Baseline neutrophils (ANC0) | CV% | 37.3 (2.6) | 37.3 (36.6–37.9) | 11.3 |
| Mean transition time (MTT) | CV% | 13.9 (6.7) | 13.9 (13.3–14.4) | 36.3 |
| Feedback (γ) | CV% | 12.2 (13) | 12.2 (11.1–13.2) | 54.7 |
| Linear drug effect (SLOPE) | CV% | 54 (4.3) | 54.0 (52.5–55.6) | 28.6 |
| Residual variability | ||||
| Proportional residual error | CV% | 49.6 (2.9) | 49.5 (48.6–50.7) | – |
Power covariate effect.
Proportional covariate effect.
Nonparametric bootstrap (n = 200), median and interquartile range. Abbreviations are as follows: G-CSF, granulocyte colony-stimulating factor; IQR, interquartile range; IIV, interindividual variability (CV%); RSE, relative standard error (%), obtained from NONMEM covariance step; SLOPE, linear drug effect coefficient.
Covariate analysis
Screening and univariate testing of covariates
Available patient demographics and laboratory values were used for a graphical evaluation of each covariate vs. individual parameter values from the base model. All parameter–covariate relationships that were selected for univariate testing are summarized in Table 4.
Table 4.
Univariate analysis results for parameters-covariate combinations evaluated
| Parameter | Covariate | Type | dOFV* | Covariate effect | RSE (%) | Significance |
|---|---|---|---|---|---|---|
| ANC0 | Prior chemotherapy | Categorical | −3.031 | 0.909 | 4.8 | – |
| Sex (male) | Categorical | −62.321 | 1.27 | 3.0 | P < 0.001 | |
| Blood transfusion | Categorical | −24.642 | 1.31 | 5.8 | P < 0.001 | |
| Pt-containing chemotherapy | Categorical | −18.924 | 1.11 | 2.6 | P < 0.001 | |
| Bodyweight | Continuous | −0.556 | 0.0358 | 145.5 | – | |
| Albumin | Continuous | −119.266 | −0.482 | 17.7 | P < 0.001 | |
| MTT | Prior chemotherapy | Categorical | −9.573 | 0.863 | NE | – |
| Albumin | Continuous | −290.1 | 0.48 | 8.6 | P < 0.001 | |
| Bilirubin | Continuous | −145.685 | −0.0997 | 21 | P < 0.001 | |
| AST | Continuous | −183.549 | −0.0993 | 15.3 | P < 0.001 | |
| ALT | Continuous | −61.198 | −0.0547 | 30.7 | P < 0.001 | |
| G-CSF | Categorical | −240.285 | 0.824 | 2.7 | P < 0.001 | |
| Lactate dehydrogenase | Continuous | −188.125 | −0.111 | 15 | P < 0.001 | |
| ALP | Continuous | −118.353 | −0.0779 | 18.5 | P < 0.001 | |
| γ | G-CSF | Categorical | −25.72 | 1.1 | 3.7 | P < 0.001 |
| SLOPE | Asian | Categorical | −9.348 | 1.11 | 3.3 | – |
| Japanese | Categorical | −0.596 | 1.02 | 3.7 | – | |
| Prior chemotherapy | Categorical | −25.31 | 1.47 | 12.6 | P < 0.001 | |
| Albumin | Continuous | −52.166 | 0.433 | 34.4 | P < 0.001 | |
| Sex (male) | Categorical | −21.761 | 0.84 | 5.3 | P < 0.001 | |
| Adjusted Child-Pugh scale | Categorical | −0.001 | 1 | NE | – | |
| Bilirubin | Continuous | −0.64 | −0.0142 | 215.5 | – | |
| AST | Continuous | −84.325 | 0.136 | 24 | P < 0.001 | |
| Bodyweight | Continuous | −11.188 | −0.183 | 41.5 | P < 0.001 | |
| G-CSF | Categorical | −58.501 | 1.27 | 6.3 | P < 0.001 |
Change from OFV of base model (OFV = −9315.974). Abbreviations are as follows: ALT, alanine aminotransferase; AST, aspartate aminotransferase; dOFV, change in objective function value; G-CSF, granulocyte colony-stimulating factor; NE, not estimated; Pt, platinum-containing; RSE, relative standard error; SLOPE, linear drug effect coefficient.
Parameter estimates and model evaluation
In order to keep the full model development feasible in terms of runtime, the full model was constructed containing all statistically significant covariates, except for covariates related to ANC0, because these were considered of less importance. Covariates that were statistically significant (Table 4) were added to a full covariate model, and subsequently a model reduction step was performed (all covariates were significant with dOFV > 32, i.e. highly statistically significant).
The final reduced covariate PK-PD model included the following parameter–covariate relationships: MTT–albumin, MTT–bilirubin, MTT–ALP, MTT–lactate dehydrogenase (LDH), SLOPE–albumin, SLOPE–aspartate transaminase (AST) and SLOPE–granulocyte colony-stimulating factor (G-CSF).
When comparing the base model and the final covariate model, IIV was reduced for MTT from 23.2 to 13.9%, for feedback from 19.9 to 12.2%, and for SLOPE from 56.8 to 54.0%. The parameter estimates of the final covariate model are given in Table 3. All parameters were estimated with adequate precision of typical estimates (RSE < 30.3%). Shrinkage was between 11.3 and 54.7%. No relevant trends in observed vs. predicted neutrophil counts were observed. The visual predictive check described the observed data adequately (Figure 2).
Figure 2.
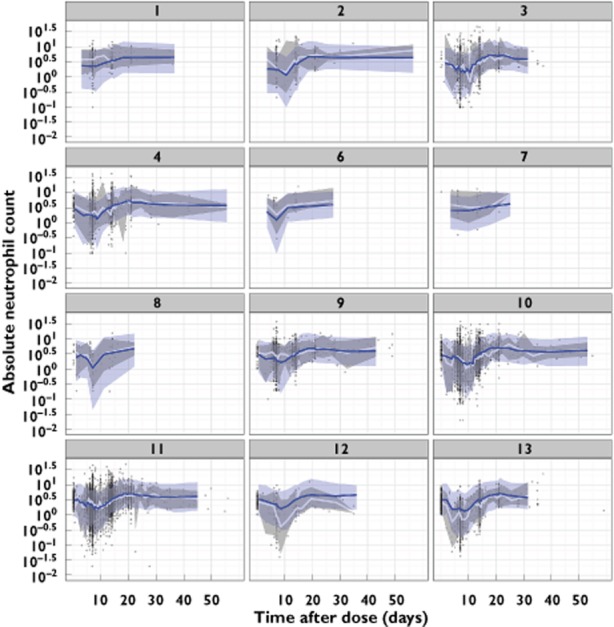
Visual predictive checks of absolute neutrophil count (×109 cells l−1) vs. time after dose for the final covariate model, by study number. The blue line and area represent the model-predicted median and 90% prediction interval. The white line and grey area represent the observed median and 90% prediction interval
Below are the equations providing the population predicted parameter values for MTT and SLOPE in the final model:
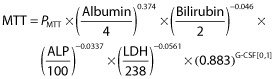 |
(11) |
| (12) |
where PMTT and PSLOPE represent the typical population parameter values, and the depicted biochemical parameter values have the same units as defined in Table 2.
Evaluation of the impact of covariates on risk for neutropenia
Covariates that were identified as significant in the univariate covariate analysis for ANC0 and in the final covariate model were evaluated for their clinical relevance using a simulation study that quantified the change in the incidence of grade 3 and 4 neutropenia (Table 5).
Table 5.
Incidence of grade 3 and 4 neutropenia for different covariates, obtained using stochastic simulations (n = 2000) using the registered eribulin dose of 1.4 mg m−2
| Parameter | Covariate | Neutropenia incidence (%) | Change in neutropenia incidence* (%) | ||
|---|---|---|---|---|---|
| Grade 3 | Grade 4 | Grade 3 | Grade 4 | ||
| Base model | 26.6 | 16.85 | 0 | 0 | |
| ANC0 | Albumin 50% reduced | 25.05 | 40.15 | −1.55 | 23.3 |
| Received blood transfusion | 19.95 | 11.20 | −6.65 | −5.65 | |
| Received prior platinum-containing chemotherapy | 25 | 14.70 | −1.60 | −2.15 | |
| Sex (male) | 21.6 | 12.05 | −5.00 | −4.80 | |
| MTT | Albumin 50% reduced | 23.15 | 55.95 | −3.45 | 39.10 |
| Alkaline phosphatase 50% reduced | 25.65 | 13.55 | −0.95 | −3.30 | |
| Bilirubin 50% reduced | 25.45 | 13.15 | −1.15 | −3.70 | |
| Received G-CSF | 27.05 | 15.1 | 0.45 | −1.75 | |
| Lactate dehydrogenase 50% reduced | 27.3 | 16.50 | 0.70 | −0.35 | |
| SLOPE | Albumin 50% reduced | 29.6 | 35.80 | 3.00 | 18.95 |
| Aspartate transaminase 50% reduced | 24.65 | 12.45 | −1.95 | −4.4 | |
| Received G-CSF | 28.95 | 20.35 | 2.35 | 3.50 | |
Compared with the base model incidences. Abbreviations are as follows: G-CSF, granulocyte colony-stimulating factor; SLOPE, linear drug effect coefficient.
The most pronounced effect was that of albumin, a covariate for ANC0, MTT and SLOPE. If albumin is decreased by 50%, changes in ANC0, MTT and SLOPE will increase grade 4 incidence by 23.3, 39.1 and 18.95%, respectively. The increases seen for hepatic function markers (AST, ALP and bilirubin) were minimal, with <4.4% increase in grade 4 neutropenia. For prior platinum-containing chemotherapy, changes were also minimal at <2.15%. Males showed a decreased incidence of 4.8% in neutropenia compared with females. Patients who received blood transfusions experienced an expected decrease in grade 4 neutropenia of 5.65%. Finally, patients who received G-CSF treatment showed an increased grade 4 incidence (3.5%) with respect to SLOPE.
Evaluation of the impact of covariates on dosing guidelines
In Figure 3, dose adjustments are depicted for a range of relative deviations in relevant PD parameter values (SLOPE, MTT and ANC0), for which covariates were identified. Based on combinations of patient-specific covariate values, the expected deviations in change of model parameter values can be computed (e.g. using Eq. 10–12), and the predicted dose adjustment necessery to match the typical naidr value can be computed.
Figure 3.

Relative change in mean transition time (MTT) vs. baseline absolute neutrophil count (ANC0), stratified by relative change in slope parameter, depicting the change in dose (in milligrams per square metre) needed to match the nadir obtained in the typical patient (i.e. when all parameters are 1) receiving the registered eribulin mesilate dose of 1.4 mg m−2. Doses higher then 1.4 mg m−2 (grey areas) are not depicted because these have not been evaluated in clinical studies and also do not take into account other toxicities
For instance, an individual with a ANC0 of 3.2 × 109 cells l−1 (i.e. 20% reduction from typical value) and, based on a hypothetical patient laboratory values, an expected SLOPE of 0.0496 μg l−1 (10% increase) and an expected MTT of 98.1 h (10% decrease), will lead to a recommended dose of 0.9 mg m−2.
The quantitative relationship between the ANC–time profile for different parameter combinations is depicted in Figure 4.
Figure 4.
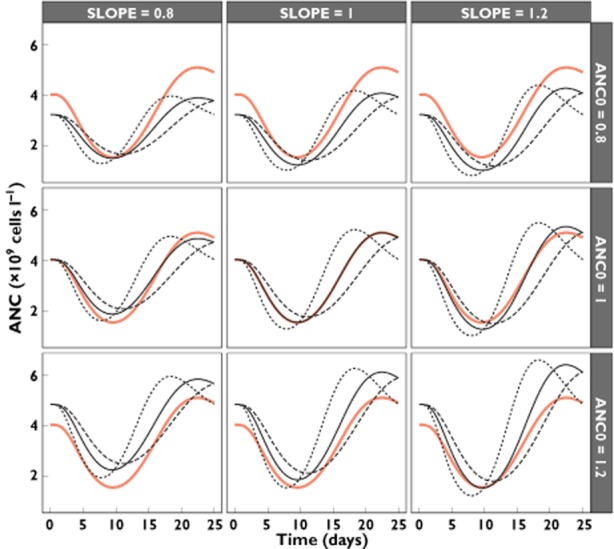
Absolute neutrophil count (ANC) vs. time for the typical patient receiving 1.4 mg m−2 (continuous red line), stratified for different relative deviations from the typical parameter values for SLOPE, ANC0 and MTT (continuous line, MTT = 1; dotted line, MTT = 0.8; dashed line, MTT = 1.2)
Evaluation of the relationship between exposure and risk for neutropenia
In order to illustrate the direct relationship between exposure (i.e. AUC) and incidence of grade 3 or 4 neutropenia further, a simulation study was conducted using the base model (Figure 5). In this figure, the fraction of patients experiencing grade 3 or 4 neutropenia is depicted vs. exposure, demonstrating a clear exposure–response relationship. The incidence of grade 3 neutropenia gradually reaches a maximum near the approved dose levels of 1.4 mg m−2, while grade 4 increases in a linear fashion for higher exposures.
Figure 5.
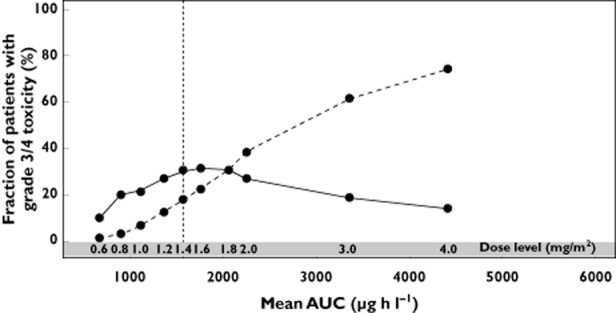
Simulation of exposure (mean AUC) vs. fraction of grade 3 (continous line) and grade 4 neutropenia (dashed line) incidence after 200 simulations for different dose levels
Discussion
Eribulin mesilate is an important drug for the treatment of heavily pretreated patients with metastatic breast cancer, as has been shown in the EMBRACE trial [2]. There is large variability in the clinical condition of this patient population considering characteristics such as organ function, pretreatment, disease burden, age and co-morbidity. It is, therefore, of importance to understand quantitatively the occurrence and determinants of neutropenia, the major dose-limiting adverse effect of eribulin mesilate. In this population PK-PD analysis, data from patients from all available clinical studies (n = 1579) that were available at the time of analysis were included, in order to obtain quantitative insight into the occurrence of neutropenia in different groups of patients. The time course for eribulin-associated neutropenia was adequately described using the developed PK-PD model and could provide insight into the exposure–response relationship (Figure 5). System-specific parameters were found to be consistent with values reported previously for other compounds [3, 5]. Patient characteristics predictive of increased sensitivity to develop neutropenia were identified, and their impact was illustrated using a simulation framework.
We first performed a univariate covariate analysis to identify potential covariates predictive of IIV in model parameters (Table 4). The clinical relevance of the parameters identified in the univariate analysis on ANC0, and the statistically significant covariates in the final covariate model, was supported by a simulation study in which the incidences of grade 3 and grade 4 neutropenia were computed (Table 5). Full model development was performed, including all identified covariates except for any covariates identified on ANC0. We excluded covariates on ANC0 in order to keep runtimes manageable during model development. Moreover, unlike other model parameters, the ANC0 is a parameter which is known in individual patients prior to start of therapy and hence is of less importance. The final full covariate model included the effect of the covariates albumin, bilirubin, ALP and LDH on the parameter MTT, and the effect of the covariates albumin, AST and G-CSF treatment on the drug effect parameter SLOPE.
During full model building, G-CSF effect on γ was removed, ALT, AST and G-CSF effects on MTT were removed, and effects of prior chemotherapy and sex on SLOPE were removed. The removal of G-CSF effect on γ and MTT could be related to confounding effects with the retained effect of G-CSF on SLOPE. The removal of ALT and AST effect on MTT could be related to confounding effects because of the inclusion of related covariates ALP and bilirubin. The removal of prior chemotherapy could be related to the generally impaired health status of patients who have received prior treatments, but in addition the number of patients without pretreatment (4%) was also low. The effect of sex on the incidence of neutropenia was also found in other analyses of conducted clinical trials, but was, however, not retained in the final model. The effect of sex on baseline was not included by choice, whereas the effect on SLOPE dropped out due to lack of significance, possibly related to other confounding factors. Nonetheless, for topotecan and docetaxel, an effect of sex on ANC0 has also been reported [13].
Kloft et al. [13] have investigated patient-related predictors of interindividual variability on model parameters of the same semi-physiological population model as was used in the present analysis, for four different anticancer agents.
With respect to the identified covariates in our final model on MTT, Kloft et al. [13] also identified bilirubin as a covariate on MTT, but did not identify albumin, ALP and LDH as covariates related to MTT. In addition, Kloft et al. [13] did identify LDH to be a covariate on γ, i.e. confirming the relevance of this covariate to its relationship with interindividual variation in the neutrophil time course.
The analysis by Kloft et al. [13] did not investigate eribulin but rather four other anticancer agents, including two taxanes (e.g. also mitotic inhibitors). In our analysis, we identified albumin, AST and G-CSF as significant parameters for SLOPE. The relevance of albumin was also found by Kloft et al. [13], and G-CSF was not included in their analysis.
The present analysis focused on the identification of determinants of neutropenia. Therefore, a previously developed PK model including covariates predictive for PK was used to generate the PK profiles as input for the PD model. For most of the patients in the present data set, no PK data were available, and only typical covariate adjusted PK parameters were used for these patients. It should be stressed that the influence of the population PK model used did not directly influence the outcome of the PD analysis, because the only aim of the PK model was to provide predictions of eribulin concentration–time profiles.
Due to the long runtimes, only the first-order estimation method could be used, which is regarded as a suboptimal estimation method. The employment of data splitting to reduce runtimes and enable external validation of the results was considered; however, as the aim of this analysis was mainly the identification of determinants of neutropenia, the use of data splitting would decrease the informativeness of the covariate and thereby the power of the analysis to identify relevant covariates.
Overall, the developed PK-PD model described the observed data adequately (Figure 2), and parameters could be estimated with good precision, which was confirmed by the bootstrap analysis conducted (Table 3). In addition, the system-specific parameters identified in the semi-physiological model for haematological toxicity were in agreement with previously published values for other drugs [3].
Our analysis indicates that the overall disease state and/or liver function-related covariates (e.g. albumin, bilirubin, ALP, LDH and AST) are the most relevant predictors for interindividual variability in the time course of neutropenia, which is consistent with covariates identified by others [13]. Eribulin is moderately protein bound (49–65%) [14].
Using the developed PK-PD model, quantitative investigation of optimal treatment strategies across different patient groups with respect to neutropenia was performed. However, as some of the identified covariate values are expected to be highly correlated (such as albumin and LDH), it it not feasible to provide concrete dosing guidelines based on specific deviations in covariate values, because of the large range of possible combinations. We therefore used a more general approach, in which we computed dose adjustments needed based on the magnitude of deviation as can be easily computed for specific patients and their associated combination of covariate values (Figure 3).
Figure 3 does not provide doses higher than the registered eribulin mesilate dose of 1.4 mg m−2, because no doses higher than 1.4 mg m−2 were evaluated in either phase II or phase III clinical trials. In addition, this analysis did not take into account any other dose-limiting toxicities that may occur. Any dose increments should therefore be considered only when initial doses appear to be well tolerated.
In the registration trial for eribulin [2] and the associated summary of product characteristics for eribulin, several dose reductions have been suggested after the occurence of adverse events. The suggested initial dose adaptations in the present analysis can potentially reduce the number of such dose reductions that are only implemented after the occurence of an adverse event using the registered dose of 1.4 mg m−2.
In conclusion, we have successfully described the exposure–response relationship for eribulin-associated neutropenia in a large number of patients. Moreover, we have provided a general approach to support dose adaptations in case of complex covariate models. The developed PK-PD model could be used to guide dose optimization, also with respect to infusion duration and timing of dose administration, with respect to elibulin-induced neutropenia.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: all authors had no support from any organization for the submitted work; A.D.R.H., J.H.B. and J.H.M.S. had no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years; the PhD programme of J.G.C.v.H. was financially supported by Eisai Ltd; A.G. and Z.H. are paid employees of Eisai Ltd; no other relationships or activities that could appear to have influenced the submitted work.
References
- 1.Gonzalez-Angulo AM, Morales-Vasquez F, Hortobagyi GN. Overview of resistance to systemic therapy in patients with breast cancer. Adv Exp Med Biol. 2007;608:1–22. doi: 10.1007/978-0-387-74039-3_1. [DOI] [PubMed] [Google Scholar]
- 2.Cortes J, O'Shaughnessy J, Loesch D, Blum JL, Vahdat LT, Petrakova K, Chollet P, Manikas A, Diéras V, Delozier T, Vladimirov V, Cardoso F, Koh H, Bougnoux P, Dutcus CE, Seegobin S, Mir D, Meneses N, Wanders J, Twelves C. Eribulin monotherapy versus treatment of physician's choice in patients with metastatic breast cancer (EMBRACE): a phase 3 open-label randomised study. Lancet. 2011;377:914–923. doi: 10.1016/S0140-6736(11)60070-6. [DOI] [PubMed] [Google Scholar]
- 3.Friberg LE, Henningsson A, Maas H, Nguyen L, Karlsson MO. Model of chemotherapy-induced myelosuppression with parameter consistency across drugs. J Clin Oncol. 2002;20:4713–4721. doi: 10.1200/JCO.2002.02.140. [DOI] [PubMed] [Google Scholar]
- 4.Friberg LE, Sandström M, Karlsson MO. Scaling the time-course of myelosuppression from rats to patients with a semi-physiological model. Invest New Drugs. 2010;28:744–753. doi: 10.1007/s10637-009-9308-7. [DOI] [PubMed] [Google Scholar]
- 5.Soto E, Keizer RJ, Trocóniz IF, Huitema ADR, Beijnen JH, Schellens JHM, Wanders J, Cendrós JM, Obach R, Peraire C, Friberg LE, Karlsson MO. Predictive ability of a semi-mechanistic model for neutropenia in the development of novel anti-cancer agents: two case studies. Invest New Drugs. 2011;29:984–995. doi: 10.1007/s10637-010-9437-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zandvliet AS, Siegel-Lakhai WS, Beijnen JH, Copalu W, Etienne-Grimaldi M-C, Milano G, Schellens JHM, Huitema ADR. PK/PD model of indisulam and capecitabine: interaction causes excessive myelosuppression. Clin Pharmacol Ther. 2008;83:829–839. doi: 10.1038/sj.clpt.6100344. [DOI] [PubMed] [Google Scholar]
- 7.Van Hasselt JGC, Schellens JHM, Mac Gillavry MR, Beijnen JH, Huitema ADR. Model-Based Evaluation and Optimization of Cardiac Monitoring Protocols for Adjuvant Treatment of Breast Cancer with Trastuzumab. Pharm Res. 2012;29:3499–3511. doi: 10.1007/s11095-012-0845-y. [DOI] [PubMed] [Google Scholar]
- 8.Van Hasselt JGC, Boekhout AH, Beijnen JH, Schellens JHM, Huitema ADR. Population pharmacokinetic-pharmacodynamic analysis of trastuzumab-associated cardiotoxicity. Clin Pharmacol Ther. 2011;90:126–132. doi: 10.1038/clpt.2011.74. [DOI] [PubMed] [Google Scholar]
- 9.Zandvliet AS, Schellens JHM, Copalu W, Beijnen JH, Huitema ADR. Covariate-based dose individualization of the cytotoxic drug indisulam to reduce the risk of severe myelosuppression. J Pharmacokinet Pharmacodyn. 2009;36:39–62. doi: 10.1007/s10928-009-9111-2. [DOI] [PubMed] [Google Scholar]
- 10.R Development Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: 2011. [Google Scholar]
- 11.Keizer RJ, Van Benten M, Beijnen JH, Schellens JHM, Huitema ADR. Piraña and PCluster: a modeling environment and cluster infrastructure for NONMEM. Comput Methods Programs Biomed. 2011;101:72–79. doi: 10.1016/j.cmpb.2010.04.018. [DOI] [PubMed] [Google Scholar]
- 12.Beal SL, Boeckman AJ, Sheiner LB. 1988. NONMEM user guides.
- 13.Kloft C, Wallin J, Henningsson A, Chatelut E, Karlsson MO. Population Pharmacokinetic-Pharmacodynamic Model for Neutropenia with Patient Subgroup Identification: comparison across Anticancer Drugs. Clin Cancer Res. 2006;12:5481–5490. doi: 10.1158/1078-0432.CCR-06-0815. [DOI] [PubMed] [Google Scholar]
- 14.Summary of product characteristics HALAVEN. EMA. 2012.
- 15.Goel S, Mita AC, Mita M, Rowinsky EK, Chu QS, Wong N, Desjardins C, Fang F, Jansen M, Shuster DE, Mani S, Takimoto CH. A phase I study of eribulin mesylate (E7389), a mechanistically novel inhibitor of microtubule dynamics, in patients with advanced solid malignancies. Clin Cancer Res. 2009;15:4207–4212. doi: 10.1158/1078-0432.CCR-08-2429. [DOI] [PubMed] [Google Scholar]
- 16.Tan AR, Rubin EH, Walton DC, Shuster DE, Wong YN, Fang F, Ashworth S, Rosen LS. Phase I study of eribulin mesylate administered once every 21 days in patients with advanced solid tumors. Clin Cancer Res. 2009;15:4213–4219. doi: 10.1158/1078-0432.CCR-09-0360. [DOI] [PubMed] [Google Scholar]
- 17.Vahdat LT, Pruitt B, Fabian CJ, Rivera RR, Smith DA, Tan-Chiu E, Wright J, Tan AR, Dacosta NA, Chuang E, Smith J, O'Shaughnessy J, Shuster DE, Meneses NL, Chandrawansa K, Fang F, Cole PE, Ashworth S, Blum JL. Phase II study of eribulin mesylate, a halichondrin B analog, in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. J Clin Oncol. 2009;27:2954–2961. doi: 10.1200/JCO.2008.17.7618. [DOI] [PubMed] [Google Scholar]
- 18.Spira AI, Iannotti NO, Savin MA, Neubauer M, Gabrail NY, Yanagihara RH, Zang EA, Cole PE, Shuster D, Das A. A Phase II Study of Eribulin Mesylate (E7389) in Patients With Advanced, Previously Treated Non-Small-Cell Lung Cancer. Clin Lung Cancer. 2012;13:31–38. doi: 10.1016/j.cllc.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 19.Dubbelman AC, Rosing H, Jansen RS, Mergui-Roelvink M, Huitema ADR, Koetz B, Lymboura M, Reyderman L, Lopez-Anaya A, Schellens JHM, Beijnen JH. Mass Balance Study of 14C-eribulin in Patients with Advanced Solid Tumours. Drug Metab Dispos. 2012;40:313–321. doi: 10.1124/dmd.111.042762. [DOI] [PubMed] [Google Scholar]
- 20.Devriese LA, Witteveen PO, Marchetti S, Mergui-Roelvink M, Reyderman L, Wanders J, Jenner A, Edwards G, Beijnen JH, Voest EE, Schellens JHM. Pharmacokinetics of eribulin mesylate in patients with solid tumors and hepatic impairment. Cancer Chemother Pharmacol. 2012;70:823–832. doi: 10.1007/s00280-012-1976-x. [DOI] [PubMed] [Google Scholar]
- 21.Devriese LA, Witteveen PEO, Wanders J, Law K, Edwards G, Reyderman L, Copalu W, Peng F, Marchetti S, Beijnen JH, Huitema ADR, Voest EE, Schellens JHM. Pharmacokinetics of eribulin mesylate in patients with solid tumours receiving repeated oral rifampicin. Br J Clin Pharmacol. 2013;75:507–515. doi: 10.1111/j.1365-2125.2012.04381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Devriese LA, Mergui-Roelvink M, Wanders J, Jenner A, Edwards G, Reyderman L, Copalu W, Peng F, Marchetti S, Beijnen JH, Schellens JHM. Eribulin mesylate pharmacokinetics in patients with solid tumors receiving repeated oral ketoconazole. Invest New Drugs. 2013;31:381–389. doi: 10.1007/s10637-012-9829-3. [DOI] [PubMed] [Google Scholar]
- 23.Lesimple T, Edeline J, Carrothers TJ, Cvitkovic F, Darpo B, Delord J-P, Léna H, Penel N, Edwards GJ, Law K, Wanders J, Kristensen A, Reyderman L. A phase I, open-label, single-arm study for QT assessment of eribulin mesylate in patients with advanced solid tumors. Invest New Drugs. 2012 doi: 10.1007/s10637-012-9893-8. DOI: 0.1007/s10637-012-9893-8. [DOI] [PubMed] [Google Scholar]
- 24.De Bono JS, Molife LR, Sonpavde G, Maroto JP, Calvo E, Cartwright TH, Loesch DM, Feit K, Das A, Zang EA, Wanders J, Agoulnik S, Petrylak DP. Phase II study of eribulin mesylate (E7389) in patients with metastatic castration-resistant prostate cancer stratified by prior taxane therapy. Ann Oncol. 2012;23:1241–1249. doi: 10.1093/annonc/mdr380. [DOI] [PubMed] [Google Scholar]
- 25.Cortes J, Vahdat L, Blum JL, Twelves C, Campone M, Roché H, Bachelot T, Awada A, Paridaens R, Goncalves A, Shuster DE, Wanders J, Fang F, Gurnani R, Richmond E, Cole PE, Ashworth S, Allison MA. Phase II study of the halichondrin B analog eribulin mesylate in patients with locally advanced or metastatic breast cancer previously treated with an anthracycline, a taxane, and capecitabine. J Clin Oncol. 2010;28:3922–3928. doi: 10.1200/JCO.2009.25.8467. [DOI] [PubMed] [Google Scholar]
- 26.Mukohara T, Nagai S, Mukai H, Namiki M, Minami H. Eribulin mesylate in patients with refractory cancers: a Phase I study. Invest New Drugs. 2012;30:1926–1933. doi: 10.1007/s10637-011-9741-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aogi K, Iwata H, Masuda N, Mukai H, Yoshida M, Rai Y, Taguchi K, Sasaki Y, Takashima S. A phase II study of eribulin in Japanese patients with heavily pretreated metastatic breast cancer. Ann Oncol. 2012;23:1441–1448. doi: 10.1093/annonc/mdr444. [DOI] [PubMed] [Google Scholar]