

Abstract
A multi-dimensional, iterative parallel synthesis effort identified a series of highly selective mGlu3 NAMs with sub-micromolar potency and good CNS penetration. Of these, ML337 resulted (mGlu3 IC50 = 593 nM, mGlu2 IC50 >30 μM) with B:P ratios of 0.92 (mouse) to 0.3 (rat). DMPK profiling and shallow SAR led to the incorporation of deuterium atoms to address a metabolic soft spot, which subsequently lowered both in vitro and in vivo clearance by >50%.
Keywords: Metabotropic glutamate receptor, mGlu3, negative allosteric modulator (NAM), ML337, MLPCN probe
Introduction
G-protein-coupled metabotropic glutamate receptors (mGluRs) have emerged as new drug targets with potential for treatment of a range of CNS disorders.1-4 Highly subtype-selective allosteric ligands have previously been developed for mGlu1, mGlu4, mGlu5 and mGlu7.1-9 While the group II mGluRs (mGlu2 and mGlu3) are among the most highly studied of the mGluR subgroups, previous efforts were limited to group II mGluR ligands that act at both mGlu2 and mGlu3.1-4,6 Recently, selective positive allosteric modulators for mGlu2 have emerged, and demonstrated that mGlu2 activation is responsible for the antipsychotic efficacy of mGlu2/3 agonists.6 However, despite major advances in understanding the functions of mGlu2, mGlu3 remains one of the least understood mGluR subtypes, due in large part to the lack of selective ligands.1-9 Despite this, numerous studies indicate that mGlu3 is the key mGluR subtype involved in glialneuronal communication, and inhibition of mGlu3 is hypothesized to have therapeutic utility in the treatment of cognitive disorders, schizophrenia, depression and Alzheimer's disease.1-12 Therefore, our laboratory focused attention on the development of selective mGlu3 negative allosteric modulators (NAMs) as probes to elucidate the role of mGlu3 in vivo.
To date, only three mGlu3 NAMs have been reported (Figure 1).11-13 The first, RO4491533 (1), a dual mGlu2/mGlu3 NAM (mGlu2 IC50 = 296 nM, mGlu3 IC50 = 270 nM) was efficacious in cognition and depression models.11 About the same time, Lilly disclosed LY2389575 (2), displaying ∼4-fold selectivity for mGlu3 over mGlu2 (mGlu2 IC50 = 17 μM, mGlu3 IC50 = 4.2 μM).12 In 2012, we disclosed a potent (IC50 = 649 nM), selective (>15-fold vs. mGlu2) and CNS-penetrant mGlu3 NAM (3, ML289), derived from a 0.37 μM mGlu5 positive allosteric modulator (PAM).13 Once again, a subtle ‘molecular switch’,15 in the form of a p-methoxy moiety, conferred selective mGlu3 inhibition over mGlu5 potentiation. While this was a notable advance, we continued to seek an mGlu3 NAM probe that was devoid of mGlu2 activity (IC50 >30 μM) in order to enable proof of concept studies.
Figure 1.

Structures and activities of reported mGlu3 NAMs 1-3.
Results and Discussion
Chemistry
3 became our lead compound from which to develop a more potent and selective mGlu3 NAM.13 As we have previously reported, due to the steep nature of allosteric modulator SAR (especially in series prone to ‘molecular switches’), we pursued an iterative parallel synthesis approach for the chemical optimization of 3, 2,12,13 which was divided into five quadrants for SAR exploration (Figure 2). First, we wanted to identify replacements for the metabolically labile p-OMe moiety to improve improve disposition.13 Second, we hoped to employ the wealth of acetylene replacements from previous mGlu5 NAM discovery efforts to replace this less than optimal moiety.8 Third, we desired to perform a broader amide scan to identify novel amide congeners that eliminate mGlu2 activity. Finally, we wanted to see if the ‘fluorine walk’ approach2 would offer advantages in terms of potency, selectivity or DMPK profiles.
Figure 2.

Library optimization strategy for 3 to improve mGlu3 NAM activity, eliminate mGlu2 activity and improve the DMPK profile.
The first libraries were aimed at identifying a replacement for the p-methoxy moiety or electronically perturbing the aryl ring, rendering P450-mediated O-dealkylation less facile.13 Following the synthetic route depicted in Scheme 1, a library of 24 analogs was readily prepared via standard amide and Sonogashira couplings, and screened against both mGlu3 and mGlu2 in kinetic assays (See supplemental information). All compounds possessed purity exceeding 95% as judged by 1H NMR and analytical LCMS (214 nM, 254 nM and ELSD). SAR in this region was found to be shallow, as all attempts to increase steric bulk on the ether or electronically deactivate the aromatic ring (Figure 3) led to a significant loss of mGlu3 activity (IC50s >10 μM); thus the p-methoxy moiety was discovered to be an essential component of the biarylacetylene pharmacophore.
Scheme 1. Synthesis of Aryl Analogues 6a.

aReagents and conditions: (a) (R)-3-hydroxymethyl piperidine, EDC, DMAP, DCM, DIPEA, 95%; (b) 20 mol% CuI, 5 mol% Pd(PPh3)4, arylacetylene (1.1 equiv.), DMF, DIEA, 60 °C, 1 h, 15-90%.
Figure 3.

Representative Ar moieties surveyed to replace the p-OMe phenyl group. All lost significant activity against mGlu3 (IC50s >10 μM).
From the literature regarding acetylene replacements in related mGlu5 NAM biaryl acetylene ligands, we synthesized and screened a diverse array of reported bioisosteres (Figure 4);8 unfortunately, only a few weak NAMs were identified, with most inactive (mGlu3 IC50s >10 μM). Therefore, the p-methoxy phenyl acetylene component was crucial for mGlu3 activity. Based on these data, we elected to survey alternative amide moieties in an effort to improve mGlu3 NAM activity and selectivity while holding the p-OMe phenyl acetylene pharmacophore constant. Key acid 7 was readily prepared by Sonogashira coupling as shown in Scheme 1, and amide analogues were prepared in high yield under standard conditions (Scheme 2).13,15 This library proved far more productive, yielding a number of active analogues, and for the first time, robust SAR and a general lack of activity at mGlu2 (Table 1).
Figure 4.

Representative acetylene biosiosteres surveyed to replace the p-OMe phenyl acetylene group.8 All were weak to inactive on mGlu3 (IC50s >10 μM).
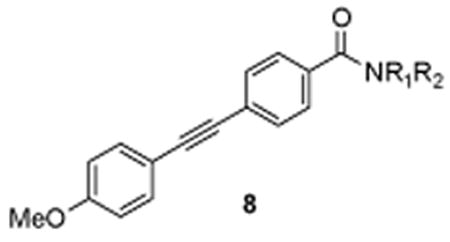
Scheme 2. Synthesis of Amide Analogues 8a.

aReagents and conditions: (a) HNR1R2, EDC, DMAP, DIPEA, CH2Cl2, rt, 16 h, 70-95%.
Table 1.
Structures and Activities of Analogues 8.
| ||||
|---|---|---|---|---|
|
| ||||
| Entry | NR1R2 | mGlu3 pIC50* | Glu Min* (%) | mGlu2 IC50 (μM) |
| 8a |

|
5.87 ±0.04 | 0.4±3.0 | >30 |
| 8b |

|
5.26±0.05 | 0.0±3.2 | >30 |
| 8c |

|
5.77 ±0.04 | 1.7±3.2 | >30 |
| 8d |

|
6.18±0.02 | 2.0±1.5 | >30 |
| 8f |
|
5.12±0.11 | 1.6±39.8 | >30 |
| 8g |

|
4.56±1.61 | --- | >30 |
| 8h |
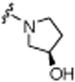
|
4.99±0.09 | 0.0±16.1 | >30 |
| 8i |
|
5.12±0.11 | 0.0±10.3 | >30 |
| 8j |

|
5.96±0.06 | -0.1 ±4.4 | >30 |
| 8k |

|
5.56±0.07 | 1.6±5.9 | >30 |
| 8l |

|
5.26±0.11 | 1.7±11.2 | >30 |
mGlu3 pIC50 and Glu Min data reported as averages ±SEM from our calcium mobilization assay; n = 3
A racemic 3-hydroxy piperidine congener (8a) showed significant activity (mGlu3 IC50 = 760 nM), and upon synthesis of the pure enantiomers, enantioselective inhibition was noted. Here, the (R)-enantiomer (8d) was more potent (mGlu3 IC50 = 650 nM) than the (S)-enantiomer (8c, mGlu3 IC50 = 1.1 μM). When the hydroxy group was capped as a methyl ether in 8b, mGlu3 NAM activity was lost. Interestingly, the [3.3.0] piperidine mimetic was active (8k and 8l), and was a reasonably effective surrogate for the piperidine ring. Contraction to a pyrrolidine ring, as in 8g-i, led to a significant diminution in potency, as did an acyclic congener 8f. Based on the potency of the tertiary hydroxyl analogue 8j (IC50 = 711 nM), we prepared the ethyl and allyl congeners as well, and resolved the enantiomers via chiral SFC.15 Only modest ∼2-fold increases in mGlu3 NAM potency were noted for the (+)-enantiomers (Supplemental Figure 1). Finally, following the synthetic routes depicted in scheme 1 and 2, we incorporated fluorine atoms into the benzoic acid moiety of 8d, and discovered two additional sub-micromolar mGlu3 NAMs 9 and 10 worthy of further profiling (Figure 5).15
Figure 5.

Potent and selective mGlu3 NAMs for further profiling.
Molecular Pharmacology
The four leading mGlu3 NAMs 8d, 8j, 9 and 10 proved to be potent and highly selective versus mGlu2 (Figure 6). Based on DMPK and ancillary pharmacology profiles (vide infra), 9 was favored for further characterization. As shown in Figure 6C, 9 displayed classical non-competitive antagonism with respect to the orthosteric agonist glutamate in a progressive fold shift assay.2,3,13,15 For certain electrophysiology studies, an exogenous agonist may be required in order to engender selective group II mGluR activation; we therefore examined the probe dependence of 9, and noted no differences between glutamate and LY37926816 (Figure 6D). Considering 9 was inactive against the remaining mGluRs (no activity at mGlu1,2,4,5,6,7,8 up to 30 μM) we declared ML337 an MLPCN probe.17
Figure 6.

Molecular pharmacology profile of 9 and related mGlu3 NAMs. A) mGlu3 EC80 antagonist CRC. All four compounds are potent and fully efficacious mGlu3 NAMs (n = 3). B) mGlu2 EC80 CRC. All four compounds are inactive up to 30 μM. C) Progressive fold shift analysis with 9 and glutamate displayed a non-competitive decrease in the EC80, indicating 9 is acting allosterically. D) Evaluating probe dependence. 9 is equipotent and efficacious in inhibiting mGlu3 activation by both glutamate and LY379268 (Supplemental Figure 2).
DMPK Disposition Attributes
9 was subsequntly profiled in a battery of in vitro and in vivo DMPK assays to assess its utility as in vivo probe (Table 2). Although 9 was found to be unstable in rat and human microsomes, it possessed free fractions in both mouse and human plasma approaching 0.03 (97% PPB), as well as a favorable P450 inhibition profile and solubility (7.8 μM in PBS). In a Ricerca radioligand binding panel of 68 GPCRs, ion channels and transporter,18 displayed significant activity (>50% inhibition @10 μM) at only 2 targets (DAT, 71% and 5-HT2B, 74%), but no functional activity at these targets. To rapidly assess the extent of CNS penetration, we performed a mouse tissue distribution study in which 8b, 8j, 9, and 10 were administered as a cassette via an IP route, followed by LC/MS/MS analysis of plasma and brain tissue. All four compounds afforded acceptable CNS exposure, producing brain-to-plasma ratios (B:P) ranging from 0.59 to 0.92 in mice (Supplemental Table 1). 9 demonstrated a B:P ratio approaching unity (B:P, 0.92), with a BrainAUC of 3.37 μM and a corresponding plasmaAUC of 3.71 μM. A subsequent rat study demonstrated a good overall CNS exposure for 9, producing a B:P ratio of 0.3 with high plasma exposures (Supplemental Table 2).
Table 2. DMPK Characterization of 9.
| Parameter | 9 |
|---|---|
| MW | 353.38 |
| TPSA | 59.7 |
| cLogP | 3.51 |
|
| |
| In Vitro Pharmacology | IC50(μM) |
|
| |
| CYP (1A2, 2C9, 3A4, 2D6) | >30, >30, >30, >30 |
|
| |
| In Vitro PK | |
|
| |
| Rat CLHEP (mL/min/kg) | 54.1 |
| Human CLHEP (mL/min/kg) | 18.9 |
| Rat PPB (fu) | 0.005 |
| mPPB (fu) | 0.027 |
|
| |
| In Vivo Rat PK (IP, 10 mg/kg, 0-6 h) | |
|
| |
| Plasma AUC0-6 (μM*h) | 33.1 |
| Brain AUC0-6 (μM*h) | 9.6 |
| Brain:Plasma | 0.3 |
The major metabolite of 9, as with 3, was P450-mediated O-demethylation.14 As mentioned above, all efforts to replace this group synthetically proved futile, resulting in inactive compounds. In an attempt to improve the PK in rodents, we elected to introduce deuterium atoms into the methoxy substituent (D3) of both 8d and 9 in order to increase the metabolic stability of these mGlu3 NAMs (providing 11 and 12, respectively).19 As shown in Table 3, introduction of the D3CO moitety led to an analog with a substantially lower intrinsic clearance (CLint) and predicted hepatic clearance value (CLhep) in vitro. Indeed, the deuteration strategy resulted in an approximate 50% lowering of the plasma clearance (CLp) in rats while providing mGlu3 NAMs of comparable potency and selectivity (Supplemental Figure 3). Importantly, identification of the principal metabolites of the deuterated analogs revealed there to be no metabolic shunt from P450-mediated O-demethylation (data not shown). Thus, employing the apparent kinetic isotope effect as a means to combat the shallow SAR of these allosteric modulators led to improved disposition in vivo. 19
Table 3.
Effect of deuterium incorporation on in vitro and in vivo rat PK with 8d and 9.

| ||||
|---|---|---|---|---|
| R = CH3,8d | R = CD3, 11 | R = CH3, 9 | R = CD3, 12 | |
| Rat CLINT (mL/min/kg) | 214 | 97.3 | 239 | 73.7 |
| Rat CLHEP (mL/min/kg) | 52.7 | 40.7 | 54.1 | 35.9 |
| Rat lV PK CLp (mL/min/kg) | 6.2 | 3.3 | 5.2 | 2.9 |
| Rat IV PK Vss (L/kg) | 0.22 | 0.21 | 0.21 | 0.18 |
| mGIU3 IC50 (μM) | 0.65 | 0.31 | 0.59 | 0.45 |
Conclusion
In summary, we have developed the most potent (mGlu3 IC50 = 593 nM, 1.9% Glu min) and selective (>30 μM versus mGlu1,2,4,5,6,7,8) mGlu3 NAM, 9, described to date. ML337 possesses a favorable DMPK and ancillary pharmacology profile, and is centrally penetrant. The major metabolic soft spot was identified to be P450-mediated O-demethylation, a fate that could not be overcome through standard steric or electronic perturbations, due to extremely shallow allosteric ligand SAR. However, by exploiting apparent kinetic isotope effects, we were able to combat the shallow SAR within this allosteric modulator series and discover an mGlu3 NAM with improved disposition. Electrophysiology and in vivo studies with 9, and its deuterated analogue 12, are in progress and will be reported in due course.
Experimental Section
Chemistry
The general chemistry, experimental information, and syntheses of all other compounds are supplied in the Supporting Information. (R)-(2-Fluoro-4-((4-methoxyphenyl)ethynyl)(3-hydroxypiperdin-1-yl)methanone, 9: To a solution of 2-fluoro-4-((4-methoxyphenyl) ethynyl) benzoic acid (675 mg, 2.5 mmol) in 20 mL DMF, was added DIPEA (1.07 g, 8.25 mmol) while stirring. EDC (560 mg, 3 mmol), HOBt (337 mg, 2.5 mmol), and (R)-3-hydroxypiperidine hydrochloride (342 mg, 2.5 mmol) were then added. The reaction was allowed to stir for 4 hours at room temperature, then quenched with a solution of saturated NaHCO3 (20 mL), washed with 5% LiCl (aqueous, 2 × 20 mL), and brine (20 mL). The reaction was extracted into dichloromethane (50 mL), and solvent was removed under vacuum. HPLC purification afforded 9 as an ivory solid (420 mg, 47%). 1H NMR (500 MHz, d6-DMSO, 75° C) δ (ppm): 7.50 (m, 2H); 7.39 (m, 3H); 6.99 (m, 2H); 4.06 (s, 1H); 3.82 (s, 3H); 3.53 (s, 1H); 3.29 (m, 2H); 2.93 (m, 1H); 1.87 (m, 1H); 1.74 (s, 1H); 1.44 (m, 2H). 13C NMR (125 MHz, d6-DMSO, 75° C) δ (ppm): 163.3, 159.7, 158.0, 156.0, 132.7, 127.2, 125.2 (d, J = 9.3 Hz), 124.3 (d, J = 16.7 Hz), 117.7 (d, J = 22.7 Hz), 114.2, 113.4, 91.1, 85.9, 64.7, 55.0, 53.2, 48.2, 32.2, 28.9. [α]D23 = -27.6° (c = 1, MeOH). LC (254 nm) 0.704 min (>99%); MS (ESI) m/z = 354.1. HRMS (TOF, ES+) C21H20FNO3.[M+H]+ calc. mass 354.1505, found 354.1507.
Supplementary Material
Acknowledgments
Funding Sources: This work was generously supported by the NIH/MLPCN U54 MH084659 (C.W.L.) and NIMH R01MH099269 (K.A.E).
Abbreviations Used
- mGlu3
metabotropic glutamate receptor subtype 3
- CRC
concentration-response-curve
- IP
intra-peritoneal
- MLPCN
Molecular Libraries Probe Production Centers Network
- RCF
relative centrifugal force
Footnotes
Supporting Information. Experimental procedures and spectroscopic data for selected compounds, detailed pharmacology and DMPK methods. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 2.Melancon BJ, Hopkins CR, Wood MR, Emmitte KA, Niswender CM, Christopoulos A, Conn PJ, Lindsley CW. Allosteric modulation of seven transmembrane spanning receptors: theory, practice, and opportunities for central nervous system drug discovery. J Med Chem. 2012;55:1445–1464. doi: 10.1021/jm201139r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Conn PJ, Christopolous A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Conn PJ, Lindsley CW, Jones C. Activation of metabotropic glutamate receptors as a novel approach for the treatment of schizophrenia. Trends in Pharm Sci. 2009;30:25–31. doi: 10.1016/j.tips.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robichaud AJ, Engers DW, Lindsley CW, Hopkins CR. Recent progress on the identification of metabotropic glutamate 4 receptor ligands and their potential utility as CNS therapeutics. ACS Chem Neurosci. 2011;2:433–449. doi: 10.1021/cn200043e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sheffler DJ, Pinkerton AB, Dahl R, Markou A, Cosford NDP. Recent Progress in the Synthesis and Characterization of Group II Metabotropic Glutamate Receptor Allosteric Modulators. ACS Chem Neurosci. 2011;2:382–393. doi: 10.1021/cn200008d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Emmitte KA. Recent advances in the design and development of novel negative allosteric modulators of mGlu5. ACS Chem Neurosci. 2011;2:411–432. doi: 10.1021/cn2000266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stauffer SR. Progress toward Positive Allosteric Modulators of the Metabotropic Glutamate Receptor Subtype 5 (mGlu5) ACS Chem Neurosci. 2011;2:450–470. doi: 10.1021/cn2000519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suzuki G, Tsukamoto N, Fushiki H, Kawagishi A, Nakamura M, Kurihara H, Mitsuya M, Ohkubo M, Ohta H. In vitro pharmacological characterization of novel isoxazolopyridone derivatives as allosteric metabotropic glutamate receptor 7 antagonists. J Pharmacol Exp Ther. 2007;323:147–156. doi: 10.1124/jpet.107.124701. [DOI] [PubMed] [Google Scholar]
- 10.Harrision PJ, Lyon L, Sartorius LJ, Burnet PWJ, Lane TA. The group II metabotropic glutamate receptor 3 (mGlu3, GRM3): expression, function and involvement in schizophrenia. J Psychopharm. 2008;22:308–322. doi: 10.1177/0269881108089818. [DOI] [PubMed] [Google Scholar]
- 11.Campo B, Kalinichev M, Lambeng N, El Yacoubi M, Royer–Urios I, Schneider M, Legarnd C, Parron D, Girard F, Bessif A, Poli S, Vaugeois JM, Le Poul E, Celanire S. Characterization of an mGluR2/3 negative allosteric modulator in rodent models of depression. J Neurogenetics. 2011;24:152–166. doi: 10.3109/01677063.2011.627485. [DOI] [PubMed] [Google Scholar]
- 12.Caraci F, Molinaro G, Battaglia G, Giuffrida ML, Riozzi B, Traficante A, Bruno V, Cannella M, Mero S, Wang X, Heinz BA, Nisenbaum ES, Britton TC, Drago F, Sortino MA, Copani A, Nicoletti F. Targeting group II metabotropic glutamate (mGlu) receptors for the treatment of psychosis associated with Alzheimer's disease: selective activation of mGlu2 receptors amplifies beta-amyloid toxicity in cultured neurons, whereas dual activation of mGlu2 and mGlu3 receptors is neuroprotective. Mol Pharm. 2011;79:618–626. doi: 10.1124/mol.110.067488. [DOI] [PubMed] [Google Scholar]
- 13.Sheffler DJ, Wenthur CJ, Bruner JA, Carrington SJS, Blobaum AL, Morrison RD, Daniels JS, Niswender CM, Conn PJ, Lindsley CW. Development of a novel, CNS penetrant metabotropic glutamate receptor 3 (mGlu3) NAM probe (ML289) derived from a closely related mGlu5 PAM. Bioorg Med Chem Lett. 2012;22:3921–3925. doi: 10.1016/j.bmcl.2012.04.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wood MR, Hopkins CR, Brogan JT, Conn PJ, Lindsley CW. Molecular switches' on allosteric ligands that modulate modes of pharmacology. Biochemistry. 2011;50:2403–2410. doi: 10.1021/bi200129s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.See Supporting Information for full details.
- 16.Monn JA, Valli MJ, Massey SM, Hansen MM, Kress TJ, Wepsiec JP, Harkness AR, Grutsch JL, Wright RA, Johnson BG, Andis SL, Kingston A, Tomlinson R, Lewis R, Griffey KR, Tizzano JP, Schoepp DD. Synthesis, pharmacological characterization, and molecular modeling of heterobicyclic amino acids related to (+)-2-aminobicyclo[3.1.0]hexane-2,6-dicarboxylic acid (LY354740): identification of two new potent, selective, and systemically active agonists for group II metabotropic glutamate receptors. J Med Chem. 1999;42:1027–1040. doi: 10.1021/jm980616n. [DOI] [PubMed] [Google Scholar]
- 17.For MLPCN, please see: http://mli.nih.gov/mli
- 18.For information on Ricerca, see: www.ricerca.com
- 19.Nelson SD, Trager WF. The use of deuterium isotope effects to probe the active site properties, mechanism of cytochrome P450-catalyzed reactions, and mechanisms of metabolically dependent toxicity. Drug Metab Dispos. 2003;31:1481–1498. doi: 10.1124/dmd.31.12.1481. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.