

Abstract
Primary ciliary dyskinesia (PCD) is a rare autosomal-recessive respiratory disorder resulting from defects of motile cilia. Various axonemal ultrastructural phenotypes have been observed, including one with so-called central-complex (CC) defects, whose molecular basis remains unexplained in most cases. To identify genes involved in this phenotype, whose diagnosis can be particularly difficult to establish, we combined homozygosity mapping and whole-exome sequencing in a consanguineous individual with CC defects. This identified a nonsense mutation in RSPH1, a gene whose ortholog in Chlamydomonas reinhardtii encodes a radial-spoke (RS)-head protein and is mainly expressed in respiratory and testis cells. Subsequent analyses of RSPH1 identified biallelic mutations in 10 of 48 independent families affected by CC defects. These mutations include splicing defects, as demonstrated by the study of RSPH1 transcripts obtained from airway cells of affected individuals. Wild-type RSPH1 localizes within cilia of airway cells, but we were unable to detect it in an individual with RSPH1 loss-of-function mutations. High-speed-videomicroscopy analyses revealed the coexistence of different ciliary beating patterns—cilia with a normal beat frequency but abnormal motion alongside immotile cilia or cilia with a slowed beat frequency—in each individual. This study shows that this gene is mutated in 20.8% of individuals with CC defects, whose diagnosis could now be improved by molecular screening. RSPH1 mutations thus appear as a major etiology for this PCD phenotype, which in fact includes RS defects, thereby unveiling the importance of RSPH1 in the proper building of CCs and RSs in humans.
Main Text
Primary ciliary dyskinesia (PCD [MIM 244400]) is a rare genetic disease usually transmitted in an autosomal-recessive fashion and affects 1 in 15,000–30,000 individuals.1 The disease results from functional and/or structural defects of motile cilia. These defects are responsible for impaired mucociliary transport, leading to recurrent respiratory-tract infections beginning in early childhood. Most male individuals have asthenospermia due to similar defects in their sperm flagella.2 Approximately 50% of persons with PCD display situs inversus, thereby defining Kartagener syndrome (MIM 244400).3 A number of genes playing a key role in the proper building of motile cilia have recently been involved in the pathogenesis of PCD.4–23 However, in spite of these significant results, the disease remains unexplained in more than half of affected individuals from our PCD cohort.
Cilia are evolutionarily conserved organelles that protrude from the surface of most eukaryotic cells. Their axoneme consists of nine peripheral microtubule doublets either surrounding a central pair of microtubules (i.e., “9+2” pattern) or not (i.e., “9+0” pattern). The 9+0 cilia are immotile, except in the embryonic node, a structure involved in the early establishment of the left-right asymmetry.24 The axoneme of motile cilia is characterized by the presence of inner dynein arms (IDAs) and outer dynein arms (ODAs), which are multiprotein ATPase complexes that are attached to the peripheral doublets and are essential for normal ciliary and flagellar movements.25 Radial spokes (RSs), which are present in 9+2 cilia, are T-shaped structures linking each peripheral microtubule doublet to the two central microtubules (C1 and C2), which are surrounded by a central sheath and participate in the formation of the central complex (CC),26,27 also called the central-pair complex.28,29 RSs and CCs are believed to serve as sensors that control beating, especially the waveform, both in motile cilia and in flagella.30 According to data obtained in the flagellated chlorophyte Chlamydomonas reinhardtii, premature RSs are assembled into the cytoplasm and are dimerized along the length of cilia;31 each RS dimer is composed of at least 23 proteins,32 five of which belong to the RS head (the horizontal bar of the “T”), which interacts with the CC.
In most individuals with PCD, the axonemal defect concerns the dynein arms and is related to mutations in different genes (DNAI14 [MIM 604366], DNAI25 [MIM 605483], DNAH56 [MIM 603335], NME87 [MIM 607421], DNAL18 [MIM 610062], CCDC1149,10 [MIM 615038], DNAAF111,12 [MIM 613190], DNAAF213 [MIM 612517], DNAAF314 [MIM 614566], LRRC615 [MIM 614930], HEATR216 [MIM 614864], CCDC10317 [MIM 614677], CCDC3918 [MIM 613798], and CCDC4019 [MIM 613799]). Some individuals have no detectable structural axonemal defect; mutations in DNAH1120 (MIM 603339) or DRC121 (MIM 615288) have been found in this phenotype. As for the PCD phenotype characterized by CC defects, it was present in 15% of individuals in our cohort.27,33 The molecular basis of this particular phenotype, which is also characterized by the fact that affected individuals never have laterality defects, remains unexplained in the majority of cases. So far, three genes have been involved in this phenotype. Two of them encode RS-head proteins: RSPH4A (MIM 612647), which has been found to be mutated in nine families,22,34 and RSPH9 (MIM 612648), in which the same mutation has been identified in three Bedouin families;22,35 the third gene, HYDIN (MIM 610812), has recently been involved in PCD with central-sheath defects.23 We undertook this study with the aim of identifying additional molecular defects involved in PCD with CC defects by combining homozygosity mapping and whole-exome sequencing (WES). The current study was approved by the Ile-de-France ethics committee (CPP07729), and written informed consent was obtained from all individuals and/or their parents.
Forty-eight unrelated families (containing 62 affected individuals) in our PCD cohort were affected by CC defects; none of them showed laterality defects. In all of them, the axonemal defect, which affected a variable proportion of cilia in each individual, was confirmed by transmission electronic microscopy (TEM), which showed the absence of one or both microtubules of the central pair (9+1 or 9+0 pattern), sometimes associated with the internalization of one peripheral doublet (8+1 pattern).27,33,36 Moreover, this ultrastructural phenotype is characterized by the fact that the RSs are present in cilia with a CC but absent from cilia with no CC. The ultrastructural defect was confirmed on repeated airway biopsies performed at different levels (i.e., nasal and bronchial sampling). By screening the genes already involved in this PCD phenotype, we identified mutations in 11 of these 48 families: RSPH4A mutations in seven families and RSPH9 mutations in four families (Table S1, available online). One individual (DCP940 from family DC564) of the 37 unrelated families (44 affected individuals) with no mutations in RSPH4A or RSPH9 was selected for WES. This female person, who was born to a consanguineous union, presented with a sinopulmonary syndrome beginning in early childhood, subfertility, and no situs inversus. She exhibited a very marked ultrastructural phenotype in which 60% of cilia showed CC defects (Figure 1 and Table 1); total ciliary immotility was observed in ciliated cells obtained by airway brushing. Her genomic DNA was first genotyped with the HumanCytoSNP-12 chip from Illumina, and the data were analyzed with GenomeStudio and CNVPartition 3.1.6 software (Illumina), as well as with an in-house script designed to identify homozygous regions. Three large homozygous regions spanning 13.8, 8.3, and 6 Mb, were identified on chromosomes 1, 21, and 8, respectively, and contained a total of 329 genes (Figure S1).
Figure 1.
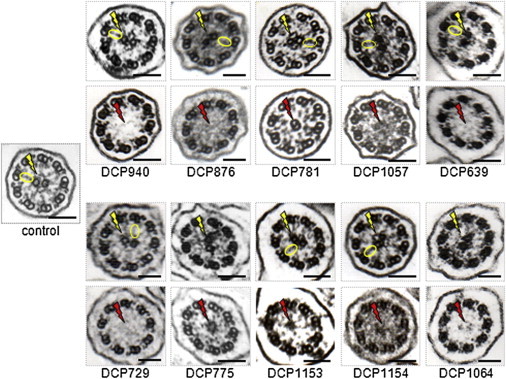
CC and RS Defects in Respiratory Cilia of Individuals with RSPH1 Mutations
The electron micrographs of cross-sections of cilia from a control and ten individuals with identified biallelic RSPH1 mutations are shown. For each affected individual, two sections are shown: one with a normal configuration showing the presence of the CC and RSs (top) and the other with an abnormal axonemal configuration characterized by CC and RS abnormalities (bottom). The yellow flashes show the presence of normal CCs in the normal control and the affected individuals; RSs are encircled in yellow in the normal control and in cilia with CCs from affected individuals. The red flashes show CC defects (including the 8+1 pattern in DCP781) in affected individuals. Black scale bars represent 0.1 μm.
Table 1.
Phenotypic Features of Individuals with Identified RSPH1 Mutations
| Family (Origin) | Individual | Known Consanguinity | Gender | Airway Disease | Fertility | NOa(nl/min) | Abnormal Cilia (TEM)b(%) | Allele 1 | Allele 2 |
|---|---|---|---|---|---|---|---|---|---|
| DC564 (North African) | DCP940 | yes | female | NRD, bronchiectasis, rhinosinusitis, otitis | subfertility | NP | 60 | c.85G>T (p.Glu29∗) | c.85G>T (p.Glu29∗) |
| DC18 (European) | DCP876 | no | male | COPD, bronchiectasis, rhinosinusitis | NR | NP | 50 | c.85G>T (p.Glu29∗) | c.366−3C>A |
| DC129 (European) | DCP781 | no | male | bronchiectasis, rhinosinusitis, otitis | asthenospermia | lowc | 58 | c.366−3C>A | c.407_410delAGTA (p.Lys136Metfs∗6) |
| DC337 (European) | DCP1057 | no | female | COPD, bronchiectasis, lobectomy, rhinosinusitis, otitis | subfertility | NP | 23 | c.275−2A>C (p.Gly92Alafs∗10) | c.727+5G>A (p.Ala244Valfs∗22) |
| DC402 (European) | DCP638 | no | male | chronic bronchitis, rhinosinusitis | NR | NP | NP | c.275−2A>C (p.Gly92Alafs∗10) | c.275−2A>C (p.Gly92Alafs∗10) |
| DCP639 | no | female | chronic bronchitis, rhinosinusitis | NR | NP | 34 | c.275−2A>C (p.Gly92Alafs∗10) | c.275−2A>C (p.Gly92Alafs∗10) | |
| DC455 (European) | DCP729 | no | female | NRD, bronchiectasis, lobectomy, rhinosinusitis, otitis | subfertility | 80 | 22 | c.85G>T (p.Glu29∗) | c.308G>A (p.Gly103Asp) |
| DC477 (North African) | DCP775 | yes | male | bronchiectasis, pneumonia, rhinosinusitis, otitis | NR | 37 | 34.5 | c.85G>T (p.Glu29∗) | c.85G>T (p.Glu29∗) |
| DC530 (European) | DCP873 | yes | male | bronchiectasis, rhinosinusitis | ND | NP | 70 | c.275−2A>C (p.Gly92Alafs∗10) | c.275−2A>C (p.Gly92Alafs∗10) |
| DC645 (European) | DCP1153 | no | male | NRD, chronic bronchitis, otitis | NR | NP | 25.5 | c.275−2A>C (p.Gly92Alafs∗10) | c.275−2A>C (p.Gly92Alafs∗10) |
| DCP1154 | no | male | bronchiectasis, otitis | NR | 45 | 19 | c.275−2A>C (p.Gly92Alafs∗10) | c.275−2A>C (p.Gly92Alafs∗10) | |
| DC651 (European) | DCP1064 | no | female | NRD, COPD, bronchiectasis, rhinosinusitis, otitis | NR | 500 | 44 | c.85G>T (p.Glu29∗) | c.366G>A (p.[Arg122Serfs∗22, p.=]) |
Abbreviations are as follows: NO, nitric oxide; TEM, transmission electron microscopy; NRD, neonatal respiratory distress; NP, not performed; COPD, chronic obstructive pulmonary disease; NR, not relevant; and ND, not determined.
Nasal NO was measured during apnea with the use of a chemiluminscence NO analyzer (NIOX Flex, Aerocrine, and Endono 8000, Seres). The mean value of the plateau was recorded. NO values above 100 nl/min were considered normal.
The method used is described in Papon et al.27
NO value not available.
Nonsense, missense, or frameshift variants identified by WES, which was performed by IntegraGen with the Agilent SureSelect All Exon v.4+UTR 70 Mb capture kit on a HiSeq 2000 machine, were filtered against several sequence-variant databases (i.e., dbSNP, 1000 Genomes, and the National Heart, Lung, and Blood Institute [NHLBI] Exome Sequencing Project Exome Variant Server [EVS]) (Table S2). Following an autosomal-recessive model of PCD, we considered further variants in genes located in homozygous regions. Among those variations (2,870), 43 were located in intergenic regions, 1,880 were found to be located outside the coding regions of genes (832 in 3′ UTR sequences, 367 in 5′ UTR sequences, and 681 in introns), seven were at an exon-intron junction, 460 corresponded to missense variations, 479 were found to be synonymous coding variants, and only one homozygous nonsense variant was found. This latter variant (c.85G>T), located in RSPH1, was not shared by internal controls (i.e., results of WES performed in individuals not affected by PCD) and was one of the 294 variants (out of the total 2,870) whose frequency was below 0.02. In humans, RSPH1 (MIM 609314) is located in chromosomal region 21q22.3 and consists of nine exons; the only predicted transcript (RefSeq accession number NM_080860.2) encodes a 309-residue protein with five N-terminal MORN (membrane occupation and recognition nexus) repeats,37 followed by a linker and a sixth MORN repeat (Figure 2A). RSPH1 is the homolog of Chlamydomonas RSP1. RSP1 is much longer than RSPH1; however, the functional domains lie in regions that have been conserved throughout evolution (sequence homology ∼63%). The c.85G>T variant leads to a premature stop codon at position 29 (p.Glu29∗). It is reported in dbSNP (rs138320978) and in EVS at an extremely low frequency (5 of 13,006 alleles, allele frequency = 0.00038) (Table S3), and the homozygous genotype is not reported in 2,276 individuals. Because the incidence of PCD ranges from 1 in 15,000 to 1 in 30,000 individuals,38 the expected frequency of a PCD allele ranges from 0.006 to 0.008 . More specifically, given the allele frequency of the c.85G>T variation (0.00038), the frequency of the homozygous genotype should be extremely low (14.44 × 10−8), which is therefore totally consistent with the fact that this genotype was identified in an individual affected by a rare disease. Therefore, this datum, together with the expected deleterious consequences of the c.85G>T variation at the protein level (i.e., premature stop codon leading to a severely truncated protein lacking 90% of the protein, including all MORN repeats, or to the absence of production of the protein through activation of the nonsense-mediated-mRNA-decay pathway39), is consistent with the implication of this allele in the disease. Sanger sequencing was performed for validation of the nonsense mutation found in DCP940 (Figure 2A and Figure S2). Parental DNA samples were not available; however, as deduced from the homozygosity mapping data (Figure S1), the c.85G>T transversion is present in the homozygous state in DCP940.
Figure 2.
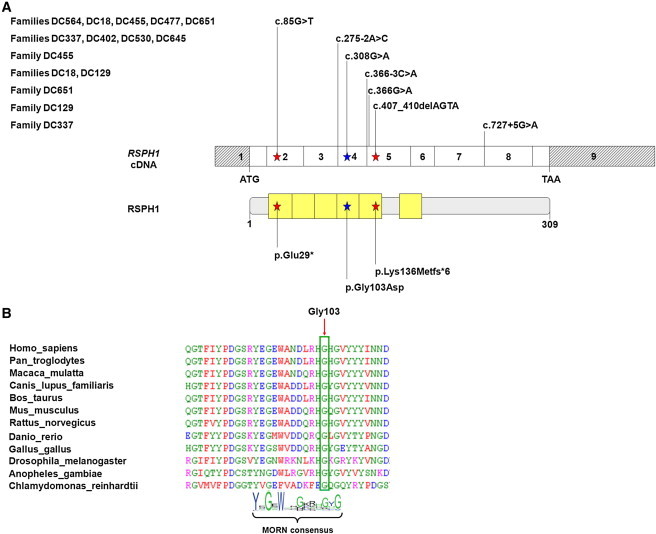
RSPH1 Mutations and Their Impact at the Protein Level in Individuals with PCD
(A) Exonic organization of the human RSPH1 cDNA, in which are shown the mutations for the ten families described in this study (top), and a domain-organization model of the corresponding protein (middle). The mutations’ impact at the protein level is shown (bottom). The nine exons are indicated by empty or hashed boxes depicting translated or untranslated sequences, respectively. According to the prediction tools provided by the NCBI and UniProt (see Web Resources), RSPH1 contains five MORN repeats, followed by a linker and a sixth MORN repeat.
(B) A partial protein alignment of RSPH1 in different species shows the evolutionary conservation of the fourth MORN repeat, which contains the amino acid substitution identified in this study.
The remaining 36 independent families (43 individuals) affected by PCD and CC defects and with no mutation in RSPH4A and RSPH9 were subsequently screened for RSPH1 mutations. Overall, 12 individuals from ten of these families were found to harbor biallelic RSPH1 mutations (Table 1, Figure 2A, Figure S2, and Table S3). We found a total of seven mutations. Two of them lead to a premature stop codon (c.85G>T [p.Glu29∗] and c.407_410delAGTA [p.Lys136Metfs∗6]). One is a missense variation (c.308G>A [p.Gly103Asp]). Three are splice-site mutations: the first affects the invariable dinucleotide of the splice acceptor site of intron 2 (c.275−2A>C), the second involves a highly conserved nucleotide of the splice acceptor site of intron 4 (c.366−3C>A), and the third involves the 5′ splice donor site of intron 7 (c.727+5G>A). The seventh mutation is an apparently synonymous variation (c.366G>A [p.=]). If translated, the p.Lys136Metfs∗6 molecular defect is predicted to result in a severely truncated protein lacking 55% of the protein (including the last MORN repeat). The p.Gly103Asp (c.308G>A) missense variation, which replaces a hydrophobic amino acid with a charged hydrophilic acidic amino acid, involves a residue that is invariant throughout evolution and that is part of the MORN consensus, as defined by Garbino et al.40 (Figure 2B). This substitution has not been described in dbSNP, Ensembl, or the EVS, and this, together with the identification of a nonsense mutation in trans in an individual with a typical PCD phenotype (DCP729), is consistent with the implication of the c.308G>A allele in the disease.
Whereas the c.308G>A, c.366−3C>A, c.366G>A, c.407_410delAGTA, and c.727+5G>A mutations are not reported in databases such as dbSNP, Ensembl, or the EVS, the c.275−2A>C splice-site mutation has been described in the EVS at an extremely low frequency in European-American populations (9 out of 8,600 alleles, allele frequency = 0.001), and in dbSNP (rs151107532) (Table S3), the homozygous genotype is never described among 2,937 individuals. Such a low frequency is compatible with the expected frequency of a PCD allele. The genotype of the 12 individuals from the ten independent families in whom we identified RSPH1 mutations is detailed in Table 1.
With the aim of assessing the functional consequences of the c.275−2A>C, c.727+5G>A, and c.366G>A variations on the processing of RSPH1 transcripts, we isolated total RNA from airway cells of the individuals harboring these variations and analyzed RSPH1 transcripts after RT-PCR amplification followed by Sanger sequencing. As shown in Figure S3, the c.275−2A>C mutation, located upstream of the fourth exon, leads to the skipping of exons 4 and 5 and to a premature stop codon (p.Gly92Alafs∗10). The c.727+5G>A mutation, involving the splice site downstream of the seventh exon, results in the incorporation of the first two intronic nucleotides in the spliced transcripts, leading to a premature stop codon (p.Ala244Valfs∗22). The apparently synonymous c.366G>A (p.=) transition involves the first base of the fifth exon; it could, therefore, affect splicing. Two transcripts were indeed coamplified (Figure S3A): the minor molecular species, which harbors the G>A transition, is the result of normal splicing; the other molecular species lacks the fifth exon, leading to a premature stop codon in exon 6 (p.Arg122Serfs∗22) (Figure S3B).
The 12 individuals with biallelic RSPH1 mutations displayed a clinical phenotype characterized by a sinopulmonary syndrome and situs solitus; four of them (DCP940, DCP729, DCP1153, and DCP1064) had unexplained neonatal respiratory distress. All adult individuals (one male and three females, who intended to have a child) had fertility problems. Ciliary motility was evaluated by optic microscopy on airway brushing in 11/12 individuals (not available for DCP873): cilia were found to be totally immotile in DCP940, whereas cilia from the same sample displayed different beating patterns (active, slow, or immotile) in the ten other individuals. High-speed-videomicroscopy (HSVM) analyses of airway cells were performed in 6 of the 11 individuals (DCP781, DCP1057, DCP729, DCP1153, DCP1154, and DCP1064) according to already described conditions.41 In brief, beating ciliated edges were recorded with a high-speed digital camera (PixeLINK A741) with a 100× objective at a rate of 355 frames per second. The percentage of beating cilia was determined, and ten cilia able to be followed during a complete beating cycle were then selected in distinct areas. Various measurements were used for determining the following parameters: the ciliary beat frequency (CBF), the beating angle, and the distance traveled by the tip of the cilium per second. The comparisons between controls evaluated in a previous study42 and individuals with RSPH1 mutations were performed with a Mann-Whitney U test (nonparametric test). A p value < 0.05 was considered significant. In the HSVM analyses, different populations of cilia were identified in each of the six individuals with biallelic RSPH1 mutations: some were immotile or displayed a very low CBF. Most importantly, others beat with a normal frequency; however, their beating pattern, characterized by movements of reduced amplitude, was abnormal: the beating angle and the distance traveled by the cilia’s tips were significantly reduced (Table 2 and Movies S1, S2, S3, S4, S5, S6, and S7). Nasal nitric oxide (NO) measurements available in five individuals (DCP781, DCP729, DCP775, DCP1154, and DCP1064) were found to be low in four cases and normal (controlled twice) in one individual (DCP1064) (Table 1). Noteworthy, the CC defects observed by TEM were found in various proportions of cilia (ranging from 19% to 70% of cilia, depending on the individual) and were never present in all cilia.
Table 2.
HSVM Parameters in Six Affected Individuals with Identified RSPH1 Mutations
| Family | Individual | Beating Cilia (%) | CBF (Hz)a | Angle (°)a | Distance Traveled (μm/sec)a | Allele 1 | Allele 2 |
|---|---|---|---|---|---|---|---|
| DC129 | DCP781 | 80 | 7 ± 2 | 36 ± 22 | 21.5 ± 19 | c.366−3C>A | c.407_410delAGTA (p.Lys136Metfs∗6) |
| DC337 | DCP1057 | 80 | 9.2 ± 3.3 | 53 ± 25 | 46.4 ± 23 | c.275−2A>C (p.Gly92Alafs∗10) | c.727+5G>A (p.Ala244Valfs∗22) |
| DC455 | DCP729 | 65 | 6.7 ± 1.6 | 76 ± 14 | 57.6 ± 25.6 | c.85G>T (p.Glu29∗) | c.308G>A (p.Gly103Asp) |
| DC645 | DCP1153 | 30 | 6.2 ± 1.6 | 58 ± 11 | 22 ± 3.8 | c.275−2A>C (p.Gly92Alafs∗10) | c.275−2A>C (p.Gly92Alafs∗10) |
| DCP1154 | 80 | 8.2 ± 1.6 | 42 ± 18 | 23.5 ± 10 | c.275−2A>C (p.Gly92Alafs∗10) | c.275−2A>C (p.Gly92Alafs∗10) | |
| DC651 | DCP1064 | 40 | 7.6 ± 2.5 | 64 ± 25 | 43 ± 30 | c.85G>T (p.Glu29∗) | c.366G>A (p.[Arg122Serfs∗22, p.=]) |
| Individuals with RSPH1 mutations (mean ± SD) | 59 ± 21 | 7.5 ± 1.1 | 54.8 ± 14.7 | 35.6 ± 15.4 | - | - | |
| Controls (mean ± SD) | 91 ± 13 | 8.9 ± 2 | 71.6 ± 6.6 | 66.7 ± 14.2 | - | - | |
| Statistical significance (p) | 0.001 | 0.13 | 0.014 | 0.002 | - | - | |
The following abbreviation is used: CBF, ciliary beat frequency.
The CBF, angle, and distance traveled were evaluated on beating cilia.
To gain further insight into the role of RSPH1, we first determined its expression by quantitative RT-PCR. The analysis revealed that RSPH1 is readily expressed in tissues with motile cilia or flagella, such as the trachea, lungs, airway brushings, and testes (Figure S4A). This expression pattern is typical of genes associated with motile cilia. We then compared its expression in ciliated tissues with the expression of RSPH4A, RSPH6A, RSPH9, and RSPH10B, whose orthologs encode the four other RS-head proteins in Chlamydomonas reinhardtii.43 We found similar expression levels of RSPH1 and RSPH4A in ciliated tissues (albeit an extremely strong expression of RSPH4A was observed in nasal brushings). The expression levels of RSPH9 and RSPH10B in those tissues were found to be lower than those of RSPH1 and RSPH4A, whereas, as shown by others,44 RSPH6A was found to be expressed in testes only (Figure S4B). These results might suggest that RSPH1, RSPH4A, RSPH9, and RSPH10B play important roles in both ciliary and flagellar axonemes, whereas RSPH6A would have a specific and so far unknown function in sperm cells.
We subsequently determined RSPH1 subcellular localization by means of immunofluorescence microscopy with an RSPH1 antibody. This was performed in human ciliated cells collected by nasal brushing from a healthy control, from DCP1064’s healthy father (with the RSPH1 c.[=];[366G>A] genotype), and from individual DCP1064 (with the RSPH1 c.[85G>T];[366G>A] genotype). Airway cells from the healthy control and the healthy father showed RSPH1 labeling within the cilia (Figures 3A–3F); by contrast, RSPH1 was not detected in cilia from individual DCP1064 (Figures 3G–3I). This result, which confirms the specificity of the labeling obtained with the RSPH1 antibody used in this experiment, is consistent with the loss-of-function mutations harbored by this individual. To test whether mutations in RSPH1 affect the subcellular localization of another RS-head protein, we determined RSPH4A subcellular localization in ciliated cells from a healthy control, individual DCP1064, and her healthy father. In airway cells from the control individual and the healthy father, we observed a ciliary labeling (Figures S5A–S5F) similar to that obtained with the RSPH1 antibody. Noteworthy, in the individual with RSPH1 mutations, the RSPH4A labeling was still localized within cilia (Figures S5G–S5I). This result is consistent with the observation that during the cytoplasmic preassembly of the RS head in Chlamydomonas, RSP4 (the ortholog of RSPH4A) is assembled prior to RSP1 (the ortholog of RSPH1).31 The ciliary labeling of RSPH4A in individual DCP1064 (with RSPH1 mutations) seems, however, to be absent from the distal tip of the cilia, suggesting that the building of RSs might be a more complex process than previously thought.
Figure 3.
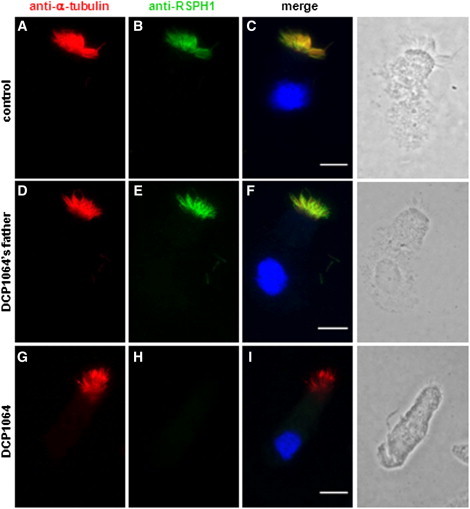
RSPH1 Localizes to Cilia and Is Absent from Airway Epithelial Cells of DCP1064
(A–F) In airway epithelial cells of a healthy control (A–C) and the healthy father of DCP1064 (D–F), RSPH1 (green) localizes within cilia (red).
(G–I) In cells from individual DCP1064, RSPH1 labeling is absent from cilia. Airway epithelial cells were examined after labeling with a rabbit RSPH1 polyclonal antibody (Sigma HPA017382, 1:100, 37°C, 1 hr) and a secondary goat anti-rabbit Alexa Fluor-488 (green) antibody (Invitrogen, A11034). For controls, we used an antibody directed against acetylated α-tubulin (mouse monoclonal [6-11B-1], Abcam ab24610, 1:700) for the visualization of microtubules revealed by a secondary goat anti-mouse Alexa Fluor-594 (red) antibody (Invitrogen, A11032). Nuclei were stained with DAPI (Sigma, 32670). White scale bars represent 10 μm.
Over the past few years, WES has become an efficient means of searching for new molecular defects, especially in recessive diseases. Last year, mutations in three genes were found by WES to be involved in PCD with different ultrastructural phenotypes: HEATR2 in one Amish family affected by dynein-arm defects,16 HYDIN in two independent families affected by central-sheath defects,23 and CCDC114 in five independent families affected by ODA defects.9,10 Although WES is a very efficient approach to uncovering variants, it could be quite challenging to point out causative mutations among an average of about, 40,000 variants per genome. To improve the efficiency of WES by reducing the number of candidate variants, we chose to combine this global approach with homozygosity mapping in selected affected individuals born to consanguineous unions. Following this strategy in an individual with a typical CC defect, we identified only 2,870 variants in homozygous regions out of a total of 43,924 variants, among which we could spot the only homozygous nonsense variant (c.85G>T) in RSPH1, an excellent candidate gene for PCD.
It has been shown in Chlamydomonas that RSP1, the ortholog of RSPH1, participates in the formation of the RS head together with RSP4, RSP6, RSP9, and RSP10, whose human homologs are RSPH4A, RSPH6, RSPH9, and RSPH10B, respectively. As shown in Chlamydomonas,31 the RS head is preassembled into the cell body prior to its attachment to the RS stalk; RSs are then dimerized within flagella. As shown through a biochemical approach, the absence of one RS-head protein (i.e., RSP4 in the pf1 mutant) or one RS-neck protein (i.e., RSP2 in the pf24 mutant) leads to a partial assembly in the cytoplasm of only RS stalks, which are also detected in flagella.31 However, to our knowledge, there is no information on the CC of these Chlamydomonas mutants. Our study shows that in humans, the absence of RSPH1 leads to an abnormal axonemal configuration with CC defects and an absence of RSs in cilia with no CCs, thereby unveiling the importance of RSPH1 in the proper building of CCs and RSs (Figure 1). Of note, the individuals (from our PCD cohort) with mutations in RSPH4A or RSPH9 have a similar ultrastructural phenotype (Figure S6). We therefore propose that this phenotype should be called PCD with CC and RS defects.
As expected for a gene associated with PCD, RSPH1 is mainly expressed in tissues with motile cilia or flagella and, as shown here, the corresponding protein localizes within cilia of airway epithelial cells. Noteworthy, RSPH1 carries six MORN repeats; the MORN motif is a repeat found in multiple copies in several proteins, including the junctophilins family. Junctophilins are believed to play a role in the regulation of intracellular-calcium-signaling pathways, especially in skeletal and cardiac muscles, as well as in neurons;40 it has also been shown that the integrity of the MORN motif is required for calcium signaling in cardiomyocytes.45 In the light of these data and given the fact that ciliary motion is a calcium-regulated event,46 in which RSs are known to play a key role,30 it is tempting to speculate that RSPH1 could participate in this regulation process. Consistently, cilia of individuals with RSPH1 mutations have an abnormal beating pattern (Movies S1, S2, S3, S4, S5, S6, and S7).
At first glance, this abnormal beating pattern appears to be similar to the one observed in individuals with RSPH4A or RSPH9 mutations. Different populations of cilia coexist in these individuals: besides immotile cilia or cilia with a slowed beat frequency, there are cilia with a normal beat frequency but with an abnormal motion, described as circular in individuals with RSPH4A or RSPH9 mutations.22,47 In individuals with RSPH1 mutations, thanks to qualitative and quantitative HSVM analysis, it was possible to analyze the ciliary motion more precisely and to show a low-amplitude movement caused by a diminished beating angle and a reduced distance traveled by cilia’s tips. Ciliary motion is also abnormal in other species with defective RS-head proteins. There is no RSP1 mutant in Chlamydomonas. However, a mutant with paralyzed flagella called pf1 is defective for RSP4,48 one of the two interactants of RSP1.49 In sea urchins, sperm flagella that have been exposed to an antibody directed against the ortholog of RSPH4A once the axoneme is properly built also have an abnormal beating pattern, which changes from a two-dimensional movement to a three-dimensional movement.50
One important consequence of our study concerns the diagnostic value of molecular studies in individuals with a suspicion of PCD. The diagnosis of PCD is indeed quite challenging in the particular case of individuals with CC defects. First, situs inversus is never present, which has been classically explained by the fact that CC defects are not expected to affect the 9+0 structure of nodal cilia. Second, although all cilia were found immotile in one individual (DCP940), we showed in individuals for whom HSVM was available that the percentage of motile cilia is highly variable, ranging from 30% to 80% (Table 2); therefore, cilia are frequently beating, and their beat frequency could even be normal (Table 2). In such a case as shown here, the identification of abnormalities in ciliary motion requires qualitative and quantitative analyses by HSVM.42 Third, the ultrastructural defect is never observed in all cilia from a given individual36,47 and can be more or less marked among individuals. In individuals with mutations in RSPH9, this difference in the observed percentage of abnormal cilia was explained by an intermittent defect along the length of cilia.22 In theory, this variability could also be explained by different RSPH1 molecular defects, but as shown in DCP1154 and DCP873, even in two individuals with the same genotype, the percentage of abnormal cilia might vary considerably (19% and 70% in DCP1154 and DCP873, respectively). Lastly, as for nasal-NO measurement, which represents a reasonable screening method for the diagnosis of PCD,51 the NO values can be normal in some individuals (from 0% to 25% of affected individuals, depending on the study).51–54 As shown here, this is the case of one CC- and RS-defect-affected individual (DCP1064) in whom NO measurement was repeated twice in our national reference center for rare lung diseases. Overall, such phenotypic features explain the difficulties in establishing the diagnosis of PCD in individuals with CC and RS defects and therefore underline the major interest of molecular analyses in confirming the diagnosis.
In summary, this study shows that mutations in RSPH1 result in a PCD phenotype with CC and RS defects. Although PCD is a rare disorder, our national recruitment makes it possible to evaluate the respective contribution of RSPH4A, RSPH9, and RSPH1 mutations to the PCD phenotype characterized by CC and RS defects. Among a total of 48 unrelated families affected by this PCD phenotype from our cohort, besides RSPH4A and RSPH9, which explain 14.6% (7/48) and 8.3% (4/48) of PCD cases, respectively, RSPH1 is involved in 20.8% (10/48) of families. RSPH1 mutations thus appear to be a major etiology for PCD with abnormal CCs and RSs.
Acknowledgments
We are grateful to the affected persons and their families, whose cooperation made this study possible, and we thank all referring physicians. This work was supported by grants from the Fondation pour la Recherche Médicale (DEQ20120323689), the Legs Poix from the Chancellerie des Universités, the Milena Carvajal ProKartagener Foundation, and the Assistance Publique – Hôpitaux de Paris (PHRC AOM06053 and P060245).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org/
Ensembl Genome Browser, http://www.ensembl.org
NCBI Conserved Domains, http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Afzelius B.A. A human syndrome caused by immotile cilia. Science. 1976;193:317–319. doi: 10.1126/science.1084576. [DOI] [PubMed] [Google Scholar]
- 2.Afzelius B.A. The immotile-cilia syndrome: a microtubule-associated defect. CRC Crit. Rev. Biochem. 1985;19:63–87. doi: 10.3109/10409238509086788. [DOI] [PubMed] [Google Scholar]
- 3.Kartagener M. Zur Pathogenese der Bronchiektasien: Bronchiektasien bei Situs viscerum inversus. Beiträge zur Klinik der Tuberkulose. 1933;83:489–501. [Google Scholar]
- 4.Pennarun G., Escudier E., Chapelin C., Bridoux A.M., Cacheux V., Roger G., Clément A., Goossens M., Amselem S., Duriez B. Loss-of-function mutations in a human gene related to Chlamydomonas reinhardtii dynein IC78 result in primary ciliary dyskinesia. Am. J. Hum. Genet. 1999;65:1508–1519. doi: 10.1086/302683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loges N.T., Olbrich H., Fenske L., Mussaffi H., Horvath J., Fliegauf M., Kuhl H., Baktai G., Peterffy E., Chodhari R. DNAI2 mutations cause primary ciliary dyskinesia with defects in the outer dynein arm. Am. J. Hum. Genet. 2008;83:547–558. doi: 10.1016/j.ajhg.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olbrich H., Häffner K., Kispert A., Völkel A., Volz A., Sasmaz G., Reinhardt R., Hennig S., Lehrach H., Konietzko N. Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left-right asymmetry. Nat. Genet. 2002;30:143–144. doi: 10.1038/ng817. [DOI] [PubMed] [Google Scholar]
- 7.Duriez B., Duquesnoy P., Escudier E., Bridoux A.-M., Escalier D., Rayet I., Marcos E., Vojtek A.-M., Bercher J.-F., Amselem S. A common variant in combination with a nonsense mutation in a member of the thioredoxin family causes primary ciliary dyskinesia. Proc. Natl. Acad. Sci. USA. 2007;104:3336–3341. doi: 10.1073/pnas.0611405104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mazor M., Alkrinawi S., Chalifa-Caspi V., Manor E., Sheffield V.C., Aviram M., Parvari R. Primary ciliary dyskinesia caused by homozygous mutation in DNAL1, encoding dynein light chain 1. Am. J. Hum. Genet. 2011;88:599–607. doi: 10.1016/j.ajhg.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Onoufriadis A., Paff T., Antony D., Shoemark A., Micha D., Kuyt B., Schmidts M., Petridi S., Dankert-Roelse J.E., Haarman E.G., UK10K Splice-site mutations in the axonemal outer dynein arm docking complex gene CCDC114 cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2013;92:88–98. doi: 10.1016/j.ajhg.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knowles M.R., Leigh M.W., Ostrowski L.E., Huang L., Carson J.L., Hazucha M.J., Yin W., Berg J.S., Davis S.D., Dell S.D., Genetic Disorders of Mucociliary Clearance Consortium Exome sequencing identifies mutations in CCDC114 as a cause of primary ciliary dyskinesia. Am. J. Hum. Genet. 2013;92:99–106. doi: 10.1016/j.ajhg.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duquesnoy P., Escudier E., Vincensini L., Freshour J., Bridoux A.-M., Coste A., Deschildre A., de Blic J., Legendre M., Montantin G. Loss-of-function mutations in the human ortholog of Chlamydomonas reinhardtii ODA7 disrupt dynein arm assembly and cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2009;85:890–896. doi: 10.1016/j.ajhg.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loges N.T., Olbrich H., Becker-Heck A., Häffner K., Heer A., Reinhard C., Schmidts M., Kispert A., Zariwala M.A., Leigh M.W. Deletions and point mutations of LRRC50 cause primary ciliary dyskinesia due to dynein arm defects. Am. J. Hum. Genet. 2009;85:883–889. doi: 10.1016/j.ajhg.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Omran H., Kobayashi D., Olbrich H., Tsukahara T., Loges N.T., Hagiwara H., Zhang Q., Leblond G., O’Toole E., Hara C. Ktu/PF13 is required for cytoplasmic pre-assembly of axonemal dyneins. Nature. 2008;456:611–616. doi: 10.1038/nature07471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mitchison H.M., Schmidts M., Loges N.T., Freshour J., Dritsoula A., Hirst R.A., O’Callaghan C., Blau H., Al Dabbagh M., Olbrich H. Mutations in axonemal dynein assembly factor DNAAF3 cause primary ciliary dyskinesia. Nat. Genet. 2012;44:381–389. doi: 10.1038/ng.1106. S1–S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kott E., Duquesnoy P., Copin B., Legendre M., Dastot-Le Moal F., Montantin G., Jeanson L., Tamalet A., Papon J.-F., Siffroi J.-P. Loss-of-function mutations in LRRC6, a gene essential for proper axonemal assembly of inner and outer dynein arms, cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2012;91:958–964. doi: 10.1016/j.ajhg.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Horani A., Druley T.E., Zariwala M.A., Patel A.C., Levinson B.T., Van Arendonk L.G., Thornton K.C., Giacalone J.C., Albee A.J., Wilson K.S. Whole-exome capture and sequencing identifies HEATR2 mutation as a cause of primary ciliary dyskinesia. Am. J. Hum. Genet. 2012;91:685–693. doi: 10.1016/j.ajhg.2012.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Panizzi J.R., Becker-Heck A., Castleman V.H., Al-Mutairi D.A., Liu Y., Loges N.T., Pathak N., Austin-Tse C., Sheridan E., Schmidts M. CCDC103 mutations cause primary ciliary dyskinesia by disrupting assembly of ciliary dynein arms. Nat. Genet. 2012;44:714–719. doi: 10.1038/ng.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Merveille A.-C., Davis E.E., Becker-Heck A., Legendre M., Amirav I., Bataille G., Belmont J., Beydon N., Billen F., Clément A. CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat. Genet. 2011;43:72–78. doi: 10.1038/ng.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Becker-Heck A., Zohn I.E., Okabe N., Pollock A., Lenhart K.B., Sullivan-Brown J., McSheene J., Loges N.T., Olbrich H., Haeffner K. The coiled-coil domain containing protein CCDC40 is essential for motile cilia function and left-right axis formation. Nat. Genet. 2011;43:79–84. doi: 10.1038/ng.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bartoloni L., Blouin J.-L., Pan Y., Gehrig C., Maiti A.K., Scamuffa N., Rossier C., Jorissen M., Armengot M., Meeks M. Mutations in the DNAH11 (axonemal heavy chain dynein type 11) gene cause one form of situs inversus totalis and most likely primary ciliary dyskinesia. Proc. Natl. Acad. Sci. USA. 2002;99:10282–10286. doi: 10.1073/pnas.152337699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wirschell M., Olbrich H., Werner C., Tritschler D., Bower R., Sale W.S., Loges N.T., Pennekamp P., Lindberg S., Stenram U. The nexin-dynein regulatory complex subunit DRC1 is essential for motile cilia function in algae and humans. Nat. Genet. 2013;45:262–268. doi: 10.1038/ng.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Castleman V.H., Romio L., Chodhari R., Hirst R.A., de Castro S.C.P., Parker K.A., Ybot-Gonzalez P., Emes R.D., Wilson S.W., Wallis C. Mutations in radial spoke head protein genes RSPH9 and RSPH4A cause primary ciliary dyskinesia with central-microtubular-pair abnormalities. Am. J. Hum. Genet. 2009;84:197–209. doi: 10.1016/j.ajhg.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olbrich H., Schmidts M., Werner C., Onoufriadis A., Loges N.T., Raidt J., Banki N.F., Shoemark A., Burgoyne T., Al Turki S., UK10K Consortium Recessive HYDIN mutations cause primary ciliary dyskinesia without randomization of left-right body asymmetry. Am. J. Hum. Genet. 2012;91:672–684. doi: 10.1016/j.ajhg.2012.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nonaka S., Yoshiba S., Watanabe D., Ikeuchi S., Goto T., Marshall W.F., Hamada H. De novo formation of left-right asymmetry by posterior tilt of nodal cilia. PLoS Biol. 2005;3:e268. doi: 10.1371/journal.pbio.0030268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Satir P., Christensen S.T. Overview of structure and function of mammalian cilia. Annu. Rev. Physiol. 2007;69:377–400. doi: 10.1146/annurev.physiol.69.040705.141236. [DOI] [PubMed] [Google Scholar]
- 26.Escalier D., Jouannet P., David G. Abnormalities of the ciliary axonemal complex in children: An ultrastructural and cinetic study in a series of 34 cases. Biol. Cell. 1982;44:271–282. [Google Scholar]
- 27.Papon J.F., Coste A., Roudot-Thoraval F., Boucherat M., Roger G., Tamalet A., Vojtek A.M., Amselem S., Escudier E. A 20-year experience of electron microscopy in the diagnosis of primary ciliary dyskinesia. Eur. Respir. J. 2010;35:1057–1063. doi: 10.1183/09031936.00046209. [DOI] [PubMed] [Google Scholar]
- 28.Heuser T., Dymek E.E., Lin J., Smith E.F., Nicastro D. The CSC connects three major axonemal complexes involved in dynein regulation. Mol. Biol. Cell. 2012;23:3143–3155. doi: 10.1091/mbc.E12-05-0357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carbajal-González B.I., Heuser T., Fu X., Lin J., Smith B.W., Mitchell D.R., Nicastro D. Conserved structural motifs in the central pair complex of eukaryotic flagella. Cytoskeleton (Hoboken) 2013;70:101–120. doi: 10.1002/cm.21094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith E.F., Yang P. The radial spokes and central apparatus: mechano-chemical transducers that regulate flagellar motility. Cell Motil. Cytoskeleton. 2004;57:8–17. doi: 10.1002/cm.10155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Diener D.R., Yang P., Geimer S., Cole D.G., Sale W.S., Rosenbaum J.L. Sequential assembly of flagellar radial spokes. Cytoskeleton (Hoboken) 2011;68:389–400. doi: 10.1002/cm.20520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang P., Diener D.R., Yang C., Kohno T., Pazour G.J., Dienes J.M., Agrin N.S., King S.M., Sale W.S., Kamiya R. Radial spoke proteins of Chlamydomonas flagella. J. Cell Sci. 2006;119:1165–1174. doi: 10.1242/jcs.02811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vallet C., Escudier E., Roudot-Thoraval F., Blanchon S., Fauroux B., Beydon N., Boulé M., Vojtek A.M., Amselem S., Clément A., Tamalet A. Primary ciliary dyskinesia presentation in 60 children according to ciliary ultrastructure. Eur. J. Pediatr. 2013;172:1053–1060. doi: 10.1007/s00431-013-1996-5. [DOI] [PubMed] [Google Scholar]
- 34.Ziętkiewicz E., Bukowy-Bieryłło Z., Voelkel K., Klimek B., Dmeńska H., Pogorzelski A., Sulikowska-Rowińska A., Rutkiewicz E., Witt M. Mutations in radial spoke head genes and ultrastructural cilia defects in East-European cohort of primary ciliary dyskinesia patients. PLoS ONE. 2012;7:e33667. doi: 10.1371/journal.pone.0033667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alsaadi M.M., Gaunt T.R., Boustred C.R., Guthrie P.A.I., Liu X., Lenzi L., Rainbow L., Hall N., Alharbi K.K., Day I.N.M. From a single whole exome read to notions of clinical screening: primary ciliary dyskinesia and RSPH9 p.Lys268del in the Arabian Peninsula. Ann. Hum. Genet. 2012;76:211–220. doi: 10.1111/j.1469-1809.2012.00704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tamalet A., Clement A., Roudot-Thoraval F., Desmarquest P., Roger G., Boulé M., Millepied M.C., Baculard T.A., Escudier E. Abnormal central complex is a marker of severity in the presence of partial ciliary defect. Pediatrics. 2001;108:E86. doi: 10.1542/peds.108.5.e86. [DOI] [PubMed] [Google Scholar]
- 37.UniProt Consortium Reorganizing the protein space at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2012;40(Database issue):D71–D75. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bush A., Chodhari R., Collins N., Copeland F., Hall P., Harcourt J., Hariri M., Hogg C., Lucas J., Mitchison H.M. Primary ciliary dyskinesia: current state of the art. Arch. Dis. Child. 2007;92:1136–1140. doi: 10.1136/adc.2006.096958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baker K.E., Parker R. Nonsense-mediated mRNA decay: terminating erroneous gene expression. Curr. Opin. Cell Biol. 2004;16:293–299. doi: 10.1016/j.ceb.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 40.Garbino A., van Oort R.J., Dixit S.S., Landstrom A.P., Ackerman M.J., Wehrens X.H.T. Molecular evolution of the junctophilin gene family. Physiol. Genomics. 2009;37:175–186. doi: 10.1152/physiolgenomics.00017.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Papon J.F., Perrault I., Coste A., Louis B., Gérard X., Hanein S., Fares-Taie L., Gerber S., Defoort-Dhellemmes S., Vojtek A.M. Abnormal respiratory cilia in non-syndromic Leber congenital amaurosis with CEP290 mutations. J. Med. Genet. 2010;47:829–834. doi: 10.1136/jmg.2010.077883. [DOI] [PubMed] [Google Scholar]
- 42.Papon J.-F., Bassinet L., Cariou-Patron G., Zerah-Lancner F., Vojtek A.-M., Blanchon S., Crestani B., Amselem S., Coste A., Housset B. Quantitative analysis of ciliary beating in primary ciliary dyskinesia: a pilot study. Orphanet J. Rare Dis. 2012;7:78. doi: 10.1186/1750-1172-7-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Piperno G., Huang B., Luck D.J. Two-dimensional analysis of flagellar proteins from wild-type and paralyzed mutants of Chlamydomonas reinhardtii. Proc. Natl. Acad. Sci. USA. 1977;74:1600–1604. doi: 10.1073/pnas.74.4.1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eriksson M., Ansved T., Anvret M., Carey N. A mammalian radial spokehead-like gene, RSHL1, at the myotonic dystrophy-1 locus. Biochem. Biophys. Res. Commun. 2001;281:835–841. doi: 10.1006/bbrc.2001.4465. [DOI] [PubMed] [Google Scholar]
- 45.Landstrom A.P., Weisleder N., Batalden K.B., Bos J.M., Tester D.J., Ommen S.R., Wehrens X.H.T., Claycomb W.C., Ko J.-K., Hwang M. Mutations in JPH2-encoded junctophilin-2 associated with hypertrophic cardiomyopathy in humans. J. Mol. Cell. Cardiol. 2007;42:1026–1035. doi: 10.1016/j.yjmcc.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schmid A., Salathe M. Ciliary beat co-ordination by calcium. Biol. Cell. 2011;103:159–169. doi: 10.1042/BC20100120. [DOI] [PubMed] [Google Scholar]
- 47.Stannard W., Rutman A., Wallis C., O’Callaghan C. Central microtubular agenesis causing primary ciliary dyskinesia. Am. J. Respir. Crit. Care Med. 2004;169:634–637. doi: 10.1164/rccm.200306-782OC. [DOI] [PubMed] [Google Scholar]
- 48.Luck D., Piperno G., Ramanis Z., Huang B. Flagellar mutants of Chlamydomonas: studies of radial spoke-defective strains by dikaryon and revertant analysis. Proc. Natl. Acad. Sci. USA. 1977;74:3456–3460. doi: 10.1073/pnas.74.8.3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kohno T., Wakabayashi K., Diener D.R., Rosenbaum J.L., Kamiya R. Subunit interactions within the Chlamydomonas flagellar spokehead. Cytoskeleton (Hoboken) 2011;68:237–246. doi: 10.1002/cm.20507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gingras D., White D., Garin J., Cosson J., Huitorel P., Zingg H., Cibert C., Gagnon C. Molecular cloning and characterization of a radial spoke head protein of sea urchin sperm axonemes: involvement of the protein in the regulation of sperm motility. Mol. Biol. Cell. 1998;9:513–522. doi: 10.1091/mbc.9.2.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marthin J.K., Nielsen K.G. Choice of nasal nitric oxide technique as first-line test for primary ciliary dyskinesia. Eur. Respir. J. 2011;37:559–565. doi: 10.1183/09031936.00032610. [DOI] [PubMed] [Google Scholar]
- 52.Narang I., Ersu R., Wilson N.M., Bush A. Nitric oxide in chronic airway inflammation in children: diagnostic use and pathophysiological significance. Thorax. 2002;57:586–589. doi: 10.1136/thorax.57.7.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mahut B., Escudier E., de Blic J., Zerah-Lancner F., Coste A., Harf A., Delclaux C. Impairment of nitric oxide output of conducting airways in primary ciliary dyskinesia. Pediatr. Pulmonol. 2006;41:158–163. doi: 10.1002/ppul.20329. [DOI] [PubMed] [Google Scholar]
- 54.Mateos-Corral D., Coombs R., Grasemann H., Ratjen F., Dell S.D. Diagnostic value of nasal nitric oxide measured with non-velum closure techniques for children with primary ciliary dyskinesia. J. Pediatr. 2011;159:420–424. doi: 10.1016/j.jpeds.2011.03.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.