

Abstract
Whole-exome sequencing and autozygosity mapping studies, independently performed in subjects with defective combined mitochondrial OXPHOS-enzyme deficiencies, identified a total of nine disease-segregating FBXL4 mutations in seven unrelated mitochondrial disease families, composed of six singletons and three siblings. All subjects manifested early-onset lactic acidemia, hypotonia, and developmental delay caused by severe encephalomyopathy consistently associated with progressive cerebral atrophy and variable involvement of the white matter, deep gray nuclei, and brainstem structures. A wide range of other multisystem features were variably seen, including dysmorphism, skeletal abnormalities, poor growth, gastrointestinal dysmotility, renal tubular acidosis, seizures, and episodic metabolic failure. Mitochondrial respiratory chain deficiency was present in muscle or fibroblasts of all tested individuals, together with markedly reduced oxygen consumption rate and hyperfragmentation of the mitochondrial network in cultured cells. In muscle and fibroblasts from several subjects, substantially decreased mtDNA content was observed. FBXL4 is a member of the F-box family of proteins, some of which are involved in phosphorylation-dependent ubiquitination and/or G protein receptor coupling. We also demonstrate that FBXL4 is targeted to mitochondria and localizes in the intermembrane space, where it participates in an approximately 400 kDa protein complex. These data strongly support a role for FBXL4 in controlling bioenergetic homeostasis and mtDNA maintenance. FBXL4 mutations are a recurrent cause of mitochondrial encephalomyopathy onset in early infancy.
Introduction
Mitochondrial respiratory chain (MRC) disorders are a large and diverse group of genetically determined multisystemic conditions characterized by faulty mitochondrial oxidative phosphorylation (OXPHOS). Because OXPHOS depends on a dual contribution of mitochondrial and nuclear genes, mitochondrial disease can be caused by mutations in either mtDNA or nuclear DNA genes. Although numerous disease-associated mutations have been identified in the 37 mtDNA-encoded genes, most are in fact rooted within the nuclear genome, particularly in nuclear genes encoding mitochondria-targeted proteins.1 The mitochondrial proteome is currently estimated to consist of approximately 1,300–1,500 members.2 Bioinformatically predicted and experimentally verified mitochondrial genes have been collated in publicly available compilations, e.g., Mitocarta or MitoP2, which represent an estimated 85% of the actual mitochondrial proteome.3,4 Clinical-molecular investigations through linkage analysis in large or consanguineous pedigrees have long contributed to the identification of several variants causing mitochondrial diseases, 96% of which encode mitochondria-localized proteins.5 More recently, massively parallel-based exome-sequencing approaches have facilitated identification of mitochondrial disease-associated mutations in simplex cases.5,6 These studies have provided substantial insight into the constituents of the mitochondrial proteome as well as into mechanisms of organelle bioenergetics and homeostasis.7 Likewise, studies on mitochondrial disease cases have allowed attribution of an OXPHOS-related role to anonymous gene entries, whose function was previously unknown or uncertain.8
Here, we harness a combined approach of whole-exome sequencing and autozygosity mapping to discover a human mitochondrial disease, caused by mutations within FBXL4 (MIM 605654) that encodes the F-box and leucine-rich repeat 4 protein. FBXL4 missense and/or nonsense recessive mutations were found in nine children from seven unrelated kindreds who presented with lactic acidemia, congenital hypotonia, and a peculiar, slowly progressive mitochondrial encephalomyopathy characterized by atrophy of the brain hemispheres and leukodystrophy. Our subjects also manifest a varying constellation of multisystemic involvement such as facial dysmorphism, renal tubular acidosis, seizures, skeletal abnormalities, and other developmental features. Functional studies clearly demonstrate that FBXL4 is a mitochondrial protein, necessary for OXPHOS proficiency and, possibly, mtDNA maintenance.
Material and Methods
Molecular Studies
Total genomic DNA was extracted by standard methods from peripheral blood lymphocytes, muscle biopsies, or skin fibroblasts (QIAamp DNA Mini Kit, QIAGEN). PCR-based quantification of mtDNA content against a standard, single-copy autosomal gene (encoding RNaseP, Actin, or COX4I1) and sequencing of the entire mtDNA were performed essentially as previously described.9,10 Total RNA was isolated from cell pellets with the RNeasy Mini Kit (QIAGEN) and reverse transcribed to cDNA with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) or GoTaq 2-Step qRT-PCR System (Promega), according to manufacturer recommendations. FBXL4 expression in DNase-treated cDNA samples was determined by reverse-transcription quantitative PCR (qPCR) either with TaqMan probes or with specific FBXL4 amplicons and SYBR-green chemistry (Table S1 available online).
Autozygosity Mapping and Linkage Analysis
DNA was processed for genome-wide SNP genotyping with the Axiom platform (Affymetrix), according to the manufacturer’s instructions. Resulting genotypes were interrogated for runs of homozygosity (ROH) with autoSNPa.11 ROH that are >2 Mb and span >107 SNPs were used as surrogates of autozygosity. Candidate disease ROH was defined based on exclusive sharing between the affected individuals. Statistical confirmation of the candidate disease ROH was achieved by running multipoint linkage analysis via the Allegro algorithm in the easyLINKAGE package.12 Combined LOD score >3 was considered significant.
Whole-Exome Sequencing Analysis
In-solution targeted enrichment of exonic sequences from index cases and also both parents in the case of subject 5 was performed with the 50Mb SureSelect Human All Exon kit from Agilent. The library was subsequently sequenced on a HiSeq2000 or HiSeq2500 (Illumina). For index cases, read alignment to the human genome assembly hg19 (UCSC Genome Browser) was done with BWA (v.0.6.2) and yielded between 9 and 12 Gb of sequence data corresponding to an average fold coverage between 96 and 154 with >92% of the target region being covered >20× (Table S2). Single-nucleotide variants and small insertions and deletions were detected with SAMtools. To extract rare potentially disease-causing variants, we excluded variants present with a frequency >0.1% in 2,250 exomes. Assuming a recessive model of inheritance, we next selected genes predicted to carry either homozygous or compound heterozygous variants, yielding 11, 82, and 7 candidates in subjects 6, 7, and 8, respectively. For subject 5, analyzed together with her parents, exome data sets were analyzed as previously described.13 Custom scripts were used to identify candidate variants that fit a recessive genetic model, revealing five genes with rare, compound heterozygous nonsynonymous variants with biparental inheritance. Variants were determined to be rare based on no more than 10 occurrences in both 536 internal exomes and 1,055 exomes from the Exome Sequencing Project (dbGaP ID: phs000288.v1.p1).
For subject 9, nucleotide sequence analysis of the seven coding exons and exon-intron boundaries of FBXL4 (NC_000006.11, NM_012160.3) was performed with suitable primers (Table S1).
Biocomputational Analyses
Prediction softwares used for pathogenicity prediction of amino acid changes were Polyphen2, SIFT, and Pmut. Prediction softwares used for mitochondrial targeting or subcellular localization were Mitoprot, TargetP, WoLF-PSORT, and MitoMiner.
A three-dimensional structure of the F-box and leucine-rich regions of FBXL4 was built based on the structures of the family members FBXL314 and SKP215 by using the MODELER comparative protein modeling program.16 The protein sequences of human FBXL4, FBXL3, and SKP2 were aligned manually with Jalview.17 The MODELER program was used to produce 50 comparative models of FBXL3 from this sequence alignment and the structures of FBXL3 and SKP2. The model with the lowest score of the MODELER objective function was taken as the representative model. The structures of the comparative models were examined and figures produced with the PyMOL molecular visualization system (The PyMOL Molecular Graphics System, v.1.4.1, Schrodinger, LLC).
Cell Cultures
Human embryonic kidney (HEK293T) cells and skin fibroblasts were cultured in 1 g/l glucose DMEM (Gibco) supplemented with 20% FBS, 1% uridine, 1% L-glutamine, and 0.2% sodium pyruvate. Cells were grown to confluence in T75 flasks in a 37°C incubator with 5% CO2 and 100% humidity. For DNA/RNA extraction and immunoblot analysis, fibroblast pellets were prepared, as previously described.18 For visualization of the mitochondrial network, the mitochondrial fluorescent dye MitoTracker RedCMXRos (Invitrogen) and the nuclear dye Hoechst33342 (Sigma) were added to the culture media at final concentrations of 50 nM and 160 nM, respectively. Cells were incubated under normal culture conditions for 30 min and then analyzed by fluorescence microscopy (Nikon-CARV2 system).
HEK293T cells were transfected with Lipofectamine 2000 (Invitrogen) with recombinant FBXL4 cDNAs and inducible stable cell lines were established with suitable selectable markers.
FACS-Based Mitochondrial Membrane Potential and Mitochondria Content Analysis
Cells were collected in HBSS, counted, and resuspended in HBSS, to obtain 20–200 × 103 cells per sample, as previously reported.19 Each sample was loaded with either 50 nM MitoTracker Green (MTG) or 20 nM TetraMethylRhodamine, Ethyl ester (TMRE) at 37°C for 30 min. Duplicate measurements were performed and unloaded control samples were run in parallel. Fluorescence activated cell sorting (FACS) was performed with Accuri C6 flow cytometer (BD Biosciencess) equipped with a 488 nm laser with 530/30 nm emission for MTG and 585/42 nm emission for TMRE. A total of 10,000 events were recorded and background-subtracted geometric means of MTG or TMRE were calculated as percent change relative to same-day controls.
Histology and Biochemistry
Cryostatic cross sections of skeletal muscle biopsies were used for histological and histochemical studies, according to standard techniques.20 Gomori trichrome staining, NADH dehydrogenase, and cytochrome c oxidase (COX) activities were performed as previously described.21 MRC enzyme activities were measured by standard spectrophotometric techniques in both muscle homogenate and digitonin-treated cultured skin fibroblasts.22 Maximum respiration rate (MRR) and extracellular acidification rate were measured in a SeaHorse FX-96 apparatus (Bioscience).23
Molecular Biology/Vectorology
FBXL4, FBXL4HA-C (C-term), and FBXL4HA-N (N-term) cDNAs were generated by PCR with adapted ends (5′ end NotI, 3′ end XhoI) and cloned by TOPO cloning (Invitrogen). Sequence-verified clones were cut with NotI/XhoI and subcloned into the pcDNA5/FRT/TO MCS. Mutant constructs were made with the QuikChange site-directed mutagenesis kit (Stratagene), with primers listed in Table S1. The FBXL4 ORF was cloned into the lentiviral expression vector pLenti6.3. The pLenti6.3/V5-TOPO vector system (Invitrogen) was used for the lentivirus-mediated expression of FBXL4 in skin fibroblast cell lines.24 Two constructs were used: the wild-type sequence of FBXL4 (FBXLwt) and as a control a construct with the c.617G>T (p.Arg206Leu) variant (FBXLmut). This mutation accidently occurred during PCR amplification and is next to the identified mutation c.614T>C (p.Ile205Thr) in individual 8, the only one not affecting the leucine-rich repeats.
Immunofluorescence Studies
Stably transfected HEK293T cells were grown on collagen-coated glass coverslips and induction of transgene expression was carried out with 5 ng/ml doxycycline for 24 hr. After fixation and permeabilization, cells were incubated with anti-HA (Roche Diagnostics) antibody, followed by a fluorescently labeled secondary antibody (Alexa Fluor 488, Invitrogen). Coverslips were mounted in ProLong Gold antifade reagent containing 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI; Invitrogen). Images were acquired with a A1R-A1 Nikon N-SIM confocal microscope.
In Vitro Import
35S-methionine-labeled proteins were generated from amplified cDNAs with a TNT Quick Coupled Transcription/Translation system (Promega). Labeled proteins were incubated with freshly prepared mouse liver mitochondria for 30 min at 37°C25 with or without 40 mg/ml trypsin, valinomycin, and 0.1% Triton X-100. Products were resolved on 10% or 12% Bis-TrisNuPAGE SDS-PAGE gels (Invitrogen). After the run, the gels were dried and exposed to phosphorscreens for 1–5 days and scanned with a Typhoon phosphorimager (GE Healthcare).
Cell Fractionation
Nuclear, cytosolic, and mitochondrial fractions from FBXL4HA-C stably transfected HEK293T cells after 5 ng/ml doxycycline induction for 24 hr were prepared as previously described. In brief, after cell homogenization in hypotonic buffer, nuclei were isolated from the low-speed centrifugation pellet.26 Mitochondria and cytosol were isolated from the supernatant by high-speed centrifugation.27 Submitochondrial localization of FBXL4 was carried out in stably transfected HEK293T cells after 0 or 5 ng/ml induction for 24 hr. Sucrose-gradient-purified mitochondria27 were resuspended in 20 mM HEPES (pH 7.8), 70 mM sucrose, 210 mM mannitol, 2 mM EDTA (1 mg/ml) and left untreated, treated with trypsin (25 ng/ml) for 30 min at room temperature or treated with increasing concentrations of digitonin (75, 150, 300, and 600 ng/ml) for 10 min at 4°C, followed by trypsin treatment, as above. Standard methods were used for the preparation of mitochondrial enriched/postmitochondrial fractions and after suborganellar (soluble and membrane) separation from mouse liver.28
Immunoblot Analyses
Samples containing 50–100 μg protein were separated by 12% or 4%–15% gradient SDS-polyacrylamide gels (NuPAGE, Invitrogen), transferred to nitrocellulose membranes, and incubated with suitable antibodies. Immune-visualization was carried out by chemiluminescence-based ECL kit (GE Healthcare). For Blue Native Gel Electrophoresis (BNGE), 106 HEK293T FBXL4HA-C cells were induced by doxycycline (2 ng/ml) for 48 hr and mitochondria harvested by differential centrifugation.27 Mitochondria were lysed in 1% digitonin, 1× NativePAGE sample buffer (Invitrogen). Samples were adjusted to 2 mM MgCl2 and treated with Benzonase (50 units) for 1 hr on ice. Lysates were then centrifuged for 30 min at 20,000 × g at 4°C. Protein complexes were resolved by NativePAGE 3%–12% Bis-Tris gels (Invitrogen) and transferred to PVDF in the presence of bicarbonate transfer buffer (10 mM NaHCO3, 3 mM Na2CO3). Immunoblot analysis was performed as reported above.
Results
Clinical Findings
Numerous clinical research centers from Europe, Saudi Arabia, and the US independently achieved and subsequently shared results from linkage and next-generation sequencing (NGS) exome analysis on families and singleton cases characterized by mitochondrial encephalomyopathy. All studies were completed according to local approval of the Institutional Review Board. Informed consent for participation in this study was obtained from the parents of all investigated subjects, in agreement with the Declaration of Helsinki and approved by the ethical committees of the centers participating in this study, where biological samples were obtained. As a result of this collaborative effort, we identified nine cases harboring recessive mutations in one gene, FBXL4.
Several children had preterm assisted delivery because of small weight or reduced fetal movements. At birth or immediately after birth, most children showed lactic acidosis, sometimes associated with hyperammonemia and signs of renal tubular acidosis. All displayed severe psychomotor delay with hypotonia, failure to thrive, and swallowing difficulty sometimes complicated by gastrointestinal dysmotility, requiring nasogastric tube feeding or PEG. Arrest and regression of neurological development was generally severe, although protracted and slowly progressive in most cases. However, three children died in infancy owing to metabolic decompensation during intercurrent infections, and most of those that have reached late childhood are nonverbal, are unable to sit autonomously, show muscle wasting and severe truncal ataxia, and, in several cases, suffer of epileptic seizures and/or choreoathetoid movements. Facial dysmorphisms and/or other developmental malformations (e.g., hypospadias, pectus excavatum) were frequent but not invariably present. The brain MRI features are dominated by supratentorial, global brain atrophy, thin corpus callosum, and altered signals in the white matter, but inconsistent involvement of the deep gray nuclei (basal ganglia and thalami) and infratentorial structures. The muscle biopsy showed variable signs of mitochondrial involvement (e.g., low COX reaction but no ragged red fibers), and variable but consistent decrease of the MRC complex activities was detected in both muscle and cultured fibroblasts. The mtDNA sequence analysis failed to reveal pathogenic mutations in all cases, but variable decrease of mtDNA amount was a consistent feature in both muscle and fibroblasts, often accompanied by reduced specific activity of citrate synthase. A clinical, biochemical, and molecular genetic synopsis is summarized in Table 1, and a more detailed outline is presented in Table S3. The pedigrees are shown in Figure 1A. The frequency of main clinical features is reported in Table S4 and detailed biochemical results are displayed in Table S5. Prototypical MRI findings in several subjects are shown in Figure 1B, and facial dysmorphisms and malformations detected in some of the subjects are illustrated in Figure 1C, with additional images of facial and body features, brain MRI, and muscle morphology displayed in Figure S1.
Table 1.
Clinical Summary Overview
| Affected Subject ID # | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|---|---|
| FBXL4 mutation(s) | c.[1703G>C; 1703G>C], p.[Gly568Ala; Gly568Ala] | c.[1444C>T; 1444C>T], p.[Arg482Trp; Arg482Trp] | c.[1444C>T; 1444C>T], p.[Arg482Trp; Arg482Trp] | c.[1444C>T; 1444C>T], p.[Arg482Trp; Arg482Trp] | c.[1790A>C; 1067del], p.[Gln597Pro; Gly356fs] | c.[1694A>G; 1694A>G], p.[Asp565Gly; Asp565Gly] | c.[1652T>A; 1652T>A], p.[Ile551Asn; Ile551Asn] | c.[614T>C; 106A>T], p.[Ile205Thr; Arg36Stop] | c.[1229C>T; 1229C>T], p.[Ser410Phe; Ser410Phe] |
| Age at presentation | 4 months | at birth | 4 months | ND | 1 month | at birth | 9 months | at birth | at birth |
| Age at last evaluation | 4 years | 4 years | 9 years | ND | 8 years | 16 months | 3 years | 3 years | 2 years |
| Deceased | − | + | − | − | − | + | − | − | + |
| Birth weighta | <10th | ND | <10th | ND | <10th | <3rd | <3rd | <3rd | <3rd |
| Last measured weighta | <3rd | 10th | 50th | ND | 42nd | <3rd (2 months) | <3rd | 3rd | 25th |
| Developmental delay | + | + | + | + | + | + | + | + | + |
| Dysmorphic facies | + | − | − | − | + | +/− (micrognathia) | + | + | − |
| Lactic acidemia | + | + | + | + | + | + | + | + | + |
| Cerebral atrophy | + | ND | + | ND | + | − | + | + | + |
| Brainstem and basal ganglia lesions | + | ND | − | ND | +/− | − | − | + | + |
| White matter lesions | + | ND | ND | ND | + | + | + | + | + |
| Hypotonia | + | + | + | + | + | + | + | + | + |
| Seizures | − | − | +/− | + | + | + | + | + | + |
| Ataxia | + | − | − | − | + | NA | + | NA | − |
| Dystonia, choreoathetosis | + | − | − | − | + | − | − | + | - |
| Swallowing dysfunction | + | ND | + | − | + | + | + | + | + |
| Failure to thrive | + | ND | − | − | − | + | + | + | + |
| MRC in muscle | ND | ND | ND | ND | multiple def. | low CI | multiple def. | multiple def. | multiple def. |
| MRC in fibros | multiple def. | normal | normal | ND | normal | normal | ND | ND | ND |
Abbreviations are as follows: ND, not done or not available; NA, not applicable; MRC, mitochondrial respiratory chain complex activities; CI, complex I.
Values as percentile.
Figure 1.
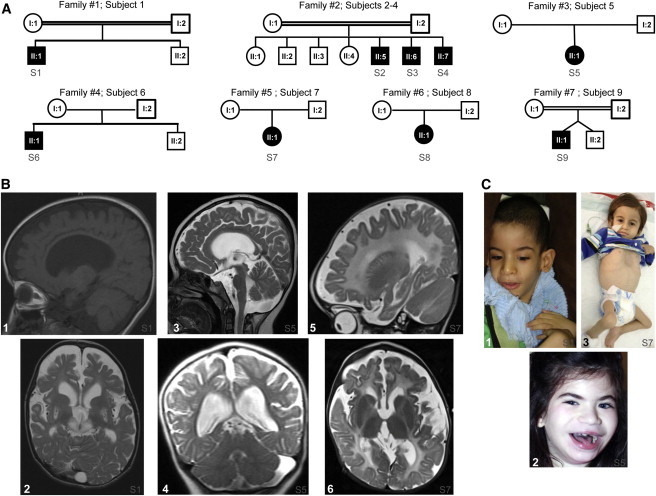
Pedigrees and Clinical Features
(A) Pedigrees. Black symbols designate affected subjects. Families and subjects are numbered according to the main text.
(B) Brain MRI. Panels 1 and 2 are subject 1 at 2 years of age; panels 3 and 4 are subject 5 at 4 years of age; and panels 5 and 6 are subject 7 at 9 months of age. Panels 1 and 2: T1-weighted sagittal interhemispheric (#1) and T2-weighted transverse (#2) sequences, displaying a thin corpus callosum, severe supratentorial brain atrophy, abnormal signals in the white matter, especially in subcortical areas, and bilateral lesions of the putamina and subthalamic region. The putaminal lesions contain scattered areas of reduced intensity possibly corresponding to calcifications. The cisternal spaces are increased. A subaracnoid cyst is visible in the occipital region. Panels 3 and 4: T2-weighted sagittal (#3) and coronal (#4) sequences displaying severe supratentorial brain atrophy and leukodystrophic changes; the infratentorial structures, including the cerebellum, are relatively spared, in spite of marked enlargement of the cisternal spaces. Panels 5 and 6: T2-weighted sagittal (#5) and transverse (#6) sequences displaying severe supratentorial brain atrophy and leukodystrophic changes; the basal nuclei, thalami, and infratentorial structures, including the cerebellum, are relatively spared.
(C) Panel 1: Subject 1 at 4 years of age; panel 2: subject 5 at 8 years of age; panel 3: subject 7 at 14 months of age. Note the facial dysmorphisms in #1 (dolichocephaly, protruded ears, narrow elongated face, and everted lower lip) and #2 (including smooth and hypotonic facies, mild synophrys, luxurious eyelashes, thick and arched eyebrows, bilateral epicanthal folds, thick lower lip with finely demarcated upper vermilion border) and pectus excavatum in #3.
Subject 1 (S1), a boy previously reported elsewhere,6 was born to healthy first-cousin parents from southern Saudi Arabia, after uneventful full-term pregnancy. Severe lactic acidosis and renal tubular acidosis (RTA) were diagnosed at 4 months of age, associated with severe global developmental delay and eventually psychomotor stagnation. Major feeding difficulties required PEG. Presently aged 4 years old (yo), he is an emaciated child with severe dystonia and cranio-facial dysmorphism (Figure 1C). Subject 2 (S2) was the first child from healthy first-cousin parents of Arabian origin (Central Province of Saudi Arabia). Only scant records are available for this child, who died at the age of 4 years with severe lactic acidosis. His younger brother is subject 3 (S3), who is now 9 yo. He shows intermittently elevated plasma lactate and severe cognitive impairment (IQ 50) and exercise intolerance, with subtle facial dysmorphism but no microcephaly. Subject 4 (S4) is a 6-month-old younger brother of subjects 2 and 3. His delivery was complicated by neonatal depression and persistent, severe lactic acidosis. He had one clinical seizure and is currently on AED. Parents refused muscle and skin biopsy. His growth parameters and facial appearance are normal. Subject 5 (S5) is a 9 yo girl, the only child from nonconsanguineous parents of European origin. She was born at the 39th week, small for gestational age (2,410 g). Apgar scores were 7–9. She has chronic lactic acidosis and RTA, global developmental delay (she is nonverbal) with truncal hypotonia, ataxia, and choreoathetoid movements, neutropenia, frequent infections, and severe GI dysmotility and swallowing difficulty, requiring PEG. She had several generalized seizures. She has dismorphic features of face and limb malformations (Figures 1A and S1A), with moderate microcephaly; her BMI is 17.4. Subject 6 (S6), the first child of European parents, was born at the 38th week, small for gestational age (2,375 g), had micrognathia, supernumerary bilateral nipples, and mild hypospadias. Apgar scores were 9–10. He had a perinatal metabolic crisis with hypoglycemia, hypotonia, and lactic acidosis (19 mM [pH 7.11], BE −24), with high plasma ammonia (363 mg%; n.v. < 80) and ketonuria. At 4 months he showed gastro-esophageal reflux and swallowing difficulty requiring nasogastric tube feeding. His head circumference remained between 3rd and 10th centile. Severe psychomotor delay and muscle wasting with ventilatory insufficiency required a positive expiratory pressure mask. He died at 16 months of acutely progressive encephalopathy during an intercurrent infection. Subject 7 (S7) was born at term as the first child of healthy first-degree cousins of Arabic origin (from Bahrain). Prenatal ultrasound revealed growth retardation and dilated lateral ventricles. His birth weight was 2,400 g (3rd percentile). Apgar score was 8–10. Clinical examination showed low-set ears, hypospadias, and undescended testes. At day 9, elevated plasma lactate (9 mM) and ammonia (125 μM) was reported. Because he did not thrive, nasogastric tube feeding was started but he continued to grow poorly, with frequent vomiting and gastro-esophageal reflux. At 9 months his growth parameters were <3rd percentile. He showed mild pectus excavatum (Figure 1C), muscle wasting, severe hypotonia, no head control, and severe psychomotor delay; repeatedly elevated plasma lactate and ammonia; slightly increased liver enzymes; and a microcytic anemia with high ferritine. At age 3 years he is bedridden with severe truncal hypotonia and severe muscle wasting. He is nonverbal. Subject 8 (S8) was born by caesarean section at 37th week, small for gestational age (1,850 g, <3rd percentile) to nonconsanguineous European parents. Her Apgar score was 8–9. She showed severe muscular hypotonia and failure to thrive. Echocardiography revealed left ventricular heart hypertrophy. Lactate was elevated (5–12 mM). Dysmorphisms included malformed ears, saddle nose, facial hypoplasia with micro-ophthalmy, long philtrum, and downward slanted eyelids. At the age of 3 years she has developed bilateral cataract and horizontal nystagmus and shows failure to thrive and a global psychomotor and developmental delay. Her body weight is 10.15 kg (3rd percentile), body length 94 cm (25th percentile), and head circumference 48.6 cm (25th percentile). Subject 9 (S9) was born at the 35th week after an uncomplicated twin pregnancy from first-cousin Turkish parents. His twin brother is healthy. During the neonatal period, persistent lactic acidosis (3–10 mM) was associated with significant generalized muscular hypotonia. In the following months the child developed persistent lactic acidosis and motor and cognitive development stopped. At age 2 years he presented with reduced head control and was unable to sit unassisted and had profound generalized muscular hypotonia, severe psychomotor delay, absence of verbal skills, and episodes of absence with no seizures. He died at 2 1/2 years of an intercurrent infection.
A Mitochondrial Encephalomyopathy Syndrome Maps to 6q16.1 and Is Caused by Recessive Mutations in FBXL4
Autozygosity mapping on a simplex case (S1, Figure 1A) showed several autozygous intervals. One, located on 6q16, overlapped with a single autozygous interval that was exclusively shared by the three affected siblings of a multiplex consanguineous family (subjects 2–4, family 2, Figure 1A) having clinical features consistent with mitochondrial encephalopathy. Combined linkage analysis of these two families revealed a single significant linkage peak with LOD of 3.67 that spanned FBXL4 (Figure S2). Consistent with the different geographic origins of these two families in Saudi Arabia, the haplotypes in the candidate interval were different. In order to identify the causal variants, the index in each family underwent exome sequencing followed by variant filtration. In each index case, a single homozygous change in FBXL4 (c.1703G>C [p.Gly568Ala] in the simplex case, c.1444C>T [p.Arg482Trp] in the multiplex case) survived the various filters within the linkage peak (Figures 2 and S2). None of these mutations was present in 242 Saudi exomes. When exome variants in the simplex case were reanalyzed based on all autozygous intervals and not just on the candidate disease interval, c.1703G>C was the only variant that survived filtration. These data indicated that the mitochondrial encephalopathy in these two unrelated Saudi kindreds linked to 6q16.1 and strongly supported of FBXL4 variants as the responsible genetic cause for the disease.
Figure 2.
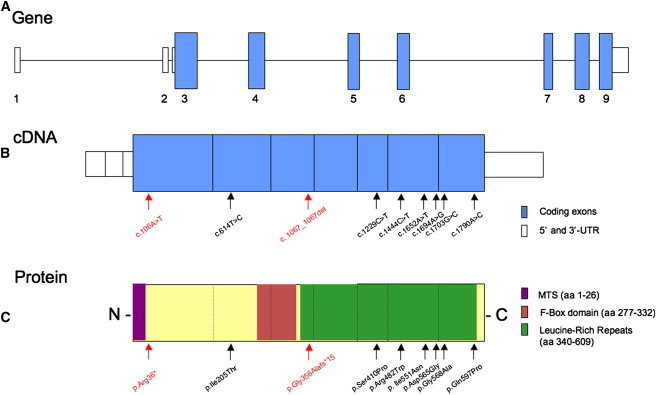
FBXL4 Gene, cDNA, and Protein
(A) Genomic structure of FBXL4 with coding (blue) and noncoding (white) exons.
(B) FBXL4 cDNA (NM_012160.3) with nucleotide changes identified in this study.
(C) FBXL4 protein with amino acid changes identified in this study. Functional domains of FBXL4 are in color. Abbreviation: MTS, mitochondrial targeting sequence. Red arrows indicate mutations that cause the premature truncation of FBXL4, therefore predicting the loss of its function.
Disease-segregating mutations in FBXL4 were independently found by exome sequencing of four additional pediatric individuals with MRC deficiencies who were recruited and analyzed at different clinical centers (subject 5–8, families 3–6 in Figure 1A). Individual exome-sequencing analysis resulted in candidate recessive gene lists of 5, 11, 82, and 7 genes each having two rare variants, for subjects 5, 6, 7, and 8, respectively. Only the long list of subject 7 contained a gene coding for a known mitochondrial protein;24 however, they included homozygous missense mutations in FBXL4 in two samples: c.1694A>G (p.Asp565Gly) in subject 6 and c.1652T>A (p.Ile551Asn) in subject 7. Subjects 5 and 8 were compound heterozygous carrying both a nonsense mutation together with a missense variant. Subject 5 harbored a c.1067del (p.Gly356Alafs∗15) nonsense mutation in the maternal FBXL4 allele and a c.1790A>C (p.Gln597Pro) missense mutation in the paternal FBXL4 allele; in subject 8, (c.[614T>C; 106A>T], p.[Ile205Thr; Arg36∗]) the nonsense variant affected the maternal allele. An additional homozygous mutation (c.1229C>T [p.Ser410Phe]) was found in a further simplex cases, subject 9 (family 7, Figure 1A), by selective FBXL4 sequencing in a cohort of clinically/biochemically suitable subjects (Figures 2 and S3). None of the variants was found in 4,500 in-house European control chromosomes or in >8,000 alleles from Americans of European descent (Exome Variant Server), nor did any control sample harbor two rare variants in FBXL4. The missense changes each involved evolutionarily conserved amino acid residues, most affecting leucine-rich repeats (Figures 2 and S3). Suitable prediction software packages assigned high scores for pathogenicity to each mutation (data available upon request). Because the p.Gly568Ala is due to a c.1703G>C change affecting the first nucleotide of the last exon, a potential splicing defect was hypothesized for the mutation in subject 1: although the retrotranscribed cDNA analysis failed to show an aberrant transcript,6 the quantification of FBXL4 transcripts by qRT-PCR revealed decreased FBXL4 cDNA amount by approximately 50% of the control level that was probably due to instability of the aberrant spliced forms. A reduction in FBXL4 levels was also found in subject 5 fibroblasts, which carry one allele with a missense mutation and one allele with a frameshift variant, probably resulting from mRNA decay of the transcripts carrying a nonsense codon. The cellular content of the FBXL4 transcript was normal in two other cases studied (subjects 6 and 7) that carry only missense mutations (results not shown).
Defective OXPHOS Is Associated with FBXL4 Mutations
Biochemical assays performed on clinical and/or research basis in muscle and/or fibroblasts obtained from several subjects confirmed the deleterious effect of FBXL4 mutations on mitochondrial bioenergetics. Muscle homogenates or isolated mitochondria from subjects with FBXL4 mutations showed variably decreased activity of MRC complexes (Table S5). Likewise, the mtDNA content was consistently, albeit variably, lower than controls in both muscle and fibroblasts of all tested samples (Figure 3A). Cultured skin fibroblasts had reduced maximal oxygen consumption rate (Table S5). Staining with MitoTracker Red, a mitochondrion-specific fluorescent marker, showed marked fragmentation of the mitochondrial network in S5 and S6 cell lines (Figure 3B) and a decrease in mitochondrial mass, as revealed by low citrate synthase (S6) or staining with MitoTracker Green (MTG) fluorescence (S5), was observed. In S6 cell lines, we found a consistent, significant (40%) reduction of the mitochondrial membrane potential (ΔΨ), as demonstrated by a membrane-potential-dependent mitochondrial fluorochrome, TMRE (Figure 3C).
Figure 3.
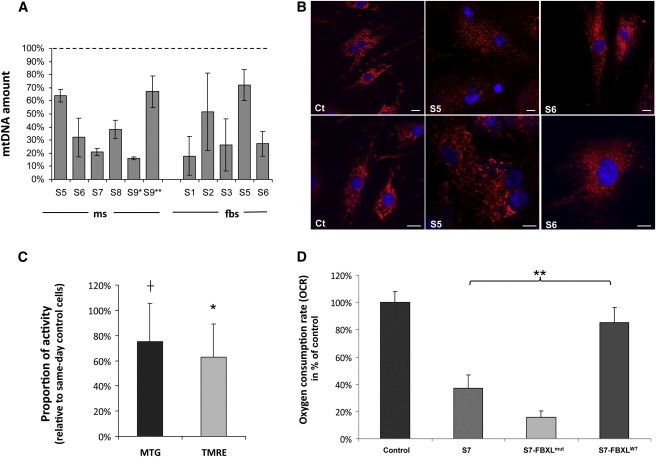
Molecular, Biochemical, and Morphological Studies in FBXL4 Mutant Muscle and Fibroblasts
(A) Quantification of mtDNA amount in muscle (ms) and fibroblasts (fbs) from available subjects (S). For subject 9, DNAs from two different muscle biopsies were analyzed (S9∗, 13 days of age; S9∗∗, 13 months of age). The bars represent the percentage of mtDNA normalized to nuclear DNA in FBXL4 mutant subjects, compared to the mean value of controls (dotted line, 100%; range of control values: 60%–160% for muscle, 70%–145% for fibroblasts). Data are represented as mean ± SD.
(B) Representative images of mitochondrial morphology in fibroblasts, showing filamentous (control, Ct) or fragmented (subjects 5 and 6) mitochondrial networks. Cells were stained with MitoTracker-Red CMX-Ros (Invitrogen) and Hoechst, a blue nuclear dye, 50 nM for 30 min. Scale bars represent 10 μm.
(C) Reductions in mitochondrial mass and mitochondrial membrane potential in mutant fibroblasts (subject 5), as indicated by reductions in MitoTracker Green (MTG) and TMRE fluorescence, respectively. The bars represent relative fluorescence values, compared to same-day measurements obtained in control fibroblasts. Fibroblasts were loaded with MTG (50 nM) or TMRE (20 nM) for 30 min at 37°C and fluorescence values determined by FACS analysis (Accuri C6). An asterisk (∗) indicates statistically significant reductions in TMRE fluorescence values (p = 0.0009, one-way ANOVA, n = 10). A cross (†) indicates a trend toward a significant reduction in MTG fluoresecence (p = 0.053, one-way ANOVA; n = 8). Data are represented as mean ± SD.
(D) Oxygen consumption rate (OCR) of nontransduced and transduced fibroblast cell lines. Fibroblasts from subject 7 (S7) and controls (NDHFneo, LONZA) were transduced with FBXL4wt- and FBXL4mut-expressing construct. OCR was determined as rotenone-sensitive respiration rate of uncoupled mitochondria (n > 7 each) and increased significantly (p < 0.001, t test) after expression of FBXL4wt only. Data are represented as mean ± SD.
FBXL4 Mutations Are Pathogenic
In order to prove the pathogenicity of identified FBXL4 mutations, we tested the biochemical rescue of OXPHOS-defective skin fibroblast cell lines from S7 after lentiviral-mediated expression of FBXL4wt cDNA by using the p.Lenti6.3/V5-TOPO vector system. In addition, we tested the activity of a mutated FBXL4 allele affecting the neighboring amino acid of the only mutation outside the conserved leucine-rich repeat domain for which no structural or functional information was available. OXPHOS activity was measured as rotenone-sensitive uncoupled oxygen consumption rate by the Seahorse system. Expression of FBXL4wt in S7 fibroblasts led to a significant increase of oxygen consumption rate (Figure 3D). Because expression of the FBXL4wt re-established rotenone-sensitive respiration to low normal levels, we further tested the functional activity of the mutated FBXL4 allele. The low oxygen consumption rate found in naive S7 cells (38% of controls) were even further reduced after expression of FBXL4mut, indicating a loss of FBXL4 function resulting from a p.Arg206Leu amino acid change, affecting the amino acid residue next to the Ile205 residue mutated in S8. This observation points to an important role of this part of the protein. Taken together, these results demonstrate the causal role of the mutations identified in FBXL4, thereby establishing FBXL4 as a gene necessary for the homeostasis of mitochondrial bioenergetics.
FBXL4 Is Targeted to Mitochondria
With different software packages for prediction of mitochondrial or subcellular localization, the FBXL4 protein sequence scored high for mitochondrial targeting, because of the presence of a potential mitochondrial targeting sequence (MTS) encompassing the first 22 to 26 amino acid residues at the N terminus. However, FBXL4 was previously proposed to interact with a nuclear endonuclease.29 In order to experimentally demonstrate the subcellular localization for FBXL4, we first used confocal immunofluorescence on HEK293T cells expressing the full-length FBXL4 tagged with the influenza-virus haemo-agglutinin (HA) epitope at the C terminus (FBXL4HA-C) under induction with doxycycline. In doxycycline-induced cells, a monoclonal HA-specific antibody showed an immunofluorescence pattern coincidental with the pattern obtained by means of the mitochondrial-targeted dye MitoTracker Red. No HA-specific staining was present in other cell compartments, including the nucleus (Figure 4A); as expected, noninduced cells were HA negative. To test whether the C-terminal HA extension of the recombinant FBXL4HA-C protein can mask a nuclear-localization signal, we then repeated the same experiment with a construct bearing the HA epitope at the N terminus of the full-length FBXL4 (FBXL4HA-N). FBXL4HA-N did localize predominantly to mitochondria; although a tenuous and diffuse immunofluorescence could be detected in the cytosol, no signal was detectable in the nucleus (Figure S4). These data suggest still robust but less efficient mitochondrial targeting of FBXL4HA-N as compared to FBXL4HA-C. Finally, to test whether translation initiation at a AUG start codon downstream from the AUG codon encoding Met1 could address the protein to the nucleus, we transfected two C terminus HA-flagged FBXL4 constructs, starting at either the second AUG, encoding Met8 (Δ8FBXL4HA-C), or the third AUG, encoding Met29 (Δ29FBXL4HA-C). The Δ8FBXL4HA-C construct, which encodes an FBXL4 variant still containing most of the predicted MTS, localized completely and exclusively to mitochondria (Figure S4). Conversely, the Δ29FBXL4HA-C construct, which encodes an FBXL4 variant lacking the predicted MTS entirely, showed a predominantly cytosolic localization (Figure S4). These results show that the N-terminal MTS is essential to target FBXL4 to mitochondria. Importantly, in neither case was nuclear localization ever observed.
Figure 4.
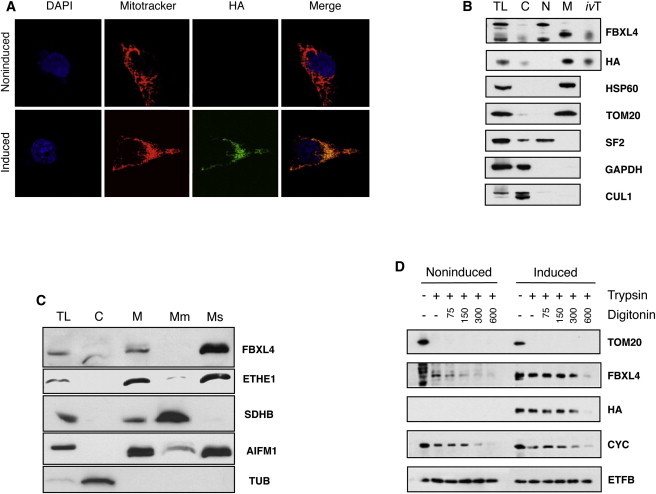
Subcellular Localization of FBXL4
(A) Recombinant protein was labeled with α-HA antibody (green), nuclei were stained blue with DAPI, and mitochondria were stained red with MitoTracker. A noninduced HEK293T cell is shown to highlight the specificity of the α-HA antibody.
(B) Total cell lysate (CL), cytosol (C), nuclei (N), and mitochondrial (M) fractions were isolated from FBXL4HA-C stably transfected HEK293T cells followed by immunoblot analysis with α-FBXL4 (Abcam) and α-HA (Roche) antibodies. In vitro synthesized human FBXL4 HA-C full-length protein (ivT) was included as control. Mitochondrial subcellular localization was confirmed by immunoblotting based on signal from HSP60 and TOM20 (mitochondrial), SF2 (nuclear), GAPDH (cytosolic). A previously suggested partner of FBXL4, cullin 1 (CUL1), is here shown to be cytosolic.
(C) Subcellular fractionations obtained from mouse liver. Mitochondrial subcellular localization was confirmed by immunoblotting based on signal from ETHE1 (mitochondrial matrix), SDHB (mitochondrial membranes), AIFM1 (mitochondrial intermembrane space), and tubulin (cytosolic).
(D) Intact mitochondria isolated from FBXL4HA-C stably transfected HEK293T cells after 0 (noninduced) or 5 ng/ml doxycycline induction for 24 hr (induced) were left untreated or incubated with trypsin or digitonin followed by trypsin digestion. Submitochondrial localization was established by comparison of FBXL4 and HA signal from immunoblots to TOM20 (outer membrane), cytochrome c, CYC (intermembrane space), and electron transfer B, ETFB (matrix).
Next, we carried out immunoblot-based visualization analysis on different HEK293T cell fractions by using both polyclonal antibodies against the native FBXL4 mammalian protein (α-FBXL4) and an anti-HA monoclonal antibody (α-HA). In the mitochondrial fraction of doxycycline-induced HEK293T cells analyzed on SDS-PAGE blots, both α-FBXL4 and α-HA detected a band of cross-reacting material (CRM) corresponding to a protein of approximately 68–70 kDa, compatible with the molecular weight of FBXL4. This band was (weakly) detected in the total cell lysate, but not in the nuclear nor in the postmitochondrial cytosolic fractions (Figure 4B). We used suitable markers to demonstrate the purity of the different cellular fractions. These results were consistently obtained in several independent experiments with HEK293T (Figure 4B) and also HeLa and 143B cells (not shown).
We then asked whether FBXL4 is bound to the mitochondrial membrane fraction or is localized in the aqueous soluble fraction of the organelle. In isolated mouse liver mitochondria, FBXL4-CRM was present only in the soluble fraction, whereas no CRM was detectable in the membrane fraction (Figure 4C); this result is in agreement with the absence of transmembrane domains in FBXL4. Finally, in order to establish which soluble compartment of mitochondria, i.e., matrix versus intermembrane compartment, FBXL4 resides in, we carried out trypsin-protection experiments in mitochondria isolated from inducible FBXL4HA-C-expressing HEK293T cells. As shown in Figure 4D, Tom20, an outer-membrane component, was completely digested by trypsin (25 μg/ml for 30 min) in intact mitochondria, whereas the 70 kDa band corresponding to FBXL4 was resistant to trypsin digestion, indicating that FBXL4 resides inside mitochondria, being protected from trypsin digestion by the outer mitochondrial membrane. However, FBXL4 band intensity was progressively reduced by trypsin treatment of mitochondria after exposure to increasing concentrations of digitonin, a detergent that solubilizes the outer, but not the inner, mitochondrial membrane. The susceptibility of FBXL4 to trypsin digestion in the presence of digitonin was identical to that of two proteins known to reside in the intermembrane space: cytochrome c (Figure 4D) and adenylate kinase 2 (Figure S5A). In contrast, two mitochondrial matrix proteins, ETF (Figure 4D) and citrate synthase (Figure S5A), were resistant to trypsin in all conditions. Convergent results were obtained by in vitro import experiments (Figure S5B). The in vitro 35S-labeled translated product of full-length FBXL4 cDNA was protected against proteinase K in naive mitochondria by translocation within mitochondria through a membrane-potential-dependent mechanism. However, we failed to detect posttranslational cleavage of the N-terminal MTS, as seen for many proteins of the mitochondrial matrix. Taken together, our findings establish FBXL4 as a protein targeted to and residing inside mitochondria, most probably within the intermembrane space.
FBXL4 Is Part of a Supramolecular Complex
FBXL4 has a leucine-rich repeat domain (Figure 2), which is typically engaged in protein-protein interactions (Figure 5A). We tested this hypothesis by immunoblot analysis on one-dimension blue-native gel electrophoresis (1D-BNGE) of the mitochondrial fraction from doxycycline-induced, FBXL4HA-C-transfected HEK293T cells. Immunovisualization by α-FBXL4 consistently revealed a CRM doublet formed by two proximal bands in the 400 kDa range. Only the lower band was immunovisualized with α-HA (Figure 5B). These results indicate that FBXL4 is present in a quaternary protein complex and suggest that the HA epitope partly interferes with the stability of the complex. Interestingly, the leucine-rich repeat domain, which is deemed to promote protein-protein interactions, is targeted by most of the mutations found in affected subjects (Figures 2A and 5A).
Figure 5.

Structural Features of FBXL4
(A) A comparative structure of the F-box and leucine-rich repeat (dark gray) domains of FBXL4, modeled on the X-ray structures of FBXL3 and SKP2. A surface representation of the CRY2 binding partner (light gray) of FBXL3 modeled in the equivalent position with FBXL4 suggests a protein-protein interaction surface is formed by the concave β-sheet of the leucine-rich repeats. Mutations of FBXL4 are shown as colored sticks: p.Gly356Alafs∗15 (red), p.Ser410Phe (orange), p.Leu433Arg (yellow), p.Arg482Trp (green), p.Ile551Asn (cyan), p.Asp565Gly (blue), p.Gly568Ala (indigo), and p.Gln597Pro (violet).
(B) Blue Native Gel Electrophoresis (BNGE) from FBXL4HA-C stably transfected HEK293T cells and induced with 2 ng/ml doxycycline for 48 hr. Samples were separated in 5%–13% gradient nondenaturating gels and immunoblotted for FBXL4 and HA in order to reveal the presence of high-molecular-weight complexes. SDS-boiled sample, NDUFB8 from complex I, and SDHB from complex II were analyzed as controls.
Discussion
The following points provide evidence that recessive FBXL4 mutations are responsible for severe, infantile-onset mitochondrial encephalomyopathy. First, two unrelated consanguineous families were both linked to the same interval in 6q16.1 by different haplotypes; exome sequencing showed them to harbor two different, probably pathogenic, homozygous FBXL4 variants as the only variants within the linkage interval in each kindred. Second, independent filtration of exome variants in four unrelated simplex families revealed six additional mutant FBXL4 alleles, including two nonsense and four missense mutations; another missense mutation was found in homozygosity by ad hoc screening of FBXL4 in a subject with clinical and biochemical features similar to the previously identified mutant subjects. Taken together, we found nine disease-associated FBXL4 mutant alleles in seven unrelated kindreds. Third, two nonsense variants predict the formation of truncated and aberrant transcripts or proteins, whereas the seven missense variants involve severe changes at evolutionarily highly conserved amino acid positions. Further, with the exception of p.Ile205Thr (S8), which is localized in a poorly structured domain, the other identified amino acid substitutions all affect the FBXL4 leucine-rich domain that is probably involved in protein-protein interactions. Fourth, albeit variable, the clinical presentation, laboratory findings, brain MRI, and disease course share several common features, including early-onset lactic academia and severe psychomotor delay and eventually stagnation, associated with predominantly supratentorial brain lesions and progressive brain atrophy. Dysmorphic facial traits and other malformations were present in some individuals. Fifth, affected subjects had consistent, albeit variable, biochemical and morphological features indicating significant impairment of mitochondrial OXPHOS, in both muscle and cultured fibroblasts. We also detected variable but consistent reduction in the mtDNA copy number. Sixth, expression of naive FBXL4 cDNA rescued the OXPHOS defect of FBXL4 mutant fibroblasts. Taken together, these results concordantly converge to indicate mutant FBXL4 as the cause of a previously unreported early-onset mitochondrial disease syndrome. With the exception of S6 and S9, who died in early infancy from metabolic crisis consequent to infection, the other children are all alive, some having entered late childhood, with protracted, slowly progressive multisystemic evolution of their clinical course. This observation suggests that FBXL4 function is important for normal health and development but not essential for survival into late childhood and possibly early adulthood.
Because a common hallmark of this syndrome consisted of impaired mitochondrial bioenergetics, we first asked whether FBXL4 is a mitochondrial protein. F-box proteins are versatile regulators of a variety of cellular functions.30,31 These proteins usually contain a wide range of other motifs including zinc fingers, leucine zipper, ring fingers, tetratricopeptide repeats (TPR), and proline-rich regions. Typically, they serve as substrate adaptors in Skp1-Cullin-F-box (SCF) E3 ubiquitin ligases, where they are required to target proteins to ubiquitination and degradation by the 26S proteasome. However, many F-box proteins may also function independently of the SCF complex and the 26S proteasome.32 A previous report suggested that FBXL4 is an E3 ubiquitin ligase interacting with JMJD2A, a histone lysine demethylase.29 However, we notice that no evidence was provided that FBXL4 is a SCF complex member. Further, JMJD2A-CRM was immunoprecipitated in the reported study by an anti-FBXL4 antibody incubated overnight with ethidium-bromide-treated whole HEK293T cell extracts; this experiment cannot exclude nonspecific interactions between the two proteins and no further evidence was provided on the subcellular localization of FBXL4. In contrast, by applying a number of different experimental approaches that were supported by in silico predictions, we clearly demonstrated here that FBXL4 is indeed localized in mitochondria, and the predicted full-length FBXL4, or variants of it, consistently failed to be detected in the nucleus of (1) human naive cultured cells, (2) cells expressing a recombinant full-length, HA-tagged FBXL4, or (3) mouse liver cell fractions. The presence of cryptic nuclear-localization signals was excluded by using several FBXL4 recombinant variants expressed in cultured cells. Notably, the localization of FBXL4 CRM was clearly separated from that of cullin-1 (see Figure 4B). Although we cannot exclude in principle that in some (untested) conditions or tissues FBXL4 may also localize in other cellular compartments, we provide compelling evidence in multiple cell lines that this protein is predominantly, if not exclusively, targeted to and localized within mitochondria. Further experimental evidence indicates that FBXL4 is present in the intermembrane space and is stably incorporated within an approximately 400 kDa quaternary complex. Interestingly, this demonstration has been made possible by the link between FBXL4 mutations and mitochondrial disease and underscores the impact that mitochondrial medicine has in the elucidation of fundamental mechanisms relevant to mitochondrial biology. In fact, our observation opens several important, and somewhat unexpected, questions concerning the precise biological function, mechanism of action, and physical status of FBXL4. In silico reconstruction of FBXL4 by means of the known structures of human FBXL3 and SKP2, which belong to the same protein family, suggests that the concave β-sheet of the leucine-rich repeats form a protein-protein interaction surface (CRY2 in case of FBXL3) (Figure 5A). Interestingly, nearly all of the missense mutations found in our subjects are contained within the leucine-rich domain (Figures 2A and 5A), suggesting that they could potentially compromise the formation of a functioning supramolecular complex.
In budding yeast, at least 21 proteins contain a discernible F-box motif.30 Two of these proteins, Mdm30 and Mfb1, have nonredundant, F-box-independent roles, because they are required for proper mitochondrial distribution and morphology during mitotic growth. F-box leucine-rich repeat proteins provide subunits to the enzymatic complex carrying out phosphorylation-dependent ubiquitination of proteins that are then disposed by proteasome degradation. Although a complete ubiquitination system has not been reported to exist within mitochondria, two ubiquitin ligases (MITOL/MARCH-V and MULAN), as well as a deubiquitinating enzyme (Ubp16/USP30), are embedded in mitochondrial outer membranes and participate in mitochondrial dynamics.33 Defects in mitochondrial morphology or respiration capacity are also reported for mutations in other ubiquitination-proteasome system (UPS) components, such as the ubiquitin ligases parkin and Rsp5, as well as proteasome subunits. These examples are likely to reflect a pervasive involvement of the UPS in recycling mitochondria-associated proteins. The flux of imported proteins and the proximity of mitochondrial import across the inner mitochondrial membrane to oxidative phosphorylation probably result in protein damage or misfolding that require a responsive quality control system. It is tempting to speculate that FBXL4 might act as a protein quality control system, connecting to other similar pathways that control OXPHOS proficiency and mitochondrial biogenesis. For instance, PINK1, a kinase predominantly found in the outer compartment of mitochondria, is rapidly degraded in ΔΨ-proficient organelles.34 However, in energetically spent mitochondria having impaired ΔΨ, PINK1 is stabilized to attract parkin, an E3 ligase that in turn promotes the disposal of the organelle, by a specialized macro-autophagic process termed mitophagy. Activation of the mitophagic processes could underlie the decreased mitochondrial mass and explain, at least to some extent, the reduced mtDNA copy number and citrate synthase activity that we observed in muscle and cultured fibroblasts from FBXL4 mutant subjects. Consistent with a possible role of FBXL4 in quality control and modulation of mitochondrial dynamics, we found that the mitochondrial network was clearly more fragmented in some mutant fibroblasts compared to controls (Figure 3C).
Future work is warranted to elucidate the composition, interactions, and mechanistic role of this mitochondrial protein/complex that controls bioenergetics homeostasis and causes a previously unrecognized infantile-onset mitochondrial disease.
Acknowledgments
The study received financial support from Fondazione Telethon grants GGP11011 and GPP10005; the CARIPLO Foundation, Italy, grant 2011/0526; the Italian Ministry of Health (GR2010-2316392); the Pierfranco and Luisa Mariani Foundation of Italy, the Italian Association of Mitochondrial Disease Patients and Families (Mitocon); the German Federal Ministry of Education and Research (BMBF) funded Systems Biology of Metabotypes grant (SysMBo #0315494A); the German Network for Mitochondrial Disorders (mitoNET #01GM0867 and 01GM1113C); E-rare grant GenoMit (JTC2011, 01GM1207, and FWF I 920-B13); the EU FP7 Mitochondrial European Educational Training project (Meet); the European Research Council (grant “Mitcare” FP7-322424); the Medical Research Council (UK); the Deanship of Scientific Research at King Saud University, Riyadh, through the Research Group Project no 301 (M.A.S.); Penn Genome Frontiers Institute (E.A.P. and X.G.); the National Institutes of Health (R03-DK082446 to M.J.F., RO1-EY012910 to E.A.P., P30EY014104-MEEI core support, and 1G20RR030939 to X.G.); the Foerderer Award for Excellence from the Children’s Hospital of Philadelphia Research Institute (X.G. and M.J.F.); institutional support from Loyola University Stritch School of Medicine (X.G.); the Clinical and Translational Research Center at the Children’s Hospital of Philadelphia (UL1-RR-024134); the Angelina Foundation Fund from the Division of Child Development and Metabolic Disease at the Children’s Hospital of Philadelphia (M.J.F.); The Tristan Mullen Fund (M.J.F.); Canadian Institutes of Health Research (C.A.S.); DHFMR Collaborative Research Grant (F.S.A.); and the Center for Mitochondrial and Epigenomic Medicine (CMEM) at The Children’s Hospital of Philadelphia. The cell lines and DNA bank of Paediatric Movement Disorders and Neurodegenerative Diseases, member of the Telethon Network of Genetic Biobanks (project no. GTB12001), funded by Telethon Italy, provided us with specimens. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The content of the article has not been influenced by the sponsors.
Contributor Information
Fowzan S. Alkuraya, Email: falkuraya@kfshrc.edu.sa.
Massimo Zeviani, Email: mdz21@mrc-mbu.cam.ac.uk.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
MitoMiner, http://mitominer.mrc-mbu.cam.ac.uk
MitoProt, http://ihg.gsf.de/ihg/mitoprot.html
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
PolyPhen-2, http://www.genetics.bwh.harvard.edu/pph2/
UCSC Genome Browser, http://genome.ucsc.edu
WoLF-PSORT, http://wolfpsort.org
References
- 1.McCormick E., Place E., Falk M.J. Molecular genetic testing for mitochondrial disease: from one generation to the next. Neurotherapeutics. 2013;10:251–261. doi: 10.1007/s13311-012-0174-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang W., Cui H., Wong L.J. Application of next generation sequencing to molecular diagnosis of inherited diseases. Top. Curr. Chem. 2012 doi: 10.1007/128_2012_325. Published online May 11, 2012. [DOI] [PubMed] [Google Scholar]
- 3.Pagliarini D.J., Calvo S.E., Chang B., Sheth S.A., Vafai S.B., Ong S.E., Walford G.A., Sugiana C., Boneh A., Chen W.K. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elstner M., Andreoli C., Klopstock T., Meitinger T., Prokisch H. The mitochondrial proteome database: MitoP2. Methods Enzymol. 2009;457:3–20. doi: 10.1016/S0076-6879(09)05001-0. [DOI] [PubMed] [Google Scholar]
- 5.Calvo S.E., Compton A.G., Hershman S.G., Lim S.C., Lieber D.S., Tucker E.J., Laskowski A., Garone C., Liu S., Jaffe D.B. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci. Transl. Med. 2012;4:118ra110. doi: 10.1126/scitranslmed.3003310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shamseldin H.E., Alshammari M., Al-Sheddi T., Salih M.A., Alkhalidi H., Kentab A., Repetto G.M., Hashem M., Alkuraya F.S. Genomic analysis of mitochondrial diseases in a consanguineous population reveals novel candidate disease genes. J. Med. Genet. 2012;49:234–241. doi: 10.1136/jmedgenet-2012-100836. [DOI] [PubMed] [Google Scholar]
- 7.Haack T.B., Danhauser K., Haberberger B., Hoser J., Strecker V., Boehm D., Uziel G., Lamantea E., Invernizzi F., Poulton J. Exome sequencing identifies ACAD9 mutations as a cause of complex I deficiency. Nat. Genet. 2010;42:1131–1134. doi: 10.1038/ng.706. [DOI] [PubMed] [Google Scholar]
- 8.Spinazzola A., Viscomi C., Fernandez-Vizarra E., Carrara F., D’Adamo P., Calvo S., Marsano R.M., Donnini C., Weiher H., Strisciuglio P. MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat. Genet. 2006;38:570–575. doi: 10.1038/ng1765. [DOI] [PubMed] [Google Scholar]
- 9.He L., Chinnery P.F., Durham S.E., Blakely E.L., Wardell T.M., Borthwick G.M., Taylor R.W., Turnbull D.M. Detection and quantification of mitochondrial DNA deletions in individual cells by real-time PCR. Nucleic Acids Res. 2002;30:e68. doi: 10.1093/nar/gnf067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dimmock D., Tang L.Y., Schmitt E.S., Wong L.J. Quantitative evaluation of the mitochondrial DNA depletion syndrome. Clin. Chem. 2010;56:1119–1127. doi: 10.1373/clinchem.2009.141549. [DOI] [PubMed] [Google Scholar]
- 11.Carr I.M., Flintoff K.J., Taylor G.R., Markham A.F., Bonthron D.T. Interactive visual analysis of SNP data for rapid autozygosity mapping in consanguineous families. Hum. Mutat. 2006;27:1041–1046. doi: 10.1002/humu.20383. [DOI] [PubMed] [Google Scholar]
- 12.Hoffmann K., Lindner T.H. easyLINKAGE-Plus—automated linkage analyses using large-scale SNP data. Bioinformatics. 2005;21:3565–3567. doi: 10.1093/bioinformatics/bti571. [DOI] [PubMed] [Google Scholar]
- 13.Falk M.J., Zhang Q., Nakamaru-Ogiso E., Kannabiran C., Fonseca-Kelly Z., Chakarova C., Audo I., Mackay D.S., Zeitz C., Borman A.D. NMNAT1 mutations cause Leber congenital amaurosis. Nat. Genet. 2012;44:1040–1045. doi: 10.1038/ng.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xing W., Busino L., Hinds T.R., Marionni S.T., Saifee N.H., Bush M.F., Pagano M., Zheng N. SCF(FBXL3) ubiquitin ligase targets cryptochromes at their cofactor pocket. Nature. 2013;496:64–68. doi: 10.1038/nature11964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schulman B.A., Carrano A.C., Jeffrey P.D., Bowen Z., Kinnucan E.R., Finnin M.S., Elledge S.J., Harper J.W., Pagano M., Pavletich N.P. Insights into SCF ubiquitin ligases from the structure of the Skp1-Skp2 complex. Nature. 2000;408:381–386. doi: 10.1038/35042620. [DOI] [PubMed] [Google Scholar]
- 16.Sali A., Blundell T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 17.Waterhouse A.M., Procter J.B., Martin D.M., Clamp M., Barton G.J. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tiranti V., Galimberti C., Nijtmans L., Bovolenta S., Perini M.P., Zeviani M. Characterization of SURF-1 expression and Surf-1p function in normal and disease conditions. Hum. Mol. Genet. 1999;8:2533–2540. doi: 10.1093/hmg/8.13.2533. [DOI] [PubMed] [Google Scholar]
- 19.Dingley S., Chapman K.A., Falk M.J. Fluorescence-activated cell sorting analysis of mitochondrial content, membrane potential, and matrix oxidant burden in human lymphoblastoid cell lines. Methods Mol. Biol. 2012;837:231–239. doi: 10.1007/978-1-61779-504-6_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heckmatt J.Z., Moosa A., Hutson C., Maunder-Sewry C.A., Dubowitz V. Diagnostic needle muscle biopsy. A practical and reliable alternative to open biopsy. Arch. Dis. Child. 1984;59:528–532. doi: 10.1136/adc.59.6.528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sciacco M., Bonilla E. Cytochemistry and immunocytochemistry of mitochondria in tissue sections. Methods Enzymol. 1996;264:509–521. doi: 10.1016/s0076-6879(96)64045-2. [DOI] [PubMed] [Google Scholar]
- 22.Bugiani M., Invernizzi F., Alberio S., Briem E., Lamantea E., Carrara F., Moroni I., Farina L., Spada M., Donati M.A. Clinical and molecular findings in children with complex I deficiency. Biochim. Biophys. Acta. 2004;1659:136–147. doi: 10.1016/j.bbabio.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 23.Invernizzi F., D’Amato I., Jensen P.B., Ravaglia S., Zeviani M., Tiranti V. Microscale oxygraphy reveals OXPHOS impairment in MRC mutant cells. Mitochondrion. 2012;12:328–335. doi: 10.1016/j.mito.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Danhauser K., Iuso A., Haack T.B., Freisinger P., Brockmann K., Mayr J.A., Meitinger T., Prokisch H. Cellular rescue-assay aids verification of causative DNA-variants in mitochondrial complex I deficiency. Mol. Genet. Metab. 2011;103:161–166. doi: 10.1016/j.ymgme.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 25.Petruzzella V., Tiranti V., Fernandez P., Ianna P., Carrozzo R., Zeviani M. Identification and characterization of human cDNAs specific to BCS1, PET112, SCO1, COX15, and COX11, five genes involved in the formation and function of the mitochondrial respiratory chain. Genomics. 1998;54:494–504. doi: 10.1006/geno.1998.5580. [DOI] [PubMed] [Google Scholar]
- 26.Cooper H.M., Spelbrink J.N. The human SIRT3 protein deacetylase is exclusively mitochondrial. Biochem. J. 2008;411:279–285. doi: 10.1042/BJ20071624. [DOI] [PubMed] [Google Scholar]
- 27.Reyes A., He J., Mao C.C., Bailey L.J., Di Re M., Sembongi H., Kazak L., Dzionek K., Holmes J.B., Cluett T.J. Actin and myosin contribute to mammalian mitochondrial DNA maintenance. Nucleic Acids Res. 2011;39:5098–5108. doi: 10.1093/nar/gkr052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ghezzi D., Viscomi C., Ferlini A., Gualandi F., Mereghetti P., DeGrandis D., Zeviani M. Paroxysmal non-kinesigenic dyskinesia is caused by mutations of the MR-1 mitochondrial targeting sequence. Hum. Mol. Genet. 2009;18:1058–1064. doi: 10.1093/hmg/ddn441. [DOI] [PubMed] [Google Scholar]
- 29.Van Rechem C., Black J.C., Abbas T., Allen A., Rinehart C.A., Yuan G.C., Dutta A., Whetstine J.R. The SKP1-Cul1-F-box and leucine-rich repeat protein 4 (SCF-FbxL4) ubiquitin ligase regulates lysine demethylase 4A (KDM4A)/Jumonji domain-containing 2A (JMJD2A) protein. J. Biol. Chem. 2011;286:30462–30470. doi: 10.1074/jbc.M111.273508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Willems A.R., Schwab M., Tyers M. A hitchhiker’s guide to the cullin ubiquitin ligases: SCF and its kin. Biochim. Biophys. Acta. 2004;1695:133–170. doi: 10.1016/j.bbamcr.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 31.Petroski M.D., Deshaies R.J. Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 32.Galan J.M., Wiederkehr A., Seol J.H., Haguenauer-Tsapis R., Deshaies R.J., Riezman H., Peter M. Skp1p and the F-box protein Rcy1p form a non-SCF complex involved in recycling of the SNARE Snc1p in yeast. Mol. Cell. Biol. 2001;21:3105–3117. doi: 10.1128/MCB.21.9.3105-3117.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakamura N., Hirose S. Regulation of mitochondrial morphology by USP30, a deubiquitinating enzyme present in the mitochondrial outer membrane. Mol. Biol. Cell. 2008;19:1903–1911. doi: 10.1091/mbc.E07-11-1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Narendra D.P., Youle R.J. Targeting mitochondrial dysfunction: role for PINK1 and Parkin in mitochondrial quality control. Antioxid. Redox Signal. 2011;14:1929–1938. doi: 10.1089/ars.2010.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.