

Abstract
Terpenes are the largest class of small molecule natural products on Earth, and the most abundant by mass. Here, we summarize recent developments in elucidating the structure and function of the proteins involved in their biosynthesis. There are 6 main building blocks or modules (α,β,γ,δ,ε and ζ) that make up the structures of these enzymes: the αα and αδ head-to-tail trans-prenyl transferases that produce trans-isoprenoid diphosphates from C5 precursors; the ε head-to-head prenyl transferases that convert these diphosphates into the tri-and tetra-terpene precursors of sterols, hopanoids and carotenoids; the βγ di- and tri-terpene synthases; the ζ head-to-tail cis-prenyl transferases that produce the cis-isoprenoid diphosphates involved in bacterial cell wall biosynthesis, and finally the α, αβ and αβγ terpene synthases that produce plant terpenes, with many of these modular enzymes having originated from ancestral α and β domain proteins. We also review progress in determining the structure and function of the two 4Fe-4S reductases involved in formation of the C5 diphosphates in many bacteria, where again, highly modular structures are found.
Keywords: terpenes, isoprenoids, biosynthesis, protein structure, evolution, metalloproteins
1. Introduction
Terpenes or isoprenoids are the most diverse class of natural products and are of interest since they are found in almost all life forms where they carry out a myriad of functions ranging from primarily structural (cholesterol in cell membranes) to functional (carotenoids in photosynthesis; retinal in vision; quinones in electron transfer)[1]. Essentially all originate, at least in part, from the C5 substrates dimethylallyl diphosphate (DMAPP, 1, Scheme 1) and isopentenyl diphosphate (IPP, 2), typically by initially condensing DMAPP with one or more IPP molecules in a 1′-4 or “head-to-tail” fashion to form (C10) geranyl diphosphate (GPP, 3); (C15) farnesyl diphosphate (FPP, 4) or (C20) geranylgeranyl diphosphate (GGPP, 5)[2]. FPP and GGPP can then condense in a head-to-head” fashion[3], also termed tail-to-tail by some[4], to form e.g. dehydro-squalene (DHS, 6), squalene (7) or phytoene (8), the precursors of carotenoids such as β-carotene (9), sterols such as cholesterol (10), and hopanoids such as bacteriohopanetetrol (11)- some of the most ancient as well as abundant natural products[1] Isoprenoids can also be used to post-translationally modify proteins (of importance in cell signaling), or they can be cyclized to form the myriad terpene natural products: (C10) monoterpenes such as menthol (12); (C15) sequiterpenes such as farnesene (13) and artemisinin (14); and (C20) diterpenes that are converted to e.g. gibberellic acid (15) and taxol (16). In addition, DMAPP is converted by plants to isoprene (17) itself at a rate of ~100 megatons/year, a reaction that is of current interest as a potential source of “renewable” fuels and other products[5].
Scheme 1.

Isoprenoid biosynthesis: substrates and products
The DMAPP and IPP precursors are made in two different pathways: the mevalonate[6] and methylerythritol phosphate (MEP) pathways[7]. The mevalonate pathway is utilized by most eukaryotes (including humans) as well as Archaea[8], while the MEP pathway is found in most eubacteria. There are of course exceptions, for example the bacterium Staphylococcus aureus uses the mevalonate pathway, while malaria parasites –eukaryotes, use the MEP pathway[9]. In plants, both pathways are found[7], with the MEP pathway typically operating in plastids while the mevalonate pathway is found in the cytosol: sterols (triterpenes) are produced via mevalonate while (C5) hemi-, mono- and di-terpenes, as well as carotenoids (tetraterpenes), are produced via MEP. In the following, we review recent developments in determining the structure and function of many of the key enzymes involved in isoprenoid biosynthesis: the head-to-head and head-to-tail prenyl transferases; the terpene synthases; as well as the 4Fe-4S reductases involved in DMAPP/IPP production in most eubacteria. These structures give important new insights into how the ~65,000 terpenoid natural products[10] are made. In particular, we propose that there are 6 major protein “building blocks” or modules (α, β, γ, δ, ε and ζ) that are used-often in combination- to make the enzymes responsible for formation of most known terpenes, and isoprenoids.
2. Head-to-Tail trans-Prenyl Transferases: αα and αδ Domain Structures
DMAPP and IPP are the C5 substrates used for terpene biosynthesis. They first condense to form the all-trans isoprenoid diphosphates GPP, FPP and GGPP in reactions catalyzed by the enzymes geranyl diphosphate synthase (GPPS), farnesyl diphosphate synthase (FPPS) and geranylgeranyl diphosphate synthase (GGPPS): 1+2→3→4→5, Scheme 2. The first of these structures to be solved [11] was that of FPPS. The structure (Fig. 1a) is almost entirely α-helical and there are two highly conserved repeats containing DDXXD residues (Fig. 1a, in red). These are used to chelate 3Mg2+[12] that, in turn, are responsible for ionization of the allylic substrate (DMAPP) to form a carbo-cation, Scheme 2, which then undergoes nucleophilic attack by the olefinic double bond in IPP, followed by H+-elimination, to form GPP. The process then repeats to form FPP, then (with GGPPS), GGPP. DMAPP (and GPP) bind via Mg2+ to the catalytic Asp in the allylic binding site in FPPS, while IPP binds via a cluster of cationic residues (R57, K60 in human FPPS) in the second, homoallylic site, Fig. 1b[12a].
Scheme 2.

Carbocation mechanism for GPP, FPP, and GGPP biosynthesis
Figure 1.

a) Structure of human FPPS showing conserved DDXXD motifs (red), Mg2+ (blue) and IPP (bottom) and S-thiolo DMAPP (top) ligands. b) expansion of the active site region in a, cationic residues in red and cyan. c) Hetero-tetramer structure of M. piperata GPPS showing catalytic α (yellow) and regulatory (δ) subunits. d) Superposition of the α,δ domains in GPPS. e) Zoledronate and IPP bound to the active site of human FPPS: color code as in a. f) Zoledronate and 19 (NOV-980) bound to the allylic (ZOL) and allosteric (NOV-980) sites in human FPPS.
FPPS as well as most GGPPS molecules function as homo-dimers (αα) with, in some cases, residues from both chains making up the catalytic site[13]. However, it has recently been found that the C10 isoprenoid synthase, GPPS (which provides the GPP needed for menthol biosynthesis), found in plants such as peppermint and spearmint, is a much more complicated system since it contains not one, but two distinct subunits[14], both of which are required for activity: a large subunit (α) containing the DD(X)nD catalytic machinery and a smaller, regulatory subunit (herein called δ) that governs chain elongation. This type of hetero-dimer organization is absent in GPPS from Abies grandis[15], but is also found in human decaprenyl diphosphate synthase[16], which produces the C50 isoprenoid diphosphate required for CoQ10 biosynthesis.
Chang et al.[17] have now reported the x-ray crystallographic structure of M. x piperita GPPS, a likely prototype for other hetero-dimeric systems. The structures reveal a novel architecture in which the large and small subunits form a hetero-dimer that, in turn, dimerizes to form a tetramer (α2δ2), Fig. 1c. The structures contain Mg2+, IPP, S-thiolo-DMAPP (a non-hydrolyzable DMAPP analog) and GPP, all of which bind only to the large, catalytic α subunit, Fig.1c (in yellow). The α domain fold is quite similar to that found in FPPS, with a 2.8Å Cα rmsd. In peppermint GPPS, the regulatory subunit inhibits chain elongation beyond C10, although with FPP as substrate, GGPP can form, in vitro. In previous work[14], it was suggested that these plant GPPS evolved from GGPPS, based on the observation of much larger sequence homology with GGPPS than FPPS (75% vs. 25%), a result now supported by the smaller (0.9Å vs. 2.7Å) Cα rmsd values for the GPPS α-subunit vs. GGPPS over FPPS. What is more surprising about the new GPPS results is that there is also a remarkably close structural similarity between the catalytic (α) and regulatory (δ) subunits in the tetramer, corresponding to a 1.87Å Cα rmsd, Fig. 1d. This strong structural similarity between the α and δ domains, together with a 32% identity and 50% sequence similarity, suggests that such αδ proteins may have originated via a gene duplication, just as with the βγ proteins involved in the terpene synthase reactions discussed below. This αδ catalytic/regulatory domain organization has also now been reported in a second system, hexaprenyl diphosphate synthase from Micrococcus luteus[18]. The δ domain there is quite small (7 helices versus 17 in the α domain) and there is a 2.1Å rmsd between the α and δ domains. The small subunit helps stabilize the dimer via hydrophobic interactions, as well as directly regulating product chain length[18] and based on these results and those with GPPS, it seems likely that similar structures will be found with human DPPS as well.
3. FPPS and GGPPS as Drug Targets
FPPS is of great pharmaceutical interest since it is an important drug target. The bisphosphonates used to treat osteoporosis (and of recent interest in cancer therapy and immunotherapy[19]), such as zoledronate (18, Scheme 3) target the allylic site in FPPS, binding as with DMAPP (Fig. 1b) to the [Mg2+]3 cluster, Fig. 1e[12]. This blocks FPP and GGPP biosynthesis and, consequently, prenylation of proteins such as Ras, resulting in tumor cell killing[20], inhibition of invasiveness[21], phenotype switching in macrophages from a tumor-promoting M2 to a tumor-killing M1 phenotype[22], as well as γδ T cell activation[23], with activated γδ T cells killing tumor cells[24]. These combined effects are thought to contribute to a relative reduction of 36% in the risk of disease progression in breast cancer patients treated with an aromatase inhibitor plus the bisphosphonate zoledronate, over aromatase therapy alone[19]. Bisphosphonates are not, however, conventionally drug-like, due to their extreme polarity and high bone-binding affinity[25], so there has recently been considerable interest in developing new, more lipophilic FPPS inhibitors[26] with Jahnke et al.[27] reporting the discovery of a third, allosteric site in FPPS, together with a new generation of inhibitors (such as 19) that bind to this site[27], Fig. 1f. These inhibitors bind with their polar groups in or close to the IPP diphosphate (PPi) site (Fig. 1f), and have IC50 values as low as 80 nM[27]. Such new-generation non-bisphosphonate FPPS inhibitors lack the structural features needed to bind to bone mineral[25], so have great potential as anti-cancer agents.
Scheme 3.

FPPS and GGPPS inhibitors
In addition to FPPS, GGPPS is also a drug target. Both FPPS as well as GGPPS have αα structures and there is only a 2.4Å rmsd between human FPPS and GGPPS (Fig. 2a;[28]). Surprisingly, however, bisphosphonates such as zoledronate do not inhibit human (or yeast) GGPPS, due it is thought to the absence of one Asp in the 2nd Asp-rich cluster (DDXXN, instead of DDXXD), the absence of which inhibits binding of the 3rd Mg2+[29]. Zoledronate does, however, bind to a GGPPS that has the extra Asp, from the malaria parasite Plasmodium vivax (Fig. 2b;[29]). More lipophilic bisphosphonates such as 20, 21, Scheme 3, bind to yeast, human as well as P. vivax GGPPS [29-30], as illustrated for example in pink in Fig. 2c[30] where the long, hydrophobic side-chain binds in the same site[28b] as does the GGPP product (Fig. 2c; in cyan). These lipophilic bisphosphonates are expected to exhibit better cell/tissue penetration and weaker bone binding [25] than do conventional bisphosphonates, and indeed, they are far more effective in killing tumor cells[26a] as well as malaria parasites[31] than is e.g. zoledronate, both in vitro and in vivo.
Figure 2.

Structures of GGPPS. a) Superposition of human FPPS (green) and human GGPPS (cyan) with the Asp-rich domain (red) and Mg2+ ions (blue) high-lighted as spheres. b) Zoledronate (ZOL) and IPP bound to active site in Plasmodium vivax GGPPS. c) Compound 21 (BPH-715, pink), IPP, Mg2+ bound to S. cerevisae GGPPS superimposed on GGPP (cyan) bound to the product site. Human GGPPS has a very similar local structure and is potently inhibited by 21, but not by zoledronate.
4. The ε Head-to-Head Prenyl Transferases
The isoprenoid diphosphates produced by GPPS, FPPS and GGPPS can be cyclized by a wide variety of terpene synthases (following Sections), and the C15 and C20 diphosphates can also be condensed in a 1′-2,3 or “head-to-head” fashion[3] to form C30 and C40 hydrocarbon species. These are the precursors of sterols, carotenoids and hopanoids, whose diagenetic products are among the most abundant small molecule organic compounds on the planet (≈1012 tons present, in sediments)[32]. With FPP, the initial condensation product (Scheme 4) is the C30 diphosphate presqualene diphosphate (PSPP, 22), formed in a reaction catalyzed by either squalene synthase (SQS) or dehydrosqualene synthase (CrtM).
Scheme 4.

Converting FPP to cyclic products
In plants, animals, fungi and some bacteria, PSPP then undergoes a Mg2+-dependent ionization and loss of PPi, ring opening and reduction (by NADPH) to form squalene, the precursor for sterols such as sitosterol, cholesterol, and ergosterol, as well as many hopanoids, such as hopene (23). In the bacterium S. aureus, the reductive step is missing and the product is dehydrosqualene (6), the precursor of the carotenoid virulence factor staphyloxanthin, a target for anti-infective development[33]. In plants, the C20 diphosphate GGPP condenses in a similar manner to form (C40) prephytoene diphosphate and thence, phytoene, the precursor of carotenoids such as β-carotene[34].
Given the abundance and importance of sterols, carotenoids and hopanoids, it is surprising that, until very recently, the only known structure of a head-to-head prenyl transferase was that of human SQS[35], Fig. 3a (in orange). As with FPPS, the structure is highly α-helical (with a 3.5Å Cα rmsd versus FPPS for 189/284 residues). However, the SQS structure gave relatively little mechanistic information since the inhibitor used was not obviously substrate or product-like. More recently, the structure of the S. aureus dehydrosqualene synthase enzyme, CrtM (in the presence of the non-reactive, FPP-substrate-like inhibitor, S-thiolo-farnesyl diphosphate, FSPP) was reported[33]. The overall fold (we will call ε) is similar to that seen in SQS (a 2.7Å Cα rmsd, Fig. 3a) and there are two FSPP ligands and 3Mg2+, Fig. 3b. But which FPP ionizes to form the farnesyl cation, and which acts as the nucleophile that reacts with the carbocation? This is not clear by inspection of the FSPP x-ray structure (Fig. 3b) since the two sets of possible cation/olefin-carbon distances are both ~5.5Å[33].
Figure 3.

Structures of CrtM and SQS. a) S. aureus CrtM (green) with FSPP, Mg2+ superimposed on human SQS (orange): essential Asp residues and Mg2+ colored as in Fig. 1. b) Active site region in CrtM + FSPP (green, yellow), Mg2+. c) PSPP, Mg2+ (all in cyan) bound to CrtM, superimposed on FSPP/Mg2+ structure (in green/yellow/blue). d) Dehydrosqualene product (pink) bound to CrtM, superimposed on PSPP structure (cyan). S1 = allylic binding site; S2 = homoallylic binding site.
Fortunately, the x-ray crystallographic structure of the PSPP intermediate bound to CrtM has now been reported[36]. The results obtained, Fig. 3c (in cyan), show that FPP in the so-called S1 site is likely to ionize and then react with the double bond in the FPP in the S2 site to form the cyclopropyl carbinyl diphosphate, PSPP, Fig. 3c. The PSPP diphosphate then “flips” back to the [Mg2+]3 cluster and undergoes a second ionization, ring opening and H+ loss, forming dehydrosqualene, which has now been detected in a surface pocket, Fig. 3d (in purple). This mechanism is supported by the results of site-directed mutagenesis[36] and the observation that superimposing the FSPP/Mg2+ CrtM structure on that of prenyl synthases (FPPS, GGPPS) whose mechanisms are known places the S1 site in the “allylic” position found in those enzymes, as well as in terpene cyclases, whose mechanisms are also known[36]. In addition, on mixing CrtM, FSPP and FPP, S-thiolo-PSPP (but no dehydrosqualene) is produced, consistent with FPP ionizing in S1 and FSPP being a good nucleophile, in S2. S-thiolo-PSPP is unable to ionize in the allylic site, just as with S-thiolo-diphosphate inhibitors of other prenyl synthases[37]. The observation of the FPP substrate and PSPP intermediate binding sites, as well as the observation that potent SQS inhibitors (of interest as anti-infectives) also inhibit CrtM and have large hydrophobic interactions in both S1 and S2 sites[36] opens up new routes to developing anti-infective drug leads that target sterol biosynthesis[38], as well as targeting virulence factor formation in S. aureus[33], but work still remains to be done to solve where and how the NADPH reduction step occurs, in SQS.
5. Diterpene Cyclases: The αβγ Fold Hypothesis
Most terpenes contain ring structures and are made by terpene synthases that are generally referred to as terpene cyclases. There was a burst of activity in this area several years ago with the structures of the (C30) triterpene cyclases squalene-hopene cyclase[39] (SHC) and oxidosqualene cyclase[40], the (C15) sesquiterpene cyclases epi-aristolochene synthase (EAS[41]) and pentalene synthase[42], as well as the (C10) monoterpene synthase, bornyl diphosphate synthase (BS[43]), being reported. However, the structures of the (C20) diterpene cyclases have been much more difficult to obtain, but are of interest since they are involved in e.g. taxol and gibberellin biosynthesis. To try and circumvent this lack of direct structural information, bioinformatics and mutagenesis experiments aimed at elucidating some of the key features of diterpene cyclase structure and function were recently reported[44]. This work was stimulated by an earlier genomics study[45] which indicated that an ancestral diterpene cyclase might be the progenitor of modern plant terpene cyclases; the observation[46] that there were structural similarities between a triterpene cyclase and a sesquiterpene cyclase, and similar observations[47] that there were sequence similarities between the bifunctional diterpene cyclase abietadiene (24) synthase (whose structure has not been reported), and that of the sesquiterpene cyclase epi-aristolochene synthase, whose structure is known. The diterpene cyclases catalyze two types of reaction. In class II cyclases, GGPP (5) is protonated to form a carbocation which then cyclizes to form, e.g., copalyl diphosphate (25), Scheme 5. This reaction is known to be catalyzed by a DXDD (not a DDXXD) catalytic motif and is chemically similar to the protonation/cyclization reaction catalyzed by e.g. squalene-hopene cyclase (7→23; Scheme 4) – which also has a highly conserved DXDD catalytic domain. In the class I cyclases, catalysis is fundamentally different and involves the same type of DDXXD/3Mg2+ domains[12b] as seen in the head-to-head and trans-head-to-tail prenyl transferases: the products are the very diverse range of monoterpenes, sesquiterpenes and diterpenes found in plants. The third class of terpene cyclases are the “mixed” class I + II cyclases – such as abietadiene synthase and levopimaradiene synthase, which can carry out both protonation-initiated as well as ionization-initiated reactions. But what might the structures of these, or indeed any other, diterpene cyclases, be?
Scheme 5.

Formation of diterpenes from GGPP
To begin to investigate this question, Cao et al.[44] followed up on earlier observations that many terpene cyclases (such as EAS) contain a highly α-helical catalytic domain (DDXXD/Mg2+) linked to a vestigial N-terminal region, a pattern found in many other proteins including bornyl diphosphate synthase and more recently, isoprene synthase[48]. The DDXXD-containing catalytic domain we call α, since there is considerable 3D structural similarity between the α domain protein FPPS and this domain in such terpene cyclases, e.g. a 3.4 Å rmsd between human FPPS and IS. In the hemi-, mono- and sesqui-terpene cyclases there is also, in general, a second helical domain we call β that itself has structural homology with the barrel structure found in squalene-hopene cyclase[44], Fig. 4a. This suggested that plant diterpene cyclases might contain not only α (Fig. 4b) and β domains but also – since the β domain in the mono- and sesqui-terpene cyclases has structural similarity to the β domain in SHC and the plant diterpene cyclases are very large, a third helical γ-domain as well-since SHC itself contains two β-barrel domains[39]. The diterpene cyclases could then have originated by fusion of the genes of α and βγ domain proteins[44], as illustrated in the hypothetical αβγ structure shown in Fig. 4c. This “structure” (obtained from a SHC/EAS/FPPS alignment) lacks a covalent bond between the α and βγ domains, but the C-terminus of SHC is only ~2.5 Å from the N-terminus of FPPS, as hight-lighted in orange in Fig. 4c.
Figure 4.

Genesis and evolution of terpene cyclases. a) Genes for ancestral βγ domain proteins (like SHC) fuse with genes for ancestral α-domain species like FPPS b) to generate αβγ three-helical domain diterpene cyclase, c) Orange shading indicates close proximity (~2.5 Å) of SHC C-terminus and FPPS N-terminus (from an α/αβ/βγ FPPS/EAS/SHC alignment). d) Structure of an actual diterpene cyclase, taxadiene synthase[51]. e) Loss of the γ domain yields an αβ protein, e.g. the sesquiterpene cyclase isoprene synthase. f) Further loss of the β domain yields other cyclases, e.g., pentalenene synthase, an α domain cyclase. Ancestral α and βγ-domain species presumably produced the FPP, GGPP and squalene used to produce lipids in Archaea; the derived families are much later arrivals. Note the N-terminal helix (magenta) portion is conserved in αβ, βγ, and αβγ proteins and is known to be required for activity.
In class I diterpene cyclases such as taxadiene synthase (TXS), just the conserved DDXXD α-domain (blue) would be functional, even though the βγ domains would be present (and would likely be important for folding). In the class II diterpene cyclases, the β (green, Fig. 4c) and γ (yellow) domains would be functional, but the α domain would not be – except, again, for a likely role in folding – since the DDXXD domain is absent. In the bifunctional class I+II diterpene cyclases, all three domains would be present and involved in catalysis, with the βγ domain catalyzing cyclization of GGPP, the α domain processing the product of the first reaction.
These structural ideas received support from the observation that many bacteria[49] produce gibberellins, diterpenes, but in bacteria their biosynthesis is catalyzed by two separate enzymes: a (class II) ent-copalyl diphosphate synthase (ent-CPPS; GGPP → ent-CPP, 26) and a (class I) kaurene synthase (KS; ent-CPP → ent-kaurene, 27), with, in Bradyrhizobium japonicum, the two open reading frames coding for these different proteins overlapping by a single nucleotide – almost a mixed function enzyme! What is of interest with this ent-CPPS is that it contains not only the DXDD catalytic motif, but also two “QW” motifs or foldons, characteristic of a β-barrel. A β-barrel has 6 inner and 6 outer helices so there should be 24 helices for a βγ structure, and using JPRED[50] and COUDES[51] bioinformatics computer programs, 23 were detected[44]. But are these structural ideas correct?
6. Taxadiene Synthase: Structure of an αβγ Fold, and Evolution to the αβ proteins
The very recent solution[52] of the first single crystal x-ray crystallographic structure of a diterpene cyclase, taxadiene synthase (TXS), supports the structural proposals described above. As predicted, TXS does in fact contain a three-helical domain, αβγ structure, Fig. 4d. In TXS, only the DDXXD motif is present since TXS is a class I cyclase, and the more ancestral DXDD catalytic motif is absent. It is thus remarkable that the αβγ structure is still preserved, even though β and γ play no role in catalysis per se, though of course may be important for folding. Indeed, it was recently shown using chimeras[44] of a plant (αβγ) CPS that, —while the α-domain is required for activity, it has no effect on the stereochemical outcome of the actual βγ-domain catalyzed reaction, and is thus only likely to be important for protein folding/stability.
These TXS structural results support the evolutionary proposal put forth previously[44] that an ancestral (class II) βγ triterpene cyclase (like SHC) may have evolved to a more modern bacterial class II diterpene cyclase which then fused with an ancestral class I cyclase to form a bifunctional, abietadiene synthase-like diterpene cyclase, the progenitor (after exon loss and recombination[45],) of many modern mono-, sequi- and di-terpene cyclases, in addition to isoprene synthase itself[48]. In TXS, the α-domain is quite similar to that found in FPPS (a 3.4 Å rmsd) but the γ-domain clearly has fewer helices present than expected for a “complete” βγ barrel structure, and in many plant terpene cyclases, as well as in isoprene synthase, the γ domain is completely absent, although the β domain remains – even though it does not play a direct role in catalysis.
These are the αβ domain proteins. They contain a very long helix “bridge” that forms part of both the α and β domains (Fig. 4e) and is present in TXS as well (Fig. 4d). And, as noted above, it appears likely that this bridge may have arisen by fusion of the C-terminus of a βγ domain protein with the N-terminus of an α-domain (orange in Fig. 4c). In the case of the αβ protein isoprene synthase, there is positive cooperativity which has been attributed to formation of a dimeric, quaternary structure: α2β2. This dimer is present both in solution as well as in the crystalline solid state. Strikingly, the x-ray structure of isoprene synthase as well as two monoterpene cyclases, limonene synthase and bornyl diphosphate synthase, have almost identical α2β2 quaternary structures[48], as can be seen in Fig. 5. Mechanistically, it has been proposed that in isoprene synthase, the diphosphate group acts a general base, abstracting one of the methyl protons in the DMAPP (1) substrate to form isoprene (17), Scheme 6. The chemistry of this elimination step would then be the same as that yielding farnesene, a potentially important diesel fuel substitute, from FPP. The molecular basis of the cooperativity found in isoprene synthase remains, however, to be elucidated. After loss of the β-domain, the α-domain cyclases such as pentalenene synthase[42] form, Fig. 4f, as proposed earlier[45].
Figure 5.

Dimeric quaternary structure of three α2β2 terpene synthases. a) Limonene synthase and product limonene. b) Bornyl diphosphate synthase and product bornyl diphosphate. c) Isoprene synthase and product isoprene. The catalytic, α or C-terminal domains are in blue, the β or N-terminal domains are in green. The catalytic DDXXD domains are in red. The buried surface areas that comprise the dimerization interface are large, 1148 +/−88 A2. The Cα rmsd between three structures is 1.4-Å. This figure is adapted from Figure 8 in ref. 48 and was constructed from the Protein Data Bank entries: 2ONG, 1N1B, and 3N0F.
Scheme 6.
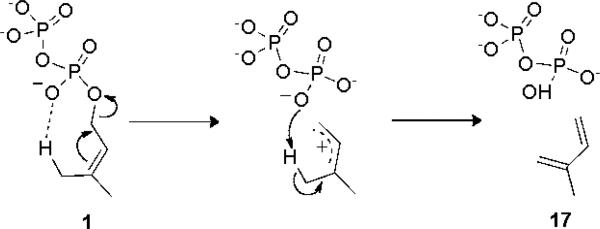
Diphosphate acts as a general base in the conversion of DMAPP to isoprene, catalyzed by isoprene synthase.
Solution of the TXS structure is thus a major development since it strongly supports previous genomics and bioinformatics-based ideas[44-45] about the genesis, as well as the evolution, of many modern plant terpene synthases, in addition to giving some confidence in the use of bioinformatics tools to correctly predict structure and function. Moreover, in more recent work, the structure of an ent-copalyl diphosphate synthase has been reported[53]. As with TXS, it is an αβγ three-helical domain protein, but in this case, has the active site at the interface between the β and γ domains.
7. The ζ (Z or cis) Prenyl Diphosphate Transferases, and Tuberculosinol Synthase
So far, we have only considered the structures and function of the trans-prenyl transferases and some terpene cyclases. There is, however, another important class of prenyl transferases, the Z or cis-prenyl transferases, which catalyze formation of isoprenoid diphosphates containing primarily cis-double bonds. These enzymes are essential for cell wall biosynthesis in bacteria, and as such are potentially important target for anti-infective development. . The protein fold, herein called the ζ–fold (for Z) is completely different to that found in the “FPPS-like” α, δ and ε prenyl transferases[54].
In undecaprenyl (C55) diphosphate synthase (UPPS) from E. coli there is a central β-sheet with 6 parallel strands and 7 surrounding α-helices, Fig. 6a[55]. The FPP and IPP substrates bind as shown in Fig. 6b and, unlike the trans-prenyl transferases and terpene cyclases, there are no conserved DDXXD motifs and no [Mg2+]3 cluster, although Mg2+ is required for catalysis[56]. These results suggest a mechanism for UPPS catalysis different from the sequential ionization-condensation-elimination mechanism observed in the trans-prenyl synthases. In recent work, Lu et al.[56] have shown that with IPP as substrate, there is no evidence for formation of a farnesyl carbocation intermediate (no 3H-farnesol formation from [3H]-FPP,) with either the trans-prenyl transferase octaprenyl diphosphate synthase (OPPS) or with UPPS, but when the reaction rate is decreased by using 3-Br IPP, 3H-farnesol forms with OPPS, but not with UPPS[56]. Since 3-Br-IPP slows down the UPPS reaction, it was proposed that cationic character develops on C3 of IPP after condensation: a concerted mechanism in which IPP attacks FPP without accumulation of a farnesyl carbocation. These results are consistent with the observation that while some of the most potent UPPS inhibitors are bisphosphonates, there is no cationic feature in the UPPS inhibitor pharmacophore[55], unlike the situation with FPPS, and indeed, the presence of a cationic feature actually reduces activity by about one order of magnitude [55]. The key inhibitor features are thus the presence of multiple hydrophobic features, in addition to the polar group, with hydrophilic bisphosphonate drugs such as risedronate having essentially no activity (IC50 ~ 660 μM; [55]).
Figure 6.

Structures and dynamics of the ζ prenyl transferase UPPS. a) Overall structure of a bisphosphonate-bound E. coli UPPS monomer. b) substrates (FPP and IPP) bound to UPPS active site. c) Structural alignment of the predicted structure of Rv3378c with UPPS. d) Molecular dynamics simulation of UPPS. Black, data taken every 10 ps; grey, every 100 ps. e) frequency of occurence of pocket versus pocket volume. The apo structure has a small pocket volume; the largest volume is close to that occupied by a large inhibitor.
The question then arises as to whether or not the ζ- fold is unique, being restricted to UPPS and closely related systems seen in Mycobacteria, or whether it might occur in other systems as well. Using the SSM program[57] to find similar folds reveals no hits. Likely candidates would be other prenyl synthase that use Mg2+, but whose structures are unknown, and the Rv3378c gene product of Mycobacterium tuberculosis, a target for anti-infective therapy[58], appears to be a likely candidate. This protein catalyzes formation of the diterpene virulence factors tuberculosinol (28) and the iso-tuberculosinols (29, 30) from tuberculosinol diphosphate (31): with in this case, H2O being the nucleophile, attacking either the C1 or C3 sites in the allylic substrate[59]. Using three structure prediction programs: I-TASSER[60], SWISS-MODEL[61], and Phyre[62], we find that Rv3378c has distant (22%) sequence homology to UPPS. The top-scoring predicted folds from each program are very similar and one is shown in Fig. 6c, superimposed on the UPPS structure, where there is a 1.93Å rmsd between the Rv3378c Cα prediction and that found in E. coli UPPS. What is particularly interesting about the models is that the DDXXD motif, known to be essential for catalysis[63], is located at the entrance to the main (UPPS) ligand-binding site, adjacent the essential D26 and Mg2+ in the UPPS structure, Fig. 6c, supporting the idea that this protein also adopts the ζ–fold — though as with the diterpene cyclases, x-ray structures are necessary to confirm these predictions.
8. The Dynamic Structure of a Prenyl Transferase, UPPS
From a drug discovery perspective with UPPS, as well as with other proteins, it is of interest to consider how ligand binding affects protein structure. In some cases, the structure of a protein may be known, but there are no substrate, product or inhibitor-bound structures, which makes discovering inhibitors using virtual screening difficult since there may be no obvious ligand-binding pocket that can be targeted. A ligand-free protein must, however, expand to accommodate substrates and products and it is this more “open” structure that is likely to enable inhibitor discovery. One approach to finding such structures is to use molecular dynamics simulations[64]. Starting with an “open” form of UPPS (the structure shown in Fig. 6a, but with the bound ligand removed), a molecular dynamics trajectory (Fig. 6d) shows that the volume (~1000 Å3) originally occupied by the bound ligand rapidly decreases, then stays constant for most of the trajectory and this volume (~430 Å3, Fig 6f) is very similar to the ~330 Å3 seen in a crystal structure of the ligand-free protein[55, 64]. There is, however, a transient opening of the protein to form the substrate/product/inhibitor-binding site, as shown in Figs. 6d-f. Remarkably, use of the rarely-sampled conformational state structure enables (using the Glide program[65]) much tighter ligand-bound poses and better IC50/docking score correlations for a series of bisphosphonate inhibitors of UPPS than found when using the closed form of the enzyme[64] . These results suggest that using MD methods to sample rare “expanded pocket” states is a potentially significant new approach to facilitate inhibitor discovery using virtual screening, which is likely to be applicable to most prenyl transferases and terpene synthases in which large pocket volumes are needed to accommodate large ligands.
9. The 4Fe-4S Reductases: Progress and Puzzles with IspG and IspH
Finally, we consider the question: how, in plant plastids and in many bacteria, are the DMAPP and IPP terpene precursors made? DMAPP and IPP biosynthesis involves initial condensation of pyruvate with glyceraldehyde phosphate to form 1-deoxyxylulose-phosphate which, after four additional reactions, forms 2-C-methyl-D-erythritol-2,4-cyclo-diphosphate (MEcPP, 32). MEcPP is then converted by E-1-hydroxy-2-methyl-but-2-enyl 4-diphosphate (HMBPP) synthase (IspG; also known as GcpE) to form HMBPP (33), which is then reduced by HMBPP reductase (IspH; also known as Lyt B) to form IPP and DMAPP, in a ~5:1 ratio. But what are the structures of these enzymes? How do they carry out these remarkable reactions?
Based on chemical analysis, bioinformatics and EPR spectroscopy, both IspG and IspH have been shown to contain 4Fe-4S clusters akin to those found in ferredoxins, but with an unusual coordination -a non-Cys residue at a unique 4th Fe atom[66], results now confirmed by x-ray structures[67]. The structure of IspH (Fig. 7a) is unusual in that it consists of a modular, clover-leaf or trefoil-like structure in which three distinct helical/sheet domains surround a central Fe/S cluster. This fold is now, however, seen to be very similar to that of two other 4Fe-4S proteins: quinolinate synthase[68] and diphthamide synthase[69] with, on average, a 2.5Å Cα rmsd amongst each of the domains, suggesting again – as with the other prenyl synthases– gene duplication and condensation. Mechanistically, it appears that the HMBPP substrate first binds to the 4th Fe of the 4Fe-4S cluster, then is reduced to an allyl reaction intermediate[67b, 70]. An intermediate lacking the HMBPP O-1 can be seen crystallographically[67b] and has Fe-C bond lengths of ~2.6-2.7 Å, which suggests a metal-ligand interaction (since the Fe-C van der Waals distance is ~3.6 Å[71]), and a detailed discussion of the IspH structure and mechanism has recently been reported in this Journal[72]. The situation with IspG is, however, more complex.
Figure 7.

Modular structures of the 4Fe-4S cluster-containing proteins IspH and IspG. a) E. coli IspH showing the three helix/sheet domains surrounding the 4Fe-4S cluster in the “closed” form (which buries the Fe/S cluster). b) E. coli IspG showing an “open” structure. The 4Fe-4S cluster from one chain is thought to interact with the TIM barrel in the second chain to form the active site (black box). c) Superposition of the TIM barrel in E. coli IspG (orange) with B. anthracis dihydropteroate synthase (cyan). d) Superposition of the 4Fe-4S cluster domain in IspG with that in spinach nitrite reductase. The Cα rmsds in c,d are 2.4, 2.1 Å, respectively.
There are two main mechanisms for IspG catalysis that seem palusible. In one, the cyclo-diphosphate ring in the MEcPP substrate opens to form a carbocation (34) that is then reduced to form an anion which is converted to the HMBPP product. In the other mechanism, the cyclo-diphosphate first isomerizes to form an epoxide (35), which is then deoxygenated by the 4Fe-4S cluster. Support for the latter mechanism is based on precedent: epoxides are known to be reduced to olefins by reduced 4Fe-4S clusters in model systems[73], plus, HMBPP epoxide is reduced by IspG to HMBPP with similar kinetics to that found with MEcPP[74]. However, it has now been reported that the rate of the MEcPP → epoxide reaction catalyzed by IspG is very slow[75] and is inconsistent with the kcat seen with both MEcPP as well as HMBPP-epoxide[76] as substrates, suggesting parallel rather than consecutive reactions, and a common reaction intermediate. Plus, there is now evidence that a carbocation forms with MEcPP+IspG[76]. What, then, might the reaction intermediate be?
When either MEcPP or HMBPP-epoxide are added to reduced IspG, the same reactive intermediate “X”[77] forms[77-78], as observed by EPR, ENDOR or HYSCORE spectroscopy. On incubation, “X” converts to the HMBPP product, which as with IspH, then binds to the 4Fe-4S cluster[78], and there have been several structures (e.g. 36-39) considered for “X”. A radical (36) is unlikely since no radical-like signals are seen in EPR spectra, plus, the resonance that is seen broadens on 57Fe-labeling[78b]. A carbanion (37) is unlikely since it would be very reactive, and a π/η complex (38) is unlikely since not only is it not an η3-oxaallyl (since the oxygen is protonated), but H3 is retained during isoprenoid biosynthesis, as evidenced by 2H-labeling studies[79]. The 13C hyperfine coupling observed (~16 MHz) is similar to that found[80] for an Fe-C bond in a Fe-Fe hydrogenase (17 MHz) as well as that computed[81] for a Mo-C single bond in a model formaldehyde-xanthine oxidase complex (~16 MHz), but is ~3x smaller than the “trans-annular” (2-bond) hyperfine coupling seen (and computed) in the square pyramidal formaldehyde-inhibited xanthine oxidase complex[81]. These results favor, then, the presence of an Fe-C bond, as in ferraoxetanes such as 39, plus, ferraoxetane itself is known to undergo a [2+2] reaction to form ethylene[82]. But what is the structure of IspG, and how might it catalyze such reactions?
In recent work, the first single crystal x-ray crystallographic structure of an IspG, from Aquifex aeolicus, was reported[83]. The structure, Fig. 7b, is of interest in that it is again modular and contains two distinct domains. The large N-terminal domain consists of a triose phosphate isomerase (TIM) barrel that is highly homologous to the structure of Bacillus anthracis dihydropteroate synthase, Fig. 7c (a 2.4Å Cα rmsd), while the C-terminal domain (which houses the 4Fe-4S cluster) is highly homologous (a 2.1Å Cα rmsd) to spinach nitrite reductase, Fig. 7d. The crystallographic results also show the presence of 3 Cys bound to the 4Fe-4S cluster, together with a highly conserved Glu, coordinated to the 4th Fe. The structure of the Thermus thermophilus protein[84] is very similar. Based on the crystallographic structures, it appears unlikely that the two domains function independently in a monomer since the DHPS and 4Fe-4S cluster regions are separated by ~45Å. However, if IspG functions as a dimer – as suggested by the observation that it crystallizes as a dimer –the C-terminus (4Fe-4S cluster) of one molecule in the dimer is then situated close to the N-terminus (TIM barrel) of the second molecule in the dimer (Fig. 6b), and Lee et al.[83] proposed that these two domains can form a “closed” conformation. This would be reminiscent of the movement of one of the three domains in IspH to form the “closed” structure that protects the reactive intermediates during catalysis[67b], as well as the closing of two domains around the 4Fe-4S cluster in acetyl-CoA synthase/carbon monoxide dehydrogenase[85].
In the closed conformation, the substrate would be sandwiched between the TIM barrel of one molecule and the 4Fe-4S cluster in the second molecule in the dimer, forming a single catalytic center in which the cyclo-diphosphate fragment in MEcPP binds to a highly conserved patch of basic residues in the TIM barrel[83]. Other highly conserved residues then catalyze ring opening, while the 4Fe-4S domain (in the closed conformation) carries out the 2H+/2e− redox reaction. This “hybrid” catalytic center would of course be reminiscent of the α2δ2 module interactions in GPPS (Fig. 1c), and is supported by the observation that there are no highly conserved basic residues in the 4Fe-4S cluster to which a diphosphate group can bind. Further support for the “open and closed” model comes from the second IspG structure (from T. thermophilus) in which Rekittke et al.[84] report an even more open, “open” structure, as well as a closed structure model in which the two domains come together to form the catalytic center- the closed structure being generated by a hinge motion between the two domains. It is, however, the TIM barrel of one molecule in the dimer that interacts with the 4Fe-4S cluster in the second molecule, as shown boxed in Fig. 7b. Closed structures with inhibitors/substrates are eagerly awaited.
10. Summary and Outlook
There have recently been numerous major developments in our understanding of the structure, function, evolution and inhibition of many of the enzymes involved in terpene and isoprenoid biosynthesis. These results are important not only from an academic perspective, but are also of practical significance because many of these proteins are targets for drug discovery. The (αα) structures of FPPS and GGPPS are of interest as anti-cancer and anti-infective drug targets, with numerous new drug leads now identified. The structures of a GPPS have been reported: they are remarkable in that GPPS (from M. piperata) consists of an α2δ2 hetero-tetramer with both catalytic (α) and regulatory (δ) subunits. The two subunits have very similar three dimensional structures, though neither domain alone has catalytic activity, and a catalytic/regulatory modular structure appears to be present in C35 and C50 prenyltransferases as well[18]. The new head-to-head synthase structures (of CrtM and SQS) are of interest since they help illuminate the first committed steps in sterol and carotenoid biosynthesis, formation of presqualene diphosphate, again of importance in drug discovery. Bioinformatics predictions about (plant) diterpene cyclase structures in which there are three domains: α, β and γ, have been confirmed experimentally, leading to added confidence in the genomics and bioinformatics proposals that many plant terpene cyclases derive from ancestral αβγ proteins, which themselves appear to have originated by fusion of α and βγ domain proteins: a schematic illustration of the different structural arrangements found with the α,β,γ and δ modules is shown in Fig. 8. The structures of several cis-prenyl transferases, some with bound inhibitors, have also been reported. These adopt the ζ-fold, and based again on bioinformatics, it appears likely that this fold may also be more widespread. The structures of the two 4Fe-4S proteins involved in C5-diphosphate production in most eubacteria have also now been solved. Both have unusual 4Fe-4S clusters with a unique Fe which appears to be involved in Fe-C bond formation during catalysis, and, again, both are modular proteins. And finally, the structures of several αβ proteins, including limonene synthase and isoprene synthase, have been solved. Their three-dimensional structures are remarkably similar, an observation that extends to their essentially identical quaternary structures, with that of isoprene synthase itself being of interest in the context of alternative fuel development.
Figure 8.

Schematic illustration summarizing the modular nature of many terpene/isoprenoid biosynthesis enzymes. FPPS and GPPS are αα dimers (human GGPPS a trimer of dimers); GPPS forms a heterotetramer α2δ2; plant diterpene cyclases (like TXS) are αβγ; many other plant terpene cyclases have lost γ and are αβ, others lack both β and γ and are purely α.
Also of general interest is the observation that while “there is no substitute“ for x-ray crystallography, the use of bioinformatics tools helped correctly predict the three-helical model for TXS; a dihydropteroate synthase (TIM barrel)-sulfite reductase like (4Fe-4S cluster) model is predicted for GcpE, and the δ subunit of heptaprenyl diphosphate synthase from Bacillus subtilis is predicted to have a fold that is very similar (2.8 Å Cα rmsd) to that recently reported for the δ subunit in Micrococcus luteus hexaprenyl diphosphate synthase (PDB ID code 3AQB), making the prediction of a ζ or UPPS-like fold in tuberculosinol synthase (Rv3378c) of considerable interest.
From a future-work perspective: noticeably absent from the structures discussed above are those of many of the enzymes involved in carotenoid biosynthesis. These include the dehydrogenases that convert e.g. dehydrosqualene and phytoene into conjugated polyenes, as well as systems such as lycopene cyclase, that catalyze ring formation. The latter is of interest since it is one of the non-redox flavoproteins in which, apparently, an anionic reduced flavin cofactor (FAD) stabilizes a cationic intermediate or transition state[86], which would be formally similar to the situation found in the class II terpene synthases. A new structure in which FAD plays a key redox role is the FAD-catalyzed reduction of GGPP chains by geranylgeranyl reductase from Thermoplasma acidophilum[87]. This enzyme catalyzes the reduction of geranylgeranyl side-chains to phytanyl side-chains in lipids in Archaea. However, how such C20 side-chains couple via their terminal methyl groups to form the C40 lipids that span the lipid bilayer in Archaea is still a mystery. Interest again is not purely academic since dehydrogenase inhibitors could act as anti-virulence factors for staph infections; carotenoid biosynthesis inhibitors are targets for bleaching herbicides; and it may be possible to use structure-based design to engineer reductases to convert e.g. β-farnesene to farnesane, a bio-fuel, or to produce lower molecular weight species such as dimethyloctane, via GPP.
We are, therefore, near the end of the beginning: the structures of many of the major proteins directly involved in terpene/isoprenoid biosynthesis are now known, and the stage is set for developing novel inhibitors that can be turned into new drugs as well as, potentially, developing new platforms for renewable, isoprene-based fuels, and other materials.
Scheme 7.

Formation of tuberculosinol virulence factors in M. tuberculosis.
Scheme 8.

Formation of HMBPP: substrate, product, and possible reactive intermediates
Acknowledgments
This work was supported by the United States Public Health Service (NIH grants GM065307, GM073216, AI074233 and CA158191). We thank Drs. Rong Cao and Ke Wang for help with the structures and graphics, and Prof. Robert Coates for helpful suggestions.
Biography
 Eric Oldfield received a BSc in Chemistry from Bristol University and a PhD in Biophysical Chemistry from Sheffield University, with Dennis Chapman. He then worked as an EMBO Fellow at Indiana University with Adam Allerhand and at MIT with John Waugh. He joined the Chemistry Department at the University of Illinois at Urbana-Champaign in 1975 and is currently the Alumni Research Scholar Professor of Chemistry. He has been the recipient of several awards including ACS's Award in Pure Chemistry, the Meldola Medal, the Colworth Medal, RSC Awards in Spectroscopy and in Soft Matter and Biophysical Chemistry, and the Biophysical Society's Avanti Award in Lipids.
Eric Oldfield received a BSc in Chemistry from Bristol University and a PhD in Biophysical Chemistry from Sheffield University, with Dennis Chapman. He then worked as an EMBO Fellow at Indiana University with Adam Allerhand and at MIT with John Waugh. He joined the Chemistry Department at the University of Illinois at Urbana-Champaign in 1975 and is currently the Alumni Research Scholar Professor of Chemistry. He has been the recipient of several awards including ACS's Award in Pure Chemistry, the Meldola Medal, the Colworth Medal, RSC Awards in Spectroscopy and in Soft Matter and Biophysical Chemistry, and the Biophysical Society's Avanti Award in Lipids.
 Fu-Yang Lin received his BS and MS degrees in Life Science from National Central University, Taiwan, and a PhD in Biophysics and Computational Biology from the University of Illinois at Urbana-Champaign with Eric Oldfield. He studied the structure, catalytic mechanism of action, and inhibition of prenyl synthases in the Oldfield Group. He is currently a postdoctoral fellow at Harvard Medical School.
Fu-Yang Lin received his BS and MS degrees in Life Science from National Central University, Taiwan, and a PhD in Biophysics and Computational Biology from the University of Illinois at Urbana-Champaign with Eric Oldfield. He studied the structure, catalytic mechanism of action, and inhibition of prenyl synthases in the Oldfield Group. He is currently a postdoctoral fellow at Harvard Medical School.
References
- 1.Chappell J. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1995;46:521–547. [Google Scholar]
- 2.a Liang PH, Ko TP, Wang AH. Eur J Biochem. 2002;269:3339–3354. doi: 10.1046/j.1432-1033.2002.03014.x. [DOI] [PubMed] [Google Scholar]; b Thulasiram HV, Erickson HK, Poulter CD. Science. 2007;316:73–76. doi: 10.1126/science.1137786. [DOI] [PubMed] [Google Scholar]
- 3.Poulter CD. Acc Chem Res. 1990;23:70–77. [Google Scholar]
- 4.Berg JM, Stryer L. Biochemistry. 5th edition W H Freeman; New York: 2002. [Google Scholar]
- 5.Bohlmann J, Keeling CI. Plant J. 2008;54:656–669. doi: 10.1111/j.1365-313X.2008.03449.x. [DOI] [PubMed] [Google Scholar]
- 6.Miziorko HM. Arch Biochem Biophys. 2011;505:131–143. doi: 10.1016/j.abb.2010.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rohmer M. Lipids. 2008;43:1095–1107. doi: 10.1007/s11745-008-3261-7. [DOI] [PubMed] [Google Scholar]
- 8.a Koga Y, Morii H. Microbiol Mol Biol R. 2007;71:97–120. doi: 10.1128/MMBR.00033-06. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lombard J, Moreira D. Mol Biol Evol. 2011;28:87–99. doi: 10.1093/molbev/msq177. [DOI] [PubMed] [Google Scholar]
- 9.Eberl M, Hintz M, Reichenberg A, Kollas AK, Wiesner J, Jomaa H. FEBS Lett. 2003;544:4–10. doi: 10.1016/s0014-5793(03)00483-6. [DOI] [PubMed] [Google Scholar]
- 10.Buckingham J. Dictionary of Natural Products on DVD. 2007 CRC Press. [Google Scholar]
- 11.Tarshis LC, Yan M, Poulter CD, Sacchettini JC. Biochemistry. 1994;33:10871–10877. doi: 10.1021/bi00202a004. [DOI] [PubMed] [Google Scholar]
- 12.a Hosfield DJ, Zhang Y, Dougan DR, Broun A, Tari LW, Swanson RV, Finn J. J Biol Chem. 2004;279:8526–8529. doi: 10.1074/jbc.C300511200. [DOI] [PubMed] [Google Scholar]; b Aaron JA, Christianson DW. Pure Appl Chem. 2010;82:1585–1597. doi: 10.1351/PAC-CON-09-09-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang TH, Guo RT, Ko TP, Wang AH, Liang PH. J Biol Chem. 2006;281:14991–15000. doi: 10.1074/jbc.M512886200. [DOI] [PubMed] [Google Scholar]
- 14.Burke C, Croteau R. J Biol Chem. 2002;277:3141–3149. doi: 10.1074/jbc.M105900200. [DOI] [PubMed] [Google Scholar]
- 15.Burke C, Croteau R. Arch Biochem Biophys. 2002;405:130–136. doi: 10.1016/s0003-9861(02)00335-1. [DOI] [PubMed] [Google Scholar]
- 16.Saiki R, Nagata A, Kainou T, Matsuda H, Kawamukai M. FEBS J. 2005;272:5606–5622. doi: 10.1111/j.1742-4658.2005.04956.x. [DOI] [PubMed] [Google Scholar]
- 17.Chang TH, Hsieh FL, Ko TP, Teng KH, Liang PH, Wang AH. Plant Cell. 2010;22:454–467. doi: 10.1105/tpc.109.071738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sasaki D, Fujihashi M, Okuyama N, Kobayashi Y, Noike M, Koyama T, Miki K. J Biol Chem. 2011;286:3729–3740. doi: 10.1074/jbc.M110.147991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gnant M, Mlineritsch B, Schippinger W, Luschin-Ebengreuth G, Postlberger S, Menzel C, Jakesz R, Seifert M, Hubalek M, Bjelic-Radisic V, Samonigg H, Tausch C, Eidtmann H, Steger G, Kwasny W, Dubsky P, Fridrik M, Fitzal F, Stierer M, Rucklinger E, Greil R, Marth C. N Engl J Med. 2009;360:679–691. doi: 10.1056/NEJMoa0806285. [DOI] [PubMed] [Google Scholar]
- 20.Senaratne SG, Pirianov G, Mansi JL, Arnett TR, Colston KW. Br J Cancer. 2000;82:1459–1468. doi: 10.1054/bjoc.1999.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Denoyelle C, Hong L, Vannier JP, Soria J, Soria C. Br J Cancer. 2003;88:1631–1640. doi: 10.1038/sj.bjc.6600925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coscia M, Quaglino E, Iezzi M, Curcio C, Pantaleoni F, Riganti C, Holen I, Monkkonen H, Boccadoro M, Forni G, Musiani P, Bosia A, Cavallo F, Massaia M. J Cell Mol Med. 2009;14:2803–2815. doi: 10.1111/j.1582-4934.2009.00926.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kunzmann V, Jomaa H, Feurle J, Bauer E, Herderich M, Wilhelm M. Blood. 1997;90:2558–2558. [Google Scholar]
- 24.Kunzmann V, Eckstein S, Lindner A, Tony HP, Wilhelm M. Blood. 2002;100:309B–309B. [Google Scholar]
- 25.Mukherjee S, Huang C, Guerra F, Wang K, Oldfield E. J Am Chem Soc. 2009;131:8374–8375. doi: 10.1021/ja902895p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.a Sanders JM, Song Y, Chan JM, Zhang Y, Jennings S, Kosztowski T, Odeh S, Flessner R, Schwerdtfeger C, Kotsikorou E, Meints GA, Gomez AO, Gonzalez-Pacanowska D, Raker AM, Wang H, van Beek ER, Papapoulos SE, Morita CT, Oldfield E. J Med Chem. 2005;48:2957–2963. doi: 10.1021/jm040209d. [DOI] [PubMed] [Google Scholar]; b Cotesta S, Glickman JF, Jahnke W, Marzinzik A, Ofner S, Rondeau J, Zoller T. p. 2010. (Novartis AG) PCT/EP2009/063257.
- 27.Jahnke W, Rondeau JM, Cotesta S, Marzinzik A, Pelle X, Geiser M, Strauss A, Gotte M, Bitsch F, Hemmig R, Henry C, Lehmann S, Glickman JF, Roddy TP, Stout SJ, Green JR. Nat Chem Biol. 2010;6:660–666. doi: 10.1038/nchembio.421. [DOI] [PubMed] [Google Scholar]
- 28.a Rondeau JM, Bitsch F, Bourgier E, Geiser M, Hemmig R, Kroemer M, Lehmann S, Ramage P, Rieffel S, Strauss A, Green JR, Jahnke W. ChemMedChem. 2006;1:267–273. doi: 10.1002/cmdc.200500059. [DOI] [PubMed] [Google Scholar]; b Kavanagh KL, Dunford JE, Bunkoczi G, Russell RG, Oppermann U. J Biol Chem. 2006;281:22004–22012. doi: 10.1074/jbc.M602603200. [DOI] [PubMed] [Google Scholar]
- 29.Artz JD, Wernimont AK, Dunford JE, Schapira M, Dong A, Zhao Y, Lew J, Russell RG, Ebetino FH, Oppermann U, Hui R. J Biol Chem. 2011;286:3315–3322. doi: 10.1074/jbc.M109.027235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y, Cao R, Yin F, Hudock MP, Guo RT, Krysiak K, Mukherjee S, Gao YG, Robinson H, Song Y, No JH, Bergan K, Leon A, Cass L, Goddard A, Chang TK, Lin FY, Van Beek E, Papapoulos S, Wang AH, Kubo T, Ochi M, Mukkamala D, Oldfield E. J Am Chem Soc. 2009;131:5153–5162. doi: 10.1021/ja808285e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singh AP, Zhang Y, No JH, Docampo R, Nussenzweig V, Oldfield E. Antimicrob Agents Chemother. 2010;54:2987–2993. doi: 10.1128/AAC.00198-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ourisson G, Albrecht P. Acc Chem Res. 1992;25:398–402. [Google Scholar]
- 33.Liu CI, Liu GY, Song Y, Yin F, Hensler ME, Jeng WY, Nizet V, Wang AH, Oldfield E. Science. 2008;319:1391–1394. doi: 10.1126/science.1153018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cunningham FX, Gantt E. Annu Rev Plant Phys. 1998;49:557–583. doi: 10.1146/annurev.arplant.49.1.557. [DOI] [PubMed] [Google Scholar]
- 35.Pandit J, Danley DE, Schulte GK, Mazzalupo S, Pauly TA, Hayward CM, Hamanaka ES, Thompson JF, Harwood HJ., Jr. J Biol Chem. 2000;275:30610–30617. doi: 10.1074/jbc.M004132200. [DOI] [PubMed] [Google Scholar]
- 36.Lin FY, Liu CI, Liu YL, Zhang Y, Wang K, Jeng WY, Ko TP, Cao R, Wang AH, Oldfield E. Proc Natl Acad Sci U S A. 2010;107:21337–21342. doi: 10.1073/pnas.1010907107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Phan RM, Poulter CD. J Org Chem. 2001;66:6705–6710. doi: 10.1021/jo010505n. [DOI] [PubMed] [Google Scholar]
- 38.Fernandes Rodrigues JC, Concepcion JL, Rodrigues C, Caldera A, Urbina JA, de Souza W. Antimicrob Agents Chemother. 2008;52:4098–4114. doi: 10.1128/AAC.01616-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wendt KU, Lenhart A, Schulz GE. J Mol Biol. 1999;286:175–187. doi: 10.1006/jmbi.1998.2470. [DOI] [PubMed] [Google Scholar]
- 40.Thoma R, Schulz-Gasch T, D'Arcy B, Benz J, Aebi J, Dehmlow H, Hennig M, Stihle M, Ruf A. Nature. 2004;432:118–122. doi: 10.1038/nature02993. [DOI] [PubMed] [Google Scholar]
- 41.Starks CM, Back K, Chappell J, Noel JP. Science. 1997;277:1815–1820. doi: 10.1126/science.277.5333.1815. [DOI] [PubMed] [Google Scholar]
- 42.Lesburg CA, Zhai GZ, Cane DE, Christianson DW. Science. 1997;277:1820–1824. doi: 10.1126/science.277.5333.1820. [DOI] [PubMed] [Google Scholar]
- 43.Whittington DA, Wise ML, Urbansky M, Coates RM, Croteau RB, Christianson DW. Proc Natl Acad Sci U S A. 2002;99:15375–15380. doi: 10.1073/pnas.232591099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cao R, Zhang Y, Mann FM, Huang C, Mukkamala D, Hudock MP, Mead ME, Prisic S, Wang K, Lin FY, Chang TK, Peters RJ, Oldfield E. Proteins. 2010;78:2417–2432. doi: 10.1002/prot.22751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Trapp SC, Croteau RB. Genetics. 2001;158:811–832. doi: 10.1093/genetics/158.2.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wendt KU, Schulz GE. Structure. 1998;6:127–133. doi: 10.1016/s0969-2126(98)00015-x. [DOI] [PubMed] [Google Scholar]
- 47.Peters RJ, Croteau RB. Biochemistry. 2002;41:1836–1842. doi: 10.1021/bi011879d. [DOI] [PubMed] [Google Scholar]
- 48.Koksal M, Zimmer I, Schnitzler JP, Christianson DW. J Mol Biol. 2010;402:363–373. doi: 10.1016/j.jmb.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morrone D, Chambers J, Lowry L, Kim G, Anterola A, Bender K, Peters RJ. FEBS Lett. 2009;583:475–480. doi: 10.1016/j.febslet.2008.12.052. [DOI] [PubMed] [Google Scholar]
- 50.Cuff JA, Clamp ME, Siddiqui AS, Finlay M, Barton GJ. Bioinformatics. 1998;14:892–893. doi: 10.1093/bioinformatics/14.10.892. [DOI] [PubMed] [Google Scholar]
- 51.Fuchs PF, Alix AJ. Proteins. 2005;59:828–839. doi: 10.1002/prot.20461. [DOI] [PubMed] [Google Scholar]
- 52.Koksal M, Jin Y, Coates RM, Croteau R, Christianson DW. Nature. 2011;469:116–120. doi: 10.1038/nature09628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koksal M, Hu H, Coates RM, Peters RJ, Christianson DW. Nat Chem Biol. 2011;7:431–433. doi: 10.1038/nchembio.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fujihashi M, Zhang YW, Higuchi Y, Li XY, Koyama T, Miki K. Proc Natl Acad Sci U S A. 2001;98:4337–4342. doi: 10.1073/pnas.071514398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guo RT, Cao R, Liang PH, Ko TP, Chang TH, Hudock MP, Jeng WY, Chen CKM, Zhang YH, Song YC, Kuo CJ, Yin FL, Oldfield E, Wang AHJ. P Natl Acad Sci USA. 2007;104:10022–10027. doi: 10.1073/pnas.0702254104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lu YP, Liu HG, Liang PH. Biochem Bioph Res Commun. 2009;379:351–355. doi: 10.1016/j.bbrc.2008.12.061. [DOI] [PubMed] [Google Scholar]
- 57.Krissinel E, Henrick K. PDBeFold. http://www.ebi.ac.uk/msd-srv/ssm.
- 58.a Pethe K, Swenson DL, Alonso S, Anderson J, Wang C, Russell DG. Proc Natl Acad Sci U S A. 2004;101:13642–13647. doi: 10.1073/pnas.0401657101. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hoshino T, Nakano C, Ootsuka T, Shinohara Y, Hara T. Org Biomol Chem. 2011;9:2156–2165. doi: 10.1039/c0ob00884b. [DOI] [PubMed] [Google Scholar]
- 59.Hoshino T, Nakano C, Ootsuka T, Shinohara Y, Hara T. Org Biomol Chem. 2011;9:2156–2165. doi: 10.1039/c0ob00884b. [DOI] [PubMed] [Google Scholar]
- 60.Roy A, Kucukural A, Zhang Y. Nat Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kiefer F, Arnold K, Kunzli M, Bordoli L, Schwede T. Nucleic Acids Res. 2009;37:D387–392. doi: 10.1093/nar/gkn750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kelley LA, Sternberg MJ. Nat Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 63.Nakano C, Ootsuka T, Takayama K, Mitsui T, Sato T, Hoshino T. Biosci Biotechnol Biochem. 2011;75:75–81. doi: 10.1271/bbb.100570. [DOI] [PubMed] [Google Scholar]
- 64.Sinko W, de Oliveira C, Williams S, Van Wynsberghe A, Durrant JD, Cao R, Oldfield E, McCammon JA. Chem Biol Drug Des. 2011;77:412–420. doi: 10.1111/j.1747-0285.2011.01101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 66.a Adam P, Hecht S, Eisenreich W, Kaiser J, Grawert T, Arigoni D, Bacher A, Rohdich F. Proc Natl Acad Sci U S A. 2002;99:12108–12113. doi: 10.1073/pnas.182412599. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hecht S, Eisenreich W, Adam P, Amslinger S, Kis K, Bacher A, Arigoni D, Rohdich F. Proc Natl Acad Sci U S A. 2001;98:14837–14842. doi: 10.1073/pnas.201399298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.a Rekittke I, Wiesner J, Rohrich R, Demmer U, Warkentin E, Xu W, Troschke K, Hintz M, No JH, Duin EC, Oldfield E, Jomaa H, Ermler U. J Am Chem Soc. 2008;130:17206–17207. doi: 10.1021/ja806668q. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Grawert T, Span I, Eisenreich W, Rohdich F, Eppinger J, Bacher A, Groll M. Proc Natl Acad Sci U S A. 2010;107:1077–1081. doi: 10.1073/pnas.0913045107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sakuraba H, Tsuge H, Yoneda K, Katunuma N, Ohshima T. J Biol Chem. 2005;280:26645–26648. doi: 10.1074/jbc.C500192200. [DOI] [PubMed] [Google Scholar]
- 69.Zhang Y, Zhu X, Torelli AT, Lee M, Dzikovski B, Koralewski RM, Wang E, Freed J, Krebs C, Ealick SE, Lin H. Nature. 2010;465:891–896. doi: 10.1038/nature09138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang W, Wang K, Liu YL, No JH, Li J, Nilges MJ, Oldfield E. Proc Natl Acad Sci U S A. 2010;107:4522–4527. doi: 10.1073/pnas.0911087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Batsanov SS. Inorg Mater. 2001;37:871–885. [Google Scholar]
- 72.Grawert T, Span I, Bacher A, Groll M. Angew Chem Int Edit. 2010;49:8802–8809. doi: 10.1002/anie.201000833. [DOI] [PubMed] [Google Scholar]
- 73.Itoh T, Nagano T, Sato M, Hirobe M. Tetrahedron Lett. 1989;30:6387–6388. [Google Scholar]
- 74.Nyland RL, 2nd, Xiao Y, Liu P, Freel Meyers CL. J Am Chem Soc. 2009;131:17734–17735. doi: 10.1021/ja907470n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xiao YL, Nyland RL, Meyers CLF, Liu PH. Chem Commun. 2010;46:7220–7222. doi: 10.1039/c0cc02594a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xiao Y, Rooker D, You Q, Free Meyers C, Liu P. Chembiochem. 2011;12:527–530. doi: 10.1002/cbic.201000716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Adedeji D, Hernandez H, Wiesner J, Kohler U, Jomaa H, Duin EC. FEBS Lett. 2007;581:279–283. doi: 10.1016/j.febslet.2006.12.026. [DOI] [PubMed] [Google Scholar]
- 78.a Wang W, Li J, Wang K, Huang C, Zhang Y, Oldfield E. Proc Natl Acad Sci U S A. 2010;107:11189–11193. doi: 10.1073/pnas.1000264107. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Xu WY, Lees NS, Adedeji D, Wiesner J, Jomaa H, Hoffman BM, Duin EC. J Am Chem Soc. 2010;132:14509–14520. doi: 10.1021/ja101764w. [DOI] [PubMed] [Google Scholar]
- 79.Hoeffler JF, Hemmerlin A, Grosdemange-Billiard C, Bach TJ, Rohmer M. Biochem J. 2002;366:573–583. doi: 10.1042/BJ20020337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Silakov A, Wenk B, Reijerse E, Albracht SP, Lubitz W. J Biol Inorg Chem. 2009;14:301–313. doi: 10.1007/s00775-008-0449-5. [DOI] [PubMed] [Google Scholar]
- 81.Shanmugam M, Zhang B, McNaughton RL, Kinney RA, Hille R, Hoffman BM. J Am Chem Soc. 2010;132:14015–14017. doi: 10.1021/ja106432h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kafafi ZH, Hauge RH, Billups WE, Margrave JL. J Am Chem Soc. 1987;109:4775–4780. [Google Scholar]
- 83.Lee M, Grawert T, Quitterer F, Rohdich F, Eppinger J, Eisenreich W, Bacher A, Groll M. J Mol Biol. 2010;404:600–610. doi: 10.1016/j.jmb.2010.09.050. [DOI] [PubMed] [Google Scholar]
- 84.Rekittke I, Nonaka T, Wiesner J, Demmer U, Warkentin E, Jomaa H, Ermler U. FEBS Lett. 2011;585:447–451. doi: 10.1016/j.febslet.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 85.Darnault C, Volbeda A, Kim EJ, Legrand P, Vernede X, Lindahl PA, Fontecilla-Camps JC. Nat Struct Biol. 2003;10:271–279. doi: 10.1038/nsb912. [DOI] [PubMed] [Google Scholar]
- 86.Yu Q, Schaub P, Ghisla S, Al-Babili S, Krieger-Liszkay A, Beyer P. J Biol Chem. 2010;285:12109–12120. doi: 10.1074/jbc.M109.091843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xu Q, Eguchi T, Mathews II, Rife CL, Chiu HJ, Farr CL, Feuerhelm J, Jaroszewski L, Klock HE, Knuth MW, Miller MD, Weekes D, Elsliger MA, Deacon AM, Godzik A, Lesley SA, Wilson IA. J Mol Biol. 2010;404:403–417. doi: 10.1016/j.jmb.2010.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]