

Abstract
The hypoxic conditions at high altitudes present a challenge for survival, causing pressure for adaptation. Interestingly, many high-altitude denizens (particularly in the Andes) are maladapted, with a condition known as chronic mountain sickness (CMS) or Monge disease. To decode the genetic basis of this disease, we sequenced and compared the whole genomes of 20 Andean subjects (10 with CMS and 10 without). We discovered 11 regions genome-wide with significant differences in haplotype frequencies consistent with selective sweeps. In these regions, two genes (an erythropoiesis regulator, SENP1, and an oncogene, ANP32D) had a higher transcriptional response to hypoxia in individuals with CMS relative to those without. We further found that downregulating the orthologs of these genes in flies dramatically enhanced survival rates under hypoxia, demonstrating that suppression of SENP1 and ANP32D plays an essential role in hypoxia tolerance. Our study provides an unbiased framework to identify and validate the genetic basis of adaptation to high altitudes and identifies potentially targetable mechanisms for CMS treatment.
Introduction
More than 140 million humans have permanently settled in high-altitude regions in various locations around the world, such as the Ethiopian plateau in East Africa, the Tibetan plateau in Asia, and the Andes Mountains in South America. These geographically distinct populations have each adapted to cope with high-altitude hypoxia. For example, a higher hemoglobin concentration and oxygen saturation were detected among Andean highlanders as compared with Tibetans at the same altitude, but there was no difference between Ethiopian highlanders and sea-level residents in these two traits.1 Furthermore, Tibetans have higher plasma concentrations of nitric oxide metabolites than do North Americans,2 and their infants have higher birth weight and higher arterial oxygen saturation than do Han Chinese infants at the same altitude.3,4 A statistical analysis of four quantitative traits (i.e., resting ventilation, hypoxic ventilatory response, oxygen saturation, and hemoglobin concentration) provided further evidence that the phenotypic adaptive responses to high-altitude hypoxia are different between the Tibetan and Andean populations.5 Such differences in the patterns of hypoxia-tolerant phenotypes suggest that distinct genetic mechanisms underlie hypoxia adaptation in different high-altitude human populations. Several studies on these populations have found high heritability in traits correlated with hypoxia tolerance (such as hemoglobin concentration,1 increased lung capacity,6 and higher birth weight7), leading to the belief that the adaptation is (at least partly) genetic in nature. Although the genetic contribution to human adaptation to high altitude has been proposed for a long time, research is still at an early stage and additional evidence is critical for our understanding adaptation at high altitude.8–14 In fact, some of the individuals living at high altitudes are maladapted and present symptoms of chronic mountain sickness (CMS).
CMS, or Monge disease, is characterized by severe polycythemia and an array of neurologic symptoms including headache, fatigue, somnolence, and depression.15 Often, people with CMS suffer from strokes and myocardial infarctions in early adulthood because of increased blood viscosity. Previous studies have shown that CMS is common in Andeans, found occasionally in Tibetans, and absent from the Ethiopian population living on the East African high-altitude plateau.16,17 Therefore, the Andean high-altitude population provides us with the opportunity to dissect the genetic mechanisms underlying high-altitude adaptation by comparing genetic variation between individuals with CMS and adapted subjects without CMS.
To address this, we sequenced the whole genomes of 20 individuals (10 with CMS and 10 without) residing in Cerro de Pasco, Peru. Unlike genotyping arrays or exome sequencing, whole-genome sequencing (WGS) captures the entire spectrum of variation in a region, thus providing nearly complete characterization of the site frequency spectrum (SFS) and allowing maximal information for discovering selective sweeps.
Material and Methods
Subjects and Clinical Characterization
All subjects are adult males residing in the Andean mountain range, in Cerro de Pasco, Peru, at an elevation of more than 4,300 m. CMS, or Monge disease, is diagnosed by CMS scores. Individuals with CMS score >15 were selected as CMS subjects, and those with CMS score <5 were chosen as non-CMS subjects (Table S1 available online). Both blood samples (for DNA samples used in WGS) and skin biopsies (for fibroblast cell cultures used in cell-based assays) were collected. Subjects were volunteers, and each subject gave informed, written consent. The UCSD institutional review board approved the protocol.
DNA Extraction, Library Construction, and Sequencing
Genomic DNA was isolated by blood DNA extraction kit (QIAGEN) and randomly fragmented. Fragments of the desired length were gel purified. Adaptor ligation and DNA cluster preparation were performed with the library preparation kit according to the manufacturer’s instruction (Illumina). WGS was performed on all 20 individuals with the Illumina HiSeq2000 platform to a mean, per-sample depth of 20×–40× (Table S2).
Read Alignment, Score Recalibration, and Variant Calling
We aligned the reads to the human reference genome (UCSC Genome Browser hg19) by BWA18 with default parameter settings. We adjusted the alignments via GATK indel realignment, Picard read duplicate marking, and GATK quality score recalibration modules19,20 under default parameter settings, as defined by the GATK manual v.2. We finally called and filtered SNVs via the GATK UnifiedGenotyper under default parameter settings. As can be seen in Table S2, the sequencing was free of mapping bias in coverage, mapping percentage, or variant counts for all subjects. See Figure S1 for an overview of the computational workflow. The variant data are available upon request.
Tests of Selection
We applied four cross-population tests of neutrality. The first two tests are based on common estimators of the scaled mutation rate θ = 4Nμ: the summed nonfixed frequencies estimator, denoted θf, and the average pairwise heterozygosity estimator, denoted θπ.21,22 For a given region, observing a high log ratio of θπ (θf) in the control relative to the case population indicates selection.23 We label these log ratio statistics as Sπ for the average heterozygosity estimator and Sf for the summed frequency estimator, where
The two remaining cross-population tests we applied are both based on the fixation index (FST) between two populations,24 where differential variant frequencies are aggregated across two populations. For instance, Hudson25 defines FST as
where πw is the within-population average heterozygosity and πb is the between-population average heterozygosity. As two populations diverge, the variability between the populations increases much more than the variability within each the population, and the statistic approaches one. One possible cause of rapid divergence observed in a specific region is positive selection. The fixation index roughly correlates to the evolutionary branch length T between two populations26 as
This approach is not directional, however. As a result, a significant statistic value may indicate a selective sweep in either the case or the control population. To address this, Shriver et al.27 and Yi et al.11 developed the concept of the population branch statistic (PBS). This combines the pairwise branch lengths of three populations as follows:
where C represents a case population, N represents an evolutionarily close control population, and O represents a distant out-group. For the four tests, we used 50 kbp sliding windows overlapping at 2 kbp intervals, with a 0.1% genome-wide FDR to determine the windows of interest.
Human Fibroblast Cell Culture, Hypoxia Treatment, and Real-Time qPCR Assay
To determine the transcriptomic impact of significant variants identified by the tests of selection, we derived primary fibroblast cells from CMS or non-CMS skin biopsies and expended them in DMEM medium supplement with 10% fetal calf serum, 2.5% penicillin/streptomycin, and 1% fungizone antibiotic (Life Technologies). When reaching 75%–80% confluence, the cells were treated with 1.5% O2 for 24 hr. Untreated cultures were used as controls. After treatment, the cells were first washed with PBS (Cellgro) and then treated with TrypLE Express (Life Technologies) for 5–10 min at 37°C to detach the cells. The detached/trypsinized cells were washed with fresh culture media and centrifuged at 200 × g for 3 min. The pelleted cells were frozen at −80°C until RNA extraction.
Total RNA was extracted with the NucleoSpin RNA II Kit (Clontech) and eluted with 40 μl of RNase-free water. RNA concentrations were measured with a NanoDrop 1000 (Thermo Scientific). cDNA was synthesized with 1 μg of total RNA with the SuperScript III First-Strand Synthesis System according to the manufacturer’s instructions (Life Technologies). Real-time qPCR was performed in duplicates in 20 μl reaction volume on a MicroAmp Fast Optical 96-Well Reaction Plate (Life Technologies). Each reaction contained 1 μl of cDNA, 2 μl of 3 μM forward and reverse primer mix, 10 μl of Power SYBR Green PCR Master Mix (Life Technologies), and 7 μl of water. The real-time PCRs were run on a 7900HT Fast Real-Time PCR System (Life Technologies) under the following conditions: 95°C for 10 min followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. GAPDH was used as internal control for normalization.
Fly Lines and Culture
We assessed the impact of observed transcriptomic changes on function under hypoxia in a model system (fruit flies). The candidate genes were obtained from the human study and their fly orthologs were identified with FlyBase. Publicly available RNAi stock lines for each candidate gene (if available, duplicate or triplicate lines per gene) were obtained from Vienna Drosophila Research Stock Center (Table S3). The w1118 was used as background control. To ubiquitously knock down the candidate gene in the F1 progeny, the da-GAL4 driver was obtained from Bloomington Drosophila Stock Center at Indiana University. All stock lines were raised at room temperature and maintained on standard cornmeal.
In Vivo Hypoxia Tolerance Test
The virgin females (n = 9) da-GAL4 were crossed to different UAS-RNAi lines (n = 6) or vice versa. Sufficient time (∼3 days) was given for the flies to mate/cross. These are referred to as “cross.” The vials were kept under ambient conditions for the flies to lay a sufficient number of fertilized eggs. After 48 hr, adults were transferred to a new vial. The original vials were then transferred to a computer-controlled hypoxia chamber, maintained at 5% oxygen on a 12/12 hr light/dark cycle at room temperature. The adults were discarded after 48 hr from the second batch of vials and these vials were then kept at ambient oxygen conditions (∼21% oxygen) to be used as “control.” After 21 days, the ratio of empty pupae (eclosed) to total pupae formed (eclosed + uneclosed) in each vial was calculated to determine the percentage eclosion rate. Simultaneously, the w1118, da-GAL4, and RNAi were “self-crossed” to be used as controls. Each set was performed in triplicate and the entire experiment was repeated to check for consistency. The differences in eclosion rate for the crosses under 21% and 5% O2 were calculated with a chi-square test and between the (UAS-RNAi × da-Gal4) cross and the UAS-RNAi alone (stock control) with an unpaired t test. A p value of <0.05 was considered significant.
Results
We sequenced the genomes with the Illumina HiSeq 2000 platform to a mean depth of 34× per individual (Table S1), mapped the reads to the UCSC Genome Browser (hg19) reference genome by BWA,18 and called SNVs with the GATK pipeline19,20 (Figure S1). Applying PLINK’s IBD test,28 we found that none of the individuals have hidden relatedness.
Lowlander Control Populations
By using cross-population tests of selection, we compared the non-CMS highlanders to the nearest 1000 Genomes (lowlander) controls.29 Applying ADMIXTURE analysis30 to 10,363 variant sites, we observed varying amounts of shared ancestry between our Andean subjects and the three American populations: CLM, MXL, and PUR (Figure S2). The closest population consists of 66 Mexican (MXL) individuals from Los Angeles, California, and was thus chosen as the lowlander control for all cross-population tests of selection. To verify this, we also used principal component analysis (PCA). In Figure S3A, we show the first two principal components of a PCA performed on the MXL samples and our Peruvian individuals. The proximity of the two populations in this space implies that MXL is an appropriate control.
We additionally performed PCA on our 20 non-CMS and CMS subjects and observed some evidence of population substructure (Figures S3B and S3C). There are many possible reasons for this, one of which is that relatively recent (and presumably less adapted) migrants to Cerro de Pasco still retain some population substructure. Therefore, in addition to comparing the non-CMS individuals to lowlander controls, we also searched for evidence of selective sweeps in the non-CMS individuals relative to the CMS individuals. Finally, as outgroup (for the PBS test), we used a distant population consisting of 67 Luhya (LWK) individuals from Webuye, Kenya. Importantly, our highlander subjects and control populations had considerable differences in read coverage (∼34× and ∼4×, respectively), leading to discrepancies in variant calling.
Variant Filters
To adjust for these differences, we filtered our call set by three steps. First, we observed several variants in clustered genomic loci that were discarded by the variant caller in the (CMS and non-CMS) study populations. This happens due to sequencing and mapping artifacts such as strand bias, low sequence complexity, or structural variations. We considered a region as suspicious if ten consecutive SNPs were filtered out by GATK in our study populations and filtered out any SNPs present in these regions in the controls. Second, following the protocol used by the 1000 Genomes project, we removed any site with a mean coverage higher than twice the median genome-wide coverage as likely to be caused by duplication.29 This removes variants found in repetitive regions, such as centromeric sequence. We also removed any site with less than 2× coverage per person in the study population as being too poorly covered to accurately call. Finally, we removed sites with an excess of heterozygotes, by using a test from Emigh31 describing the heterozygote probability as
We discarded variants with p value less than 0.05. After applying the above filters, a total of 5,937,347 variants in the subjects with CMS and 5,777,092 variants in the subjects without CMS remained.
Identifying Regions under Positive Selection
Under positive directional selection, any haplotype harboring a beneficial mutation, as well as linked “hitchhiking” mutations, rapidly increases in frequency. This leads to a characteristic loss of genetic diversity centered on the beneficial mutation known as a “selective sweep.”21 Importantly, the loss of genetic diversity and the consequent skew in the site frequency spectrum (SFS) can be used to detect loci important for adaptation to selective stress.21,22 We use cross-population tests to adjust for events shared between case and control populations (such as population bottlenecks, genetic drift, or even directional selection acting on unrelated phenotypes). These are probably due to events that took place before the divergence of our case and control populations, and are thus less likely to be related to hypoxia tolerance. Population-specific selection can be measured as a large decrease in diversity in the case population compared to controls.21
This is usually captured as a skew in the site frequency spectrum (SFS) of a region under selection. However, the SFS (and thus the performance of different tests of selection) is significantly affected by many parameters, including the selective pressure(s) affecting the beneficial allele, as well as the length of time (t) for which the allele has been under selection.23 For a complex phenotype such as hypoxia tolerance, we expect that multiple loci throughout the genome may simultaneously undergo selective sweeps, each under a distinct selection pressure and for a distinct time period. For this reason, we apply several tests of selection: Sf, Sπ, FST, and PBS. Because the tests are powerful under different regimes (weak/strong and early/late) of selection,23 and because we have no prior knowledge of the regime we are after, we consider regions found significant in any of the above tests as potentially interesting.
We also assume that the genetic basis for adaptation to hypoxia influences relatively few loci genome-wide. As a result, for a cross-population test, the null distribution of two neutrally evolving populations can be approximated by the observed case versus control distribution. We set significance thresholds corresponding to the top 0.1% genome-wide value for each statistic. For the non-CMS versus MXL tests, these values were 0.11 (PBS), 0.19 (FST), 2.93 (Sπ), and 3.87 (Sf). For the non-CMS versus CMS tests, these were 0.17 (PBS), 0.31 (FST), 2.18 (Sπ), and 3.23 (Sf). This set of analyses identified 314 regions spanning 29.67 Mbp that were significant in at least one test under a 0.1% genome-wide false discovery rate (see Figures S4–S26 and Table S5).
Region Prioritization Criteria
Because we wanted to validate potential gene candidates experimentally, we developed a series of automated prioritization criteria in order to shortlist candidates that showed the strongest signals of selection.
Frequency Block Differential Relative to Control Population
A region under strong selection should have multiple variants present with a high frequency differential between case and control populations. To identify this, we iterate over all possible case frequency values f, where f = (1/n, 2/n, …, (n − 1)/n) (for a case population sample of size n haplotypes). For a given value of f, we isolate all variants in the region with frequency within 1/n from f. From these, we define an f frequency block as a subset of ≥10 consecutive SNPs. For each f frequency block, we calculate the frequency differential, defined by the absolute difference in mean frequency between the non-CMS population and the associated control population (either CMS or the closer of the MXL/CEU populations). We prioritize regions where the maximum block differential in the region is greater than 20%. We set the threshold at 20% because this is the expected sampling variance of a variant at a given frequency when sampling 20 haplotypes (corresponding to ten CMS or ten non-CMS subjects) from a population. A total of 170 regions were considered prioritized under this criterion.
Frequency Block Differential Relative to HapMap Control Populations
In order to ensure that the prioritized regions represent selection for high-altitude adaptation (rather than other phenotypes, potentially shared with different populations), we expanded our controls to include additional populations. These controls included the CMS individuals we sequenced, the MXL/CEU 1000 Genomes populations, and other lowlander HapMap populations.32 We prioritize regions where the observed haplotype block has frequency differential of greater than 20% (our sampling error, as mentioned above) compared to all other sampled controls. A total of 35 regions were considered prioritized under this (as well as the previous) criterion.
Frequency Block Differential after Integrating Existing Genotype Data
We also used variant calls from a previous genotyping study by Bigham et al.12 to further prioritize candidate regions. The data from this study provided us with two advantages. First, the authors performed genotyping on 49 Andean highlanders with no symptoms of CMS (including 24 from the same population we sequenced, in Cerro de Pasco, Peru). This helped us refine our sample frequencies and identify any false signals caused by sampling. Second, they genotyped 39 lowlanders of Native American ancestry from Southern Mexico, providing us with an additional lowlander control population. This population is both geographically closer to our Andean highlander subjects and does not show any signs of admixture12 with Europeans.
For a given region, we extracted all variants sampled from the previously identified f frequency block that were also sampled by Bigham et al.12 For these, we refined our non-CMS frequencies by taking an average (weighted by sample size) over the highlander frequencies from both studies. Because of the increased population size (total of 59 subjects), the expected error due to sampling was reduced to less than 10%. As a result, for a given region, if the revised block frequency of the adapted population was greater than 10% compared to all controls, we considered the region prioritized. We note that previously prioritized regions that had no variants sampled by Bigham et al.12 were unaffected by this criterion. A total of 20 regions remained under consideration after this step.
RefSeq Genes Overlapping the Region
Finally, we prioritized candidate regions that had at least one gene (as defined by RefSeq release 45, downloaded January 14, 2011) within their boundaries. Although regions that do not overlap known genes may contain important regulatory variants, for an initial pass, we focused our efforts on regions for which there are more readily accessible methods to identify and validate linked genes. However, we did attempt to identify important regulatory variation in the significant nongenic regions by determining variants within transcription factor binding sites, as defined by TRANSFAC or ENCODE (see Table S5).
The 11 final regions all had haplotypes that were much higher in frequency in the non-CMS population than in many controls (including our CMS population as well as several sequenced and genotyped lowlander populations) and are presented in Table 1. These include many plausible candidates, including genes involved in oxidative stress response (DGKK [MIM 300837], DUOX1 [MIM 606758], DUOX2 [MIM 606759], DUOXA1 [MIM 612771], and DUOXA2 [MIM 612772]), response to reactive oxygen species (GABRA3 [MIM 305660]), cell metabolism, and signaling (PFKM [MIM 610681], SENP1 [MIM 612157], and ANP32D [MIM 606878]).
Table 1.
List of the Top 11 Genomic Prioritized Regions
| Genomic Region | Gene Symbol | Tests |
|---|---|---|
| chr3: 33,254,596–33,314,596 | SUSD5 | Sπ,CMS |
| chr6: 58,244,452–58,392,452 | GUSBP4 | Sπ,MXL |
| chr6: 157,504,452–157,554,452 | ARID1B | FST,MXL |
| chr10: 101,014,523–101,092,523 | CNNM1 | PBSMXL |
| chr11: 118,147,948–118,199,948 | CD3E | Sπ,CMS |
| chr12: 48,411,360–48,555,360a | SENP1, PFKM, ASB8 | Sf,MXL, Sf,CMS, Sπ,MXL, Sπ,CMS |
| chr12: 48,751,360–48,907,360a | ANP32D, C12orf54 | Sπ,MXL, Sπ,CMS |
| chr15: 45,338,058–45,436,058 | SORD, DUOX2, DUOXA2, DUOXA1, DUOX1 | FST,MXL |
| chr19: 19,665,844–19,747,844 | PBX4, LPAR2, GMIP | PBSMXL |
| chrX: 50,147,676–50,197,676 | DGKK | Sπ,CMS |
| chrX: 151,275,676–151,421,676 | MAGEA5, MAGEA10, GABRA3 | PBSCMS, FST,CMS |
Only two of these regions (both on chromosome 12) are significant in both non-CMS versus CMS and non-CMS versus MXL tests.
Two genomic regions (both on chromosome 12) appeared in the top 0.1% in both non-CMS versus MXL and non-CMS versus CMS tests. The first is a 144 kbp region at chr12: 48,411,360–48,555,360 that contains a block of 66 “differential” SNPs with mean frequencies of 99% in non-CMS, 66% in CMS, 58% in MXL, and 14% in CEU. The second region spans 156 kbp at chr12: 48,751,360–48,907,360 and contains a block of 114 “differential” SNPs with mean frequencies of 99% in non-CMS, 53% in CMS, 47% in MXL, and 5% in CEU (Figure 1). Three genes (SENP1, PFKM, and ASB8 [MIM 615053]) are located in the first region, and two genes (ANP32D and C12orf54) are located in the second region. Strikingly, some of these genes have been shown to regulate CMS-related phenotypes in mammals. Specifically, mice carrying a deficient Pfkm allele exhibit severe cardiac and hematological disorders, muscle hypoxia and hypervascularization, impaired oxidative metabolism, fiber necrosis, and exercise intolerance.33 In addition, previous studies have also found that Senp1−/− led to erythropoiesis defect in mice.34,35 SENP1 regulates the activities of several cell signaling pathways through desumoylation of key mediators. For example, SENP1 enhances ASK1-JNK activation and cell apoptosis through desumoylation of HIPK1 in a ROS-dependent manner.36 Furthermore, SENP1-dependent desumoylation also regulates the stability and activity of HIF1α and GATA1 transcription factors34,35 that play important roles in regulating physiological responses to hypoxia including erythrogenesis, angiogenesis, and metabolic adaptation.37,38
Figure 1.
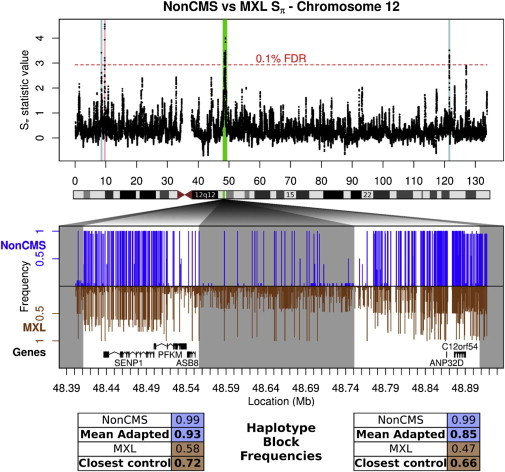
Profile of the Only Two Candidate Regions that Are Significant in Non-CMS versus CMS and Non-CMS versus MXL Tests
One of the statistics in which both of these regions are significant (Sπ,MXL) is plotted across chromosome 12. Five distinct regions exceed the 0.1% FDR threshold–the two highlighted in light blue do not have a major frequency differential between the non-CMS and MXL populations, whereas the one highlighted in pink is similar in other controls. The remaining two are considered prioritized and highlighted in green. The SNP frequencies in the area encompassing these two, part of q13.11, are plotted in the middle. In this plot, the two prioritized regions are highlighted in white, and other regions are shaded in gray. As can be seen, in both regions, there is an almost complete fixation of a haplotype in the non-CMS population that is at a much lower frequency in all lowlander and maladapted controls.
To further validate the significance of the observed signs of selection between the non-CMS and CMS subjects in these regions (see Table 1), we computed an empirical p value by using 10,000 permutations of the class labels for (1) the observed Sf value in the first region (p = 0.0006) and (2) the observed Sπ and Sf values in the second region (p = 0.0036 and 0.0005, respectively).
Frequency Validations by Sanger Sequencing
It is a well-known fact that CMS is correlated with age.39 The subjects we sequenced via WGS were chosen to have large differences in signs and symptoms. Thus, it would be unlikely for our non-CMS individuals to develop strong CMS symptoms. However, as Table S1 shows, the individuals with CMS were on average 10 years older than the individuals without CMS. To validate the frequencies of significant SNPs identified by WGS (ensuring that the signal we saw was not confounded by this age differential), we tested 20 more subjects in the two chromosome 12 regions mentioned above. Ten of these had early-onset CMS symptoms (mean CMS score = 16.4, mean age = 28), and the other ten were older residents who did not have CMS (mean CMS score = 6.9, mean age = 44). In this sampling, the non-CMS individuals are significantly older than the CMS individuals. We determined the frequencies of rs7963934 from the SENP1 region and rs72644851 from the ANP32D region by Sanger sequencing. These SNPs were in the primary differential haplotype blocks identified by WGS. For rs7963934, WGS revealed a non-CMS frequency of 100% and a CMS frequency of 70%, whereas Sanger sequencing revealed a non-CMS frequency of 90% and a CMS frequency of 60%. For rs72644851, WGS revealed a non-CMS frequency of 100% and a CMS frequency of 50%, whereas Sanger sequencing revealed a non-CMS frequency of 90% and a CMS frequency of 50%. In both cases, the frequency differential discovered by WGS was confirmed by Sanger sequencing in a larger number of samples, ruling out the effect of age.
In Vitro and In Vivo Validation of Candidate Genes
We extended our investigation to study the functional impact of the SNP variants and candidate genes identified by our current analysis. We did this with human fibroblast cells40 derived from four of the CMS and non-CMS subjects as well as in vivo with a Drosophila model. Drosophila melanogaster provides a powerful in vivo model to dissect genetic mechanisms that contribute to human disease, including aging,41,42 neurologic and cardiac disease,43–45 cancer,46,47 and mechanisms underlying hypoxia tolerance or susceptibility.48 One of the reasons fruit flies have been used so extensively is that many genes that contain human disease-causing mutations are evolutionarily conserved in Drosophila melanogaster.49–51 Of the top five candidate genes identified from our Andean samples, two genes have orthologs identified in the Drosophila genome (FlyBase gene symbol CG32110 for human gene SENP1 and Mapmodulin for human gene ANP32D).52,53
We measured the transcriptional response of the candidate genes to hypoxia challenge by real-time qPCR. Interestingly, the expression levels of SENP1 and ANP32D were significantly higher in the CMS cells as compared to the non-CMS cells after hypoxia treatment. In contrast, PFKM was downregulated in both non-CMS and CMS cells (Figure 2). These results suggested that as compared to room-air cultured cells, the suppression of SENP1, ANP32D, and/or PFKM in non-CMS cells is beneficial for high-altitude adaptation. As a corollary, when comparing the transcriptional response of CMS cells to that of non-CMS cells, the upregulation of SENP1 and ANP32D in CMS cells might reflect mechanisms that underlie maladaptation to high altitudes in the CMS individuals. We then proceeded to test this hypothesis in vivo in Drosophila melanogaster.
Figure 2.
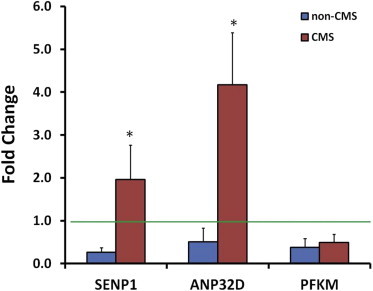
Hypoxia Response of Top Candidate Genes in Non-CMS and CMS Cells
Fibroblast cells were derived from skin biopsies obtained from the subjects with and without CMS. Two non-CMS and two CMS cell lines were treated with 1.5% O2 for 24 hr. The expression levels of SENP1, ANP32D, and PFKM were measured by quantitative real-time PCR. Compared to the normoxia controls (represented by the green line), hypoxia treatment induced a significant downregulation of SENP1, ANP32D, and PFKM in non-CMS cells (blue bars). In contrast, hypoxia treatment upregulated the expression of SENP1 and ANP32D in the CMS cells (red bars), an opposite effect to the changes observed in the non-CMS cells (∗p < 0.05, nonparametric Wilcoxon rank test). Each bar represents mean ± SEM of two measurements in duplicate.
We took advantage of a Drosophila GAL4/UAS-RNAi system54–56 to knock down the transcript levels of these orthologs individually, mimicking the transcriptional suppression of these candidate genes in the non-CMS samples in an attempt to determine their potential role in adaptation to high-altitude hypoxia. In addition, this strategy is also relevant to test the opposite hypothesis that the upregulation of SENP1 and ANP32D after hypoxia in the CMS samples is deleterious for survival in a hypoxic environment. The UAS-RNAi × GAL4 crosses were first cultured in normoxia to determine the effect of RNAi-mediated knockdown of each candidate gene on development. All the crosses developed normally with eclosion rates of more than 95%, demonstrating that downregulation of the candidate genes had no significant effect on development in normoxia. The flies resulting from these crosses were then tested under a hypoxic condition (5% O2) by scoring the eclosion rate, an index of completion of development and survival. This hypoxic condition has been previously proven to be critical for distinguishing hypoxia-tolerant flies from others.57,58 In order to minimize false positive results induced by off-target effects of RNAi or insertion effects of a particular UAS-RNAi transgene, we included only Drosophila orthologs with multiple available UAS-RNAi transgenic lines in this analysis. As shown in Figure 3, a dramatic enhancement of hypoxia tolerance was observed when both CG32110 and Mapmodulin were knocked down, demonstrating that downregulation of the orthologs of SENP1 and ANP32D is indeed beneficial for survival in severe hypoxic conditions in vivo.
Figure 3.
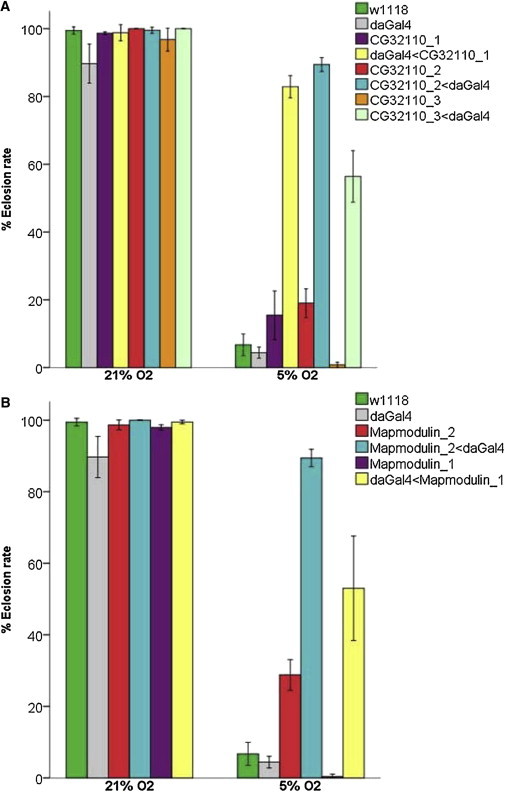
Downregulation of Human SENP1 and ANP32D Orthologs in Drosophila Enhances Survival under Hypoxic Condition
da-Gal4 driver was used for ubiquitously knocking down the candidate gene by crossing with respective UAS-RNAi lines. Respective eclosion rates were observed at 21% and 5% O2 (Table S2).
(A) CG32110-RNAi when crossed with da-Gal4 significantly increases the eclosion rate at 5% O2. The results were consistent in three RNAi lines targeting the same human SENP1 ortholog gene CG32110 (∗p < 0.005, unpaired t test).
(B) The differences were also significant for eclosion rates (%) of the F1 progeny for the two Mapmodulin-RNAi fly lines (∗p < 0.005, unpaired t test).
Each bar represents mean ± SEM of eclosion rate. The w1118 and da-Gal4 stocks were tested and used as background controls. The genotypes of each RNAi line are provided in Table S4.
Discussion
We present here a genome-wide study of genetic adaptation in humans that confirms the effect of relevant genotypes on expression and further validates their role in model organisms. With the enhanced power of WGS, we identified a number of putative regions showing strong signals of selective sweep. We find that two genes in these regions, ANP32D and SENP1, show significantly increased expression in individuals with CMS compared to those without. Consequently, we hypothesized that downregulating these genes could be beneficial in coping with hypoxia. We found that flies with these genes downregulated had a remarkably enhanced survival rate under hypoxia.
There are several implications of our study. First, many of the haplotypes found in our regions under selection are also present in lowlander controls. This implies that the beneficial mutations leading to adaptation in Andeans may be very old. This has been suggested as likely in previous studies,59 because the time under selective stress (approximately 600 generations) is relatively short for adaptation to be driven by de novo mutations. As a result, we designed our prioritization criteria to look for large frequency differentials. This type of approach has been shown to be robust to selection on standing variation.60
We also performed high-coverage WGS of 20 individuals. Traditionally, genome-wide scans for selection generally involve sampling the genome through genotyping or whole-exome sequencing. However, there is an important trade-off when using these experimental designs. Specifically, WGS provides for a complete sampling of variant sites, albeit (usually) on a much smaller number of individuals. This completeness is critical for detecting selection. For instance, consider the prioritized region found on chromosome 19 (Figure 4). With WGS, this region corresponds to the highest peak in the chromosome, with a block of 84 variants at roughly 40% frequency in individuals without CMS but only 6% in MXL. However, the Nimblegen 2.1 M exon capture array targets only two variants in this block. As for genotyping, the ∼1 M Affymetrix Genome-Wide Human SNP Array 6.0 samples only one variant site in the block. In both cases, the resulting signal is much weaker and far below the 0.1% genome-wide FDR. Importantly, this argument holds for 10 of the 11 prioritized regions identified in our study, where we find strong peaks for our tests at a low genomic FDR (Figures S27–S37) by WGS, but not when restricting to sites sampled by the alternative approaches. In fact, as Figures S27–S37 show, the genotyping array used in Bigham et al.12’s work does not capture a single variant in 5 of the 11 regions’ differential haplotype blocks.
Figure 4.
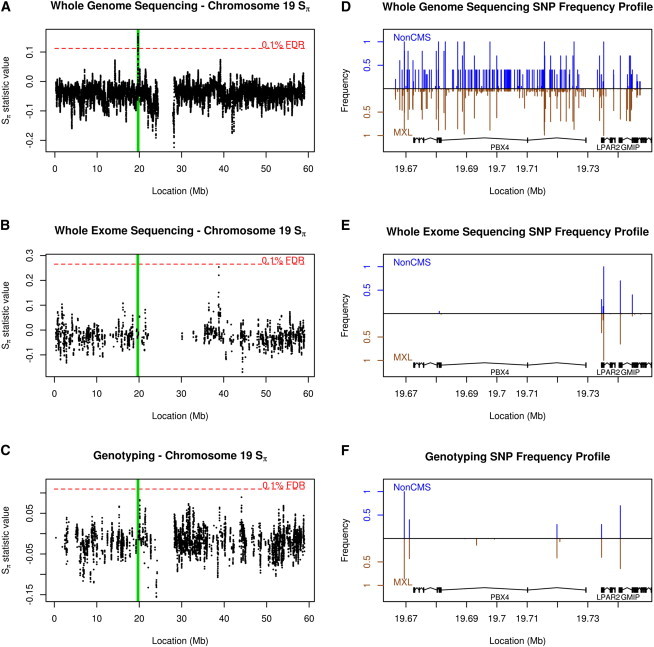
The Impact of Sequence Assay Type on Signals of Selection
(A–C) Sπ values across chromosome 19 in subjects without CMS, compared to the MXL population, when applied to (A) variants present in our WGS study, (B) variants included in targets from whole-exome sequencing, and (C) the subset of ∼1 M variants from genotyping. The red line represents a genome-wide, 0.1% FDR. Highlighted in green is the genomic region of PBX4, LPAR2, and GMIP, one of the 11 strongest candidate regions in our study.
(D–F) SNP frequency profiles of the region highlighted in green for non-CMS (blue) compared to MXL (brown, inverted) for (D) WGS, (E) whole-exome sequencing, and (F) genotyping. As can be seen, the strong signal present via WGS is reduced drastically in genotyping and entirely absent by exome sequencing.
Finally, our study reveals many mechanistic insights on human adaptation (and maladaptation) to hypoxia. Because individuals with CMS are polycythemic (with hematocrit > 65%), their blood becomes much more viscous. In turn, this increased viscosity jeopardizes blood flow to major organs, sometimes to the degree of ischemia, leading in some individuals to myocardial infarction and stroke.61–63 SENP1 is known to regulate erythropoiesis,35 and indeed Senp1−/− mice die of anemia in early life.34 This gives credence to the idea that the increased expression of SENP1 plays a role in the basic pathogenesis of polycythemia in CMS individuals. In contrast, in spite of the fact that there is little known about ANP32D and the PP32 phosphatase gene family, ANP32D functions as an oncogene. We raise here the possibility that this particular gene alters cellular metabolism in a fashion that is similar to that of cancer cells, especially given that such cells can flourish in low-oxygen conditions. In conclusion, a better understanding of the mechanisms underlying hypoxia tolerance in high-altitude human populations will, in all likelihood, elucidate the pathogenesis of other conditions at sea level, including congenital heart disease, obstructive sleep apnea, and cancer.
Acknowledgments
We thank Mary Hsiao and Orit Poulsen for technical assistance and Abigail W. Bigham for providing Latin American highlander and lowlander genotype data. This study is supported by NSF-CCF-1115206, NSF-III-1318386, 5RO1-HG004962, and U54 HL108460 to V.B. and National Institutes of Health awards 1P01HL098053 and 5P01HD32573 to G.G.H.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
FlyBase, http://flybase.org/
International HapMap Project, http://hapmap.ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Beall C.M. Andean, Tibetan, and Ethiopian patterns of adaptation to high-altitude hypoxia. Integr. Comp. Biol. 2006;46:18–24. doi: 10.1093/icb/icj004. [DOI] [PubMed] [Google Scholar]
- 2.Erzurum S.C., Ghosh S., Janocha A.J., Xu W., Bauer S., Bryan N.S., Tejero J., Hemann C., Hille R., Stuehr D.J. Higher blood flow and circulating NO products offset high-altitude hypoxia among Tibetans. Proc. Natl. Acad. Sci. USA. 2007;104:17593–17598. doi: 10.1073/pnas.0707462104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moore L.G. Human genetic adaptation to high altitude. High Alt. Med. Biol. 2001;2:257–279. doi: 10.1089/152702901750265341. [DOI] [PubMed] [Google Scholar]
- 4.Niermeyer S., Yang P., Shanmina, Drolkar, Zhuang J., Moore L.G. Arterial oxygen saturation in Tibetan and Han infants born in Lhasa, Tibet. N. Engl. J. Med. 1995;333:1248–1252. doi: 10.1056/NEJM199511093331903. [DOI] [PubMed] [Google Scholar]
- 5.Beall C.M. Tibetan and Andean patterns of adaptation to high-altitude hypoxia. Hum. Biol. 2000;72:201–228. [PubMed] [Google Scholar]
- 6.Frisancho A.R., Frisancho H.G., Albalak R., Villain M., Vargas E., Soria R. Developmental, genetic, and environmental components of lung volumes at high altitude. Am. J. Hum. Biol. 1997;9:191–203. doi: 10.1002/(SICI)1520-6300(1997)9:2<191::AID-AJHB5>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 7.Moore L.G., Shriver M., Bemis L., Vargas E. An evolutionary model for identifying genetic adaptation to high altitude. In: Roach R.C., Wagner P.D., Hackett P.H., editors. Hypoxia and Exercise. Springer; New York: 2007. pp. 101–118. [Google Scholar]
- 8.Lorenzo V.F., Yang Y., Simonson T.S., Nussenzveig R., Jorde L.B., Prchal J.T., Ge R.L. Genetic adaptation to extreme hypoxia: study of high-altitude pulmonary edema in a three-generation Han Chinese family. Blood Cells Mol. Dis. 2009;43:221–225. doi: 10.1016/j.bcmd.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bigham A.W., Mao X., Mei R., Brutsaert T., Wilson M.J., Julian C.G., Parra E.J., Akey J.M., Moore L.G., Shriver M.D. Identifying positive selection candidate loci for high-altitude adaptation in Andean populations. Hum. Genomics. 2009;4:79–90. doi: 10.1186/1479-7364-4-2-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simonson T.S., Yang Y., Huff C.D., Yun H., Qin G., Witherspoon D.J., Bai Z., Lorenzo F.R., Xing J., Jorde L.B. Genetic evidence for high-altitude adaptation in Tibet. Science. 2010;329:72–75. doi: 10.1126/science.1189406. [DOI] [PubMed] [Google Scholar]
- 11.Yi X., Liang Y., Huerta-Sanchez E., Jin X., Cuo Z.X., Pool J.E., Xu X., Jiang H., Vinckenbosch N., Korneliussen T.S. Sequencing of 50 human exomes reveals adaptation to high altitude. Science. 2010;329:75–78. doi: 10.1126/science.1190371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bigham A.W., Bauchet M., Pinto D., Mao X., Akey J.M., Mei R., Scherer S.W., Julian C.G., Wilson M.J., López Herráez D. Identifying signatures of natural selection in Tibetan and Andean populations using dense genome scan data. PLoS Genet. 2010;6:e1001116. doi: 10.1371/journal.pgen.1001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu S., Li S., Yang Y., Tan J., Lou H., Jin W., Yang L., Pan X., Wang J., Shen Y. A genome-wide search for signals of high-altitude adaptation in Tibetans. Mol. Biol. Evol. 2011;28:1003–1011. doi: 10.1093/molbev/msq277. [DOI] [PubMed] [Google Scholar]
- 14.Ji L.D., Qiu Y.Q., Xu J., Irwin D.M., Tam S.C., Tang N.L., Zhang Y.P. Genetic adaptation of the hypoxia-inducible factor pathway to oxygen pressure among eurasian human populations. Mol. Biol. Evol. 2012;29:3359–3370. doi: 10.1093/molbev/mss144. [DOI] [PubMed] [Google Scholar]
- 15.León-Velarde F., McCullough R.G., McCullough R.E., Reeves J.T., CMS Consensus Working Group Proposal for scoring severity in chronic mountain sickness (CMS). Background and conclusions of the CMS Working Group. Adv. Exp. Med. Biol. 2003;543:339–354. doi: 10.1007/978-1-4419-8997-0_24. [DOI] [PubMed] [Google Scholar]
- 16.Xing G., Qualls C., Huicho L., Rivera-Ch M., Stobdan T., Slessarev M., Prisman E., Ito S., Wu H., Norboo A. Adaptation and mal-adaptation to ambient hypoxia; Andean, Ethiopian and Himalayan patterns. PLoS ONE. 2008;3:e2342. doi: 10.1371/journal.pone.0002342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beall C.M., Decker M.J., Brittenham G.M., Kushner I., Gebremedhin A., Strohl K.P. An Ethiopian pattern of human adaptation to high-altitude hypoxia. Proc. Natl. Acad. Sci. USA. 2002;99:17215–17218. doi: 10.1073/pnas.252649199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fu Y.-X. Statistical properties of segregating sites. Theor. Popul. Biol. 1995;48:172–197. doi: 10.1006/tpbi.1995.1025. [DOI] [PubMed] [Google Scholar]
- 22.Achaz G. Frequency spectrum neutrality tests: one for all and all for one. Genetics. 2009;183:249–258. doi: 10.1534/genetics.109.104042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Udpa N., Zhou D., Haddad G.G., Bafna V. Tests of selection in pooled case-control data: an empirical study. Front Genet. 2011;2:83. doi: 10.3389/fgene.2011.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holsinger K.E., Weir B.S. Genetics in geographically structured populations: defining, estimating and interpreting F(ST) Nat. Rev. Genet. 2009;10:639–650. doi: 10.1038/nrg2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hudson R.R., Slatkin M., Maddison W.P. Estimation of levels of gene flow from DNA sequence data. Genetics. 1992;132:583–589. doi: 10.1093/genetics/132.2.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cavalli-Sforza, L. (1969). Human diversity. Proceedings of the 12th International Congress in Genetics 2, 405–416.
- 27.Shriver M.D., Kennedy G.C., Parra E.J., Lawson H.A., Sonpar V., Huang J., Akey J.M., Jones K.W. The genomic distribution of population substructure in four populations using 8,525 autosomal SNPs. Hum. Genomics. 2004;1:274–286. doi: 10.1186/1479-7364-1-4-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A.R., Bender D., Maller J., Sklar P., de Bakker P.I.W., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.The 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alexander D.H., Novembre J., Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–1664. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Emigh T.H. A comparison of tests for Hardy-Weinberg equilibrium. Biometrics. 1980;36:627–642. [PubMed] [Google Scholar]
- 32.Frazer K.A., Ballinger D.G., Cox D.R., Hinds D.A., Stuve L.L., Gibbs R.A., Belmont J.W., Boudreau A., Hardenbol P., Leal S.M., International HapMap Consortium A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.García M., Pujol A., Ruzo A., Riu E., Ruberte J., Arbós A., Serafín A., Albella B., Felíu J.E., Bosch F. Phosphofructo-1-kinase deficiency leads to a severe cardiac and hematological disorder in addition to skeletal muscle glycogenosis. PLoS Genet. 2009;5:e1000615. doi: 10.1371/journal.pgen.1000615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu L., Ji W., Zhang H., Renda M.J., He Y., Lin S., Cheng E.C., Chen H., Krause D.S., Min W. SENP1-mediated GATA1 deSUMOylation is critical for definitive erythropoiesis. J. Exp. Med. 2010;207:1183–1195. doi: 10.1084/jem.20092215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheng J., Kang X., Zhang S., Yeh E.T. SUMO-specific protease 1 is essential for stabilization of HIF1alpha during hypoxia. Cell. 2007;131:584–595. doi: 10.1016/j.cell.2007.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li X., Luo Y., Yu L., Lin Y., Luo D., Zhang H., He Y., Kim Y.O., Kim Y., Tang S., Min W. SENP1 mediates TNF-induced desumoylation and cytoplasmic translocation of HIPK1 to enhance ASK1-dependent apoptosis. Cell Death Differ. 2008;15:739–750. doi: 10.1038/sj.cdd.4402303. [DOI] [PubMed] [Google Scholar]
- 37.De Maria R., Zeuner A., Eramo A., Domenichelli C., Bonci D., Grignani F., Srinivasula S.M., Alnemri E.S., Testa U., Peschle C. Negative regulation of erythropoiesis by caspase-mediated cleavage of GATA-1. Nature. 1999;401:489–493. doi: 10.1038/46809. [DOI] [PubMed] [Google Scholar]
- 38.Prabhakar N.R., Semenza G.L. Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia-inducible factors 1 and 2. Physiol. Rev. 2012;92:967–1003. doi: 10.1152/physrev.00030.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Monge-C C., Arregui A., León-Velarde F. Pathophysiology and epidemiology of chronic mountain sickness. Int. J. Sports Med. 1992;13(Suppl 1):S79–S81. doi: 10.1055/s-2007-1024603. [DOI] [PubMed] [Google Scholar]
- 40.Boutin A.T., Weidemann A., Fu Z., Mesropian L., Gradin K., Jamora C., Wiesener M., Eckardt K.U., Koch C.J., Ellies L.G. Epidermal sensing of oxygen is essential for systemic hypoxic response. Cell. 2008;133:223–234. doi: 10.1016/j.cell.2008.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Michno K., van de Hoef D., Wu H., Boulianne G.L. Modeling age-related diseases in Drosophila: can this fly? Curr. Top. Dev. Biol. 2005;71:199–223. doi: 10.1016/S0070-2153(05)71006-1. [DOI] [PubMed] [Google Scholar]
- 42.Grotewiel M.S., Martin I., Bhandari P., Cook-Wiens E. Functional senescence in Drosophila melanogaster. Ageing Res. Rev. 2005;4:372–397. doi: 10.1016/j.arr.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 43.Lu B. Recent advances in using Drosophila to model neurodegenerative diseases. Apoptosis. 2009;14:1008–1020. doi: 10.1007/s10495-009-0347-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lessing D., Bonini N.M. Maintaining the brain: insight into human neurodegeneration from Drosophila melanogaster mutants. Nat. Rev. Genet. 2009;10:359–370. doi: 10.1038/nrg2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Diop S.B., Bodmer R. Drosophila as a model to study the genetic mechanisms of obesity-associated heart dysfunction. J. Cell. Mol. Med. 2012;16:966–971. doi: 10.1111/j.1582-4934.2012.01522.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Polesello C., Roch F., Gobert V., Haenlin M., Waltzer L. Modeling cancers in Drosophila. Prog. Mol. Biol. Transl. Sci. 2011;100:51–82. doi: 10.1016/B978-0-12-384878-9.00002-9. [DOI] [PubMed] [Google Scholar]
- 47.Vidal M., Cagan R.L. Drosophila models for cancer research. Curr. Opin. Genet. Dev. 2006;16:10–16. doi: 10.1016/j.gde.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 48.Zhou D., Visk D.W., Haddad G.G. Drosophila, a golden bug, for the dissection of the genetic basis of tolerance and susceptibility to hypoxia. Pediatr. Res. 2009;66:239–247. doi: 10.1203/PDR.0b013e3181b27275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bier E. Drosophila, the golden bug, emerges as a tool for human genetics. Nat. Rev. Genet. 2005;6:9–23. doi: 10.1038/nrg1503. [DOI] [PubMed] [Google Scholar]
- 50.Pandey U.B., Nichols C.D. Human disease models in Drosophila melanogaster and the role of the fly in therapeutic drug discovery. Pharmacol. Rev. 2011;63:411–436. doi: 10.1124/pr.110.003293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gilbert L.I. Drosophila is an inclusive model for human diseases, growth and development. Mol. Cell. Endocrinol. 2008;293:25–31. doi: 10.1016/j.mce.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 52.McQuilton P., St Pierre S.E., Thurmond J., FlyBase Consortium FlyBase 101—the basics of navigating FlyBase. Nucleic Acids Res. 2012;40(Database issue):D706–D714. doi: 10.1093/nar/gkr1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hu Y., Flockhart I., Vinayagam A., Bergwitz C., Berger B., Perrimon N., Mohr S.E. An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinformatics. 2011;12:357. doi: 10.1186/1471-2105-12-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fischer J.A., Giniger E., Maniatis T., Ptashne M. GAL4 activates transcription in Drosophila. Nature. 1988;332:853–856. doi: 10.1038/332853a0. [DOI] [PubMed] [Google Scholar]
- 55.Brand A.H., Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 56.Dietzl G., Chen D., Schnorrer F., Su K.C., Barinova Y., Fellner M., Gasser B., Kinsey K., Oppel S., Scheiblauer S. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature. 2007;448:151–156. doi: 10.1038/nature05954. [DOI] [PubMed] [Google Scholar]
- 57.Zhou D., Xue J., Chen J., Morcillo P., Lambert J.D., White K.P., Haddad G.G. Experimental selection for Drosophila survival in extremely low O(2) environment. PLoS ONE. 2007;2:e490. doi: 10.1371/journal.pone.0000490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Azad P., Zhou D., Zarndt R., Haddad G.G. Identification of genes underlying hypoxia tolerance in Drosophila by a P-element screen. G3 (Bethesda) 2012;2:1169–1178. doi: 10.1534/g3.112.003681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rupert J.L., Hochachka P.W. Genetic approaches to understanding human adaptation to altitude in the Andes. J. Exp. Biol. 2001;204:3151–3160. doi: 10.1242/jeb.204.18.3151. [DOI] [PubMed] [Google Scholar]
- 60.Chen H., Patterson N., Reich D. Population differentiation as a test for selective sweeps. Genome Res. 2010;20:393–402. doi: 10.1101/gr.100545.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Monge C.C., Whittembury J. Chronic mountain sickness. Johns Hopkins Med. J. 1976;139(Suppl):87–89. [PubMed] [Google Scholar]
- 62.Ergueta J., Spielvogel H., Cudkowicz L. Cardio-respiratory studies in chronic mountain sickness (Monge’s syndrome) Respiration. 1971;28:485–517. doi: 10.1159/000192835. [DOI] [PubMed] [Google Scholar]
- 63.West J.B. High-altitude medicine. Am. J. Respir. Crit. Care Med. 2012;186:1229–1237. doi: 10.1164/rccm.201207-1323CI. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.