

Abstract
Vitelliform macular dystrophies (VMD) are inherited retinal dystrophies characterized by yellow, round deposits visible upon fundus examination and encountered in individuals with juvenile Best macular dystrophy (BMD) or adult-onset vitelliform macular dystrophy (AVMD). Although many BMD and some AVMD cases harbor mutations in BEST1 or PRPH2, the underlying genetic cause remains unknown for many affected individuals. In a large family with autosomal-dominant VMD, gene mapping and whole-exome sequencing led to the identification of a c.713T>G (p.Leu238Arg) IMPG1 mutation, which was subsequently found in two other families with autosomal-dominant VMD and the same phenotype. IMPG1 encodes the SPACR protein, a component of the rod and cone photoreceptor extracellular matrix domains. Structural modeling indicates that the p.Leu238Arg substitution destabilizes the conserved SEA1 domain of SPACR. Screening of 144 probands who had various forms of macular dystrophy revealed three other IMPG1 mutations. Two individuals from one family affected by autosomal-recessive VMD were homozygous for the splice-site mutation c.807+1G>T, and two from another family were compound heterozygous for the mutations c.461T>C (p.Leu154Pro) and c.1519C>T (p.Arg507∗). Most cases had a normal or moderately decreased electrooculogram Arden ratio. We conclude that IMPG1 mutations cause both autosomal-dominant and -recessive forms of VMD, thus indicating that impairment of the interphotoreceptor matrix might be a general cause of VMD.
Main Text
Macular dystrophies are inherited retinal dystrophies in which various forms of deposits, pigmentary changes, and atrophic lesions are observed in the macula lutea, the cone-rich region of the human central retina. Mutations in several genes, expressed in either the photoreceptor rods and cones (ABCA4 [MIM 601691], ELOVL4 [MIM 605512], PRPH2 [MIM 179605], PROM1 [MIM 604365]) or in the retinal pigment epithelium (BEST1 [MIM 607854], CDH3 [MIM 114021], TIMP3 [MIM 188826]), the photoreceptor supporting tissue, are associated with inherited macular dystrophies.
Vitelliform macular dystrophies (VMDs) form a subset of macular dystrophies characterized by yellow, round deposits usually at the center of the macula and containing lipofuscin, a chemically heterogeneous pigment visualized by autofluorescence imaging of the fundus.1 Best macular dystrophy (BMD [MIM 153700]) is a juvenile form of VMD inherited as an autosomal-dominant trait;2 it leads to a loss of visual acuity and in many cases features a characteristic decrease in the Arden ratio obtained from the electrooculogram. Many families affected by BMD harbor a heterozygous mutation in BEST1,3,4 which is specifically expressed in the retinal pigment epithelium (RPE).5 In addition to BMD, there are cases of adult-onset vitelliform macular dystrophy (AVMD [MIM 608161]), which are frequently simplex and feature a small vitelliform disc on the fovea, a moderate loss of visual acuity, and a normal Arden ratio. In AVMD-affected families showing autosomal-dominant inheritance, mutations in BEST1 or PRPH2 are sometimes found.6,7 However, most cases of AVMD and some cases of BMD do not harbor mutations in either BEST1 or PRPH2.8 The involvement of both genes was also excluded from a form of autosomal-dominant atypical VMD described in one family (VMD1 [MIM 153840]).9 Therefore, a subset of VMD-affected individuals must harbor mutations in other genes.
Members of a large French family (MTP327) were identified as having autosomal-dominant VMD. The disease was assessed with visual-acuity testing, funduscopy, and autofluorescence imaging. A full-fields electroretinogram (ERG) was recorded with a ganzfeld apparatus (Metrovision, Pérenchies, France) with a bipolar contact lens electrode on maximally dilated pupils according to the international ISCEV protocol. Dim blue stimulation after 20 min of dark adaptation was used for eliciting rod responses, and 30 Hz flicker stimulation after 10 min of light adaptation was used for eliciting cone responses. Electrooculography (EOG) was also performed, and the Arden ratio, defined as the ratio of the amplitude of the light peak over that of the dark trough, was calculated (normal value ≥ 1.80). There were eight affected individuals in six generations examined (Figure 1A). The age of onset ranged from 20 to 45 years, and the mean visual acuity was 20/30 (Table 1). Six of eight individuals had a single subfoveal vitelliform lesion, and two individuals, V:2 and V:5, had several small vitelliform deposits in both eyes (Figure 2). In most individuals, the ERG rod responses were above the lower limit (except IV:4, who had moderately decreased responses), and the cone responses were either normal or moderately decreased. All individuals except IV:6 had a normal or a slightly decreased (V:5) EOG Arden ratio (Table 1). Informed and written consent consistent with the tenets of the Declaration of Helsinki was obtained for all participating individuals. IRB approval was obtained for this study from the Department of Ophthalmology of the Hospital of Montpellier. Direct Sanger sequencing of the coding exons and adjacent intronic sequences of BEST1 and PRPH2 (primer pairs and PCR conditions are available on request) as well as QMPSF (quantitative multiplex PCR of short fluorescent fragments) and MLPA (multiplex ligation-dependent probe amplification) analysis aimed at detecting large genomic rearrangements did not detect any disease-causing variant. To localize the gene defect responsible for VMD, we performed a genome-wide scan of the 15 living individuals of the family at the Centre National de Génotypage (CNG, Evry, France) by using a 250K Nsp Array (Affymetrix, Santa Clara, CA). The results were analyzed with TASE, a custom software program that compares every SNP between each individual in the family,10 and showed that affected persons shared a common 95.2 Mb haplotype in chromosomal region 6p12.1–q24.3, between the SNPs rs1076701 and rs6910680 (Figure 3). We confirmed the linkage at this locus with microsatellite markers; the maximum LOD score (calculated with Superlink-online) was 3.02 for D6S1622 on 6q13 (Figure 1). This 95.2 Mb region contains 705 genes. Within this region are ELOVL4, which causes dominant macular Stargardt-like dystrophy, and the MCDR1 [MIM 136550] locus known to cause North Carolina macular dystrophy (Figure 3). All exons and flanking intron regions of ELOVL4 were sequenced, but no mutation was found. The relevant gene within the MCDR1 locus remains unknown and, in addition, the phenotype of the MTP327 family differs from that of North Carolina macular dystrophy.
Figure 1.

Pedigrees of the Families Affected by Autosomal-Dominant Vitelliform Macular Dystrophy
(A–C) Pedigrees, alleles of the microsatellite markers surrounding IMPG1, and the IMPG1 c.713T>G (p.Leu238Arg) mutation are shown in families MTP327 (A), MTP1290 (B), and MD0531 (C). The haplotype common to the three families is shown in black.
(D) Electropherograms showing the normal control sequence and the affected sequence (from individual IV:2 of family MTP327) surrounding the c.713T>G mutation.
Table 1.
Summary of Clinical Data
| Sex, Age | Visual Acuity (right eye; left eye) | EOG Arden Ratio (Right Eye; Left Eye) |
ERG (Dim Blue Light; 30 Hz Flickers |
||
|---|---|---|---|---|---|
| Right Eye | Left Eye | ||||
| MTP327 | |||||
| IV:2 | F, 63 | 20/200; 20/60 | 2.49; 2.82 | 217; 98 | 208; 97 |
| IV:4 | M, 63 | 20/50; 30/50 | 2.11; 2.27 | 132; 67 | 104; 62 |
| IV:6 | M, 56 | 20/400; CF | 1.25; 1.34 | 205; 42 | 227; 49 |
| V:2 | F, 36 | 20/20; 20/20 | ND | ND | |
| V:3 | F, 31 | 20/200; 20/40 | 2.44; 2.62 | ND | |
| V:4 | M, 28 | 20/20; 20/25 | 1.94; 1.95 | ND | |
| V:5 | M, 37 | 20/20; 20/20 | 1.71; 1.54 | 274; 70 | 233; 61 |
| V:6 | F, 21 | 20/60; 20/20 | 2.20; 2.01 | 258; 51 | 243; 48 |
| MTP1290 | |||||
| III:4 | M, 64 | 20/40; 20/30 | 1.66; 1.73 | 333; 83 | 319; 72 |
| III:5 | M, 62 | 20/20; 20/20 | 2.44; 2.14 | 292; 61 | 262; 78 |
| MD0531 | |||||
| III:1 | F, 65 | 40/200; 20/200 | 2.91; 2.85 | 427; 105 | 409; 101 |
| III:5 | F, 55 | 20/30; 25/30 | 2.25; 2.35 | 385; 88 | 357; 72 |
| III:8 | M, 49 | 40/200; 20/50 | 2.31; 2.61 | ND | |
| NAL69 | |||||
| II:2 | F, 46 | 20/30; 25/30 | 1.48; 1.64 | normal sc and ph ERG | |
| II:4 | F, 43 | 20/20; 20/20 | 1.58; 1.68 | normal sc ERG, slightly decreased ph ERG | |
| NA1863 | |||||
| II:1 | M, 34 | 20/22; 20/22 | 1.53; 1.61 | decreased sc and ph ERG | |
| II:2 | F, 29 | 20/33; 20/20 | 1.67; 2.41 | decreased sc and ph ERG | |
| NAX1 | |||||
| II:2 | F, 40 | 20/60; 20/40 | 2.32; 2.55 | 121; 75 | 146; 81 |
Abbreviations are as follows: CF, counting fingers; ND, not done; EOG, electrooculography; ERG, full-field electroretinography; M, male; F, female; ERG values for dim blue stimulation are amplitudes of the b wave (normal > 150 μV) and for 30 Hz flickers the peak-to-peak amplitude (normal > 70 μV); sc, scotopic; and ph, photopic.
Figure 2.
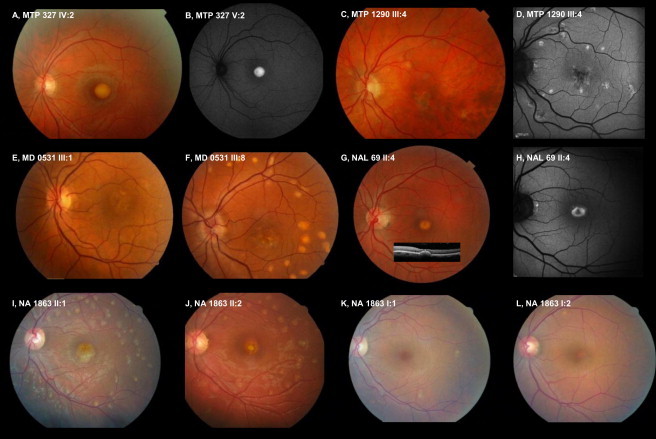
Fundus Pictures Showing the Vitelliform Macular Dystrophy Phenotypes Caused by IMPG1 Mutations
Codes for family names and member numbers refer to the family trees from Figures 1 and 4. Color pictures are fundus photographs, and black and white pictures (B, D, and H) are autofluorescence fundus imaging. Among the three families with the heterozygous, dominantly inherited missense change p.Leu238Arg, in family MTP327 (A) a macular, round vitelliform deposit containing highly autofluorescent material is found (B), whereas in family MTP1290 multifocal vitelliform deposits are encountered (C) also with autofluorescent material (D), and in family MD0531 macular (E) and multifocal (F) vitelliform deposits are found in distinct individuals. In the two families with recessive inheritance, a similar macular vitelliform deposit was found. In family NAL69, a small round macular deposit (G, insert: retinal scan showing the foveal dome-shaping accumulation of vitelliform material), which was autofluorescent (H), was seen. In family NA1863 there were additional multifocal vitelliform deposits in the two affected individuals (I and J); the heterozygous asymptomatic parents showed only tiny vitelliform spots outside the macula (K and L).
Figure 3.
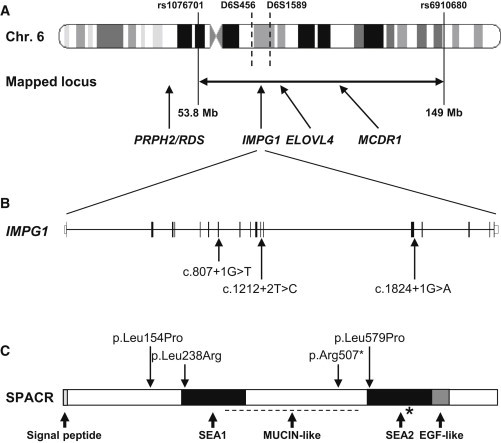
Chromosomal Localization of IMPG1, Protein-Domain Organization of SPACR, and Position of Detected Mutations
(A) The 95.2 Mb mapped disease locus in chromosomal region 6p12.1–q24.3 for family MTP327 is located between the SNPs rs1076701 and rs6910680. IMPG1, ELOVL4, and MCDR1 within the region and PRPH2/RDS outside the region are indicated. The position of D6S456 and D6S1589 markers shared by the three families with the p.Leu238Arg substitution is shown between dotted lines.
(B and C) Schematic representation of the exon-intron structure of IMPG1. Shown are (B) the location of the splice-site mutations found in this study and (C) the domain structure of its encoded SPACR protein, including the signal peptide, the N-terminal SEA1 and C-terminal SEA2 domains, the mucin-like domain (dotted line), the autoproteolytic site (∗), and the EGF-like domain. The missense and nonsense changes found in this study are indicated, as is the previously described change p.Leu579Pro in the BCAMD condition.
To identify the pathogenic mutation, we performed whole-exome sequencing (WES) of seven affected individuals of family MTP327 (individuals IV:2, IV:4, IV:6, V:3, V:4, V:5, and V:6) by using SureSelect Human All Exon Kits Version 3 in-solution enrichment methodology (Agilent, Santa Clara, CA). After excluding the common variants listed in the dbSNP 131, we filtered the 1,170 single-nucleotide variants (SNVs) and 307 indels on chromosomal region 6p12.1–q24.3 to keep only those present in coding and splice-site regions in the seven affected heterozygous individuals, reducing the number of variants to only six SNVs and one indel (Table S1). Three of the missense SNVs were found in the EVS database with a frequency ≥ 6/13,006, and the two other missense SNVs and the indel belonged to genes with wide tissue expression. We therefore retained a heterozygous missense variant, c.713T>G in exon 7 of IMPG1 (MIM 602870, RefSeq accession number NM_001563.2), that caused a leucine-to-arginine substitution (p.Leu238Arg) at codon 238 in the encoded protein SPACR, preferentially expressed in the eye (Figure 1D). This variant was not found in public human SNP databases and was not detected in 114 control chromosomes from unaffected ethnically matched individuals. The mutation cosegregated with the disease phenotype in all affected members and was absent in all unaffected members (Figure 1A).
We then sequenced exon 7 of IMPG1 in a series of 251 unrelated probands with VMD and various other forms of macular dystrophy. In two other families affected by autosomal-dominant VMD (family MTP1290 from France and family MD0531 from Spain), we found that all affected individuals were heterozygous for the same c.713T>G change, whereas nonaffected individuals did not harbor the mutation (Figure 1). In both of these families, the ERG responses were normal, and all individuals had a normal or slightly decreased (III:4 of family MTP1290) Arden ratio (Table 1). To test the origin of the c.713T>G change in the three families (MTP327, MTP1290, and MD0531), we genotyped the microsatellite markers D6S1622, D6S456, D6S1589, and D6S251, which spanned the 7.2 Mb surrounding IMPG1. We found that all affected members of the three families shared the same alleles of the two markers D6S456 (76,107,334 nt) and D6S1589 (78,456,489 nt) flanking IMPG1, suggesting that the three families could be distantly related (Figure 1). In addition, among the seven candidate genes resulting from the WES, only IMPG1 was located in this shared region, thus definitively assigning IMPG1 to VMD in these families.
Multiple-amino-acid sequence alignment of SPACR orthologs showed the conservation of the leucine at position 238 in some mammals and a substitution to phenylalanine in other orthologs (Figure S1A). The substitution p.Leu238Arg was predicted to be damaging by PolyPhen2, SIFT, and align-GVGD programs. Sequence comparison indicated that the Leu238 residue, in agreement with the presence of a conserved hydrophobic residue (Leu or Phe) at that position in known orthologs, is buried in the hydrophobic core of the N-terminal SEA domain (SEA1) of SPACR, the IMPG1 protein product. A model of the N-terminal SEA1 domain was built with ViTO software11 and the NMR structure of PDB1IVZ12 served as a template. Substitution of the hydrophobic nonpolar Leu238 for the positively charged arginine would destabilize the protein because there are many hydrophobic residues in its environment (Figure S2). Indeed, Arg238 cannot move toward a more polar environment, thus modifying the SEA1 domain of SPACR.
To further characterize the effect of the p.Leu238Arg substitution on SPACR, we performed immunoblot analysis and immunofluorescence analysis on Cos7 cells transiently transfected with pRK5 fused in-frame with wild-type or p.Leu238Arg mutant SPACR. By immunoblotting with a rabbit anti-SPACR polyclonal antibody,13 we found that both wild-type and mutant proteins migrated at the predicted molecular weight of 147 kDa (Figure S3A). By performing immunofluorescence analysis with the same antibody and using a secondary donkey anti-rabbit antibody (Alexa Fluor 488-conjugated, Invitrogen, Courtaboeuf, France), we found that both wild-type and mutant proteins localized to the cytoplasm with the same pattern (Figures S3B and S3C), suggesting that the pathogenic effect of the p.Leu238Arg substitution does not involve subcellular trafficking or severe protein unfolding.
We then sequenced the 17 exons and flanking intronic regions of IMPG1 in 144 probands, a subset of the 251 unrelated probands who had VMD and various forms of macular dystrophy and who were used for exon 7 screening. In the French family NAL69, we found that the proband (II:4) and his affected sister (II:2) were compound heterozygous for the paternal c.1519C>T mutation in exon 13, leading to the nonsense change p.Arg507∗, and for the maternal c.461T>C mutation in exon 3, leading to the missense change p.Leu154Pro (Figure 4A). The p.Arg507∗ change was found in the EVS database at a very low frequency (1/13,006 alleles). The p.Leu154Pro substitution was not found in public human SNP databases and is predicted to be damaging by PolyPhen2, SIFT, and align-GVGD programs. The Leu154 residue is conserved in mammals and is replaced by other hydrophobic aliphatic amino acids in birds and fish, in which an isoleucine or a valine, respectively, is found at this position (Figure S1A). In the consanguineous Italian family NA1863, we found that the affected brother II:1 and the affected sister II:2 were homozygous for the splice-site mutation c.807+1G>T in intron 7, which was absent from the human SNP databases, whereas the asymptomatic parents were each heterozygous for the mutation (Figure 4A). In affected individuals of families NAL69 and NA1863, the EOG Arden ratio was moderately decreased (Table 1). During fundus examination, some asymptomatic heterozygous carriers were found to have minor changes. In family NAL69, although the daughter, III:2, was normal, the son, III:1, showed a slight defect in the line between the inner and outer segments in the nasal parafovea of the left eye. In family NA1863, fundus examination showed that the parents had tiny extramacular deposits (Figure 2). This suggests that the c.461T>C and c.807+1G>T changes could be sufficient to cause subclinical retinal abnormalities and exert a mild pathogenic effect at the heterozygous state. Yet, because the macular vitelliform disc with decreased visual acuity (Table 1) was present only in the individuals who were either compound heterozygous (family NAL69) or homozygous (family NA1863), it is likely that the disease in these two families segregated as an autosomal-recessive trait.
Figure 4.
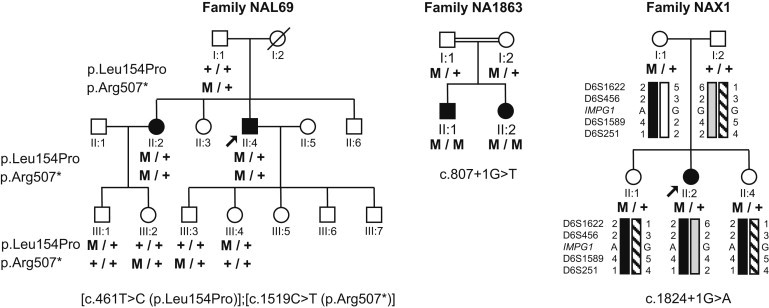
Pedigrees of Families Showing Autosomal-Recessive Inheritance and of a Family with a Simplex Case of Vitelliform Macular Dystrophy
Families NAL69, NA1863, and NAX1, affected by vitelliform macular dystrophies. A double horizontal line indicates consanguinity in family NA1863.
An additional IMPG1 variant was found in a simplex case of the French family NAX1. This female individual had a small vitelliform disk and was heterozygous for the splice-site mutation c.1824+1G>A in intron 13; this mutation was absent from the human SNP databases (Figure 4A). No other mutation was found in this proband. Her two unaffected sisters and her mother were also heterozygous for the mutation. However, the two unaffected sisters shared a paternal allele different from the paternal allele of the affected sister (Figure 4A), suggesting that the proband could harbor an unidentified paternal mutation located in the noncoding regions of IMPG1. An incomplete penetrance or a mutation in a different gene could also be considered.
The IMPG1 mutations found in the six families (three with autosomal-dominant inheritance, two with autosomal-recessive inheritance, and one simplex case) described in this study caused a consistent VMD phenotype. A heterozygous missense change (p.Leu579Pro) in SPACR was previously reported in individuals of one family affected with a retinal disease called benign concentric annular macular dystrophy (BCAMD [MIM 153870]).14 This autosomal-dominant condition was described only in this family and features a perifoveal maculopathy. Some family members progressively evolve with age toward a retinitis pigmentosa (RP [MIM 268000]) phenotype (night blindness and attenuated retinal arterioles) by the fourth and fifth decades of life.15,16 None of the family members with BCAMD showed vitelliform deposits. In addition, the six VMD families described herein did not show any annular atrophy in the macular area, and aged individuals did not show signs of RP. Therefore, the IMPG1-linked VMD phenotype of our individuals is different from that of BCAMD and thus represents a different clinical form caused by IMPG1 mutations. Other dominant or simplex cases of macular dystrophy that are presumably similar to BCAMD have been reported, but none of them have so far been linked to mutations in IMPG1.17–22 In parallel, a screening of IMPG1 mutations in other forms of inherited macular dystrophy23 or macular drusen24 failed to detect a mutation.
IMPG1 (interphotoreceptor matrix proteoglycan 1) encodes SPACR, a 150 kDa sialoprotein associated with photoreceptor cones and rods protein, a component of the interphotoreceptor matrix located in the subretinal space, in which photoreceptors and the RPE are in close apposition.13,25 This 797 amino acid secreted glycoprotein contains a central mucin-like domain and an EGF-like domain in the C-terminal region (Figure 3). Both N- and O-linked glycoconjugates are present, mainly bound in the mucin-like domain, accounting for approximately 30% of the molecular mass.13 SPACR also contains two SEA domains (after sea urchin sperm protein, enterokinase, and agrin), a highly conserved structure composed of four anti-parallel β sheets surrounded by α helices in an O-linked glycosylation-rich environment.26 The SEA domains are encountered in mucins, transmembrane serine proteases, and dystroglycan. The C-terminal SEA2 domain of SPACR contains the characteristic autoproteolytic GSVIV sequence, a cleavage site shown in mucin 1 and dystroglycan to produce two polypeptides, which remain tightly associated after proteolysis via the SEA domain.27,28 The N-terminal SEA1 domain of SPACR does not contain the cleavage site (Figure S1B). Apart from cleavage, the roles of the SEA domains remain poorly defined, although they might contribute to the adaptation of the extracellular matrices to specific physical constraints. The substitution p.Leu238Arg, which was present in the three families with the autosomal-dominant form of VMD, is situated at the beginning of the N-terminal SEA1 domain. We speculate that the p.Leu238Arg change has a dominant-negative effect. However, we could not detect any subcellular mislocalization, abnormal production, or change in gel-migration properties of the p.Leu238Arg altered protein (Figure S3), indicating that c.713T>G is not a null mutation and that the pathogenic effect of the mutant could be its abnormal behavior inside the extracellular matrix. According to this hypothesis, null mutations would lead to a full phenotype only in homozygous or compound heterozygous states. This is indeed what we have found here in the families with autosomal-recessive inheritance, in which at least one allele carried either a nonsense (family NAL69) or a presumed frameshift (family NA1863) mutation. SPACR is highly homologous to SPACRCAN (sialoprotein associated with photoreceptor cone and rod proteoglycans),29 a 200 kDa protein encoded by IMPG2 (interphotoreceptor matrix proteoglycan 2 [MIM 607056]) and also found in the interphotoreceptor matrix. Mutations in IMPG2 cause RP, another form of inherited retinal dystrophy with primary involvement of the peripheral retina,30 highlighting the critical function of the interphotoreceptor matrix in photoreceptor function and maintenance.31
The accumulation of vitelliform deposits in the retina is encountered in conditions due to impaired metabolisms of either photoreceptors or the RPE. The finding of IMPG1- and SPACR-deficient cases introduces a third actor in the causes of VMD; namely, this actor is the interphotoreceptor matrix that joins together the photoreceptors and the RPE. It will be of interest to further explore other cases of VMD, unlinked to the above mentioned genes, to test whether other components of the interphotoreceptor matrix are involved in the VMDs.
Acknowledgments
We thank the family members who participated in this study, for which informed consent for clinical examination and genetic analysis was obtained. The project was supported by Retina France (the 100-Exome Project), SOS Rétinite, Centre National de Génotypage, PI09/0459 from ISCIII (Fundaluce 2011 and ONCE 2012, Spain), the Italian Telethon Foundation, the RP Liguria Association, and INSERM. We address special thanks to UNADEV for supporting G.M. and M.H. and to CIBERER for supporting A.A.F. and M.C.. C.P.H., C.A.G., E.D.B., F.P.M.C., R.K.K., S.K., and S.B. are members of the European Retinal Disease Consortium (ERDC). At the time these studies took place, ERDC also comprised Rob W.J. Collin, Anneke I. den Hollander, Chris Inglehearn, Bart P. Leroy, Dror Sharon, Carmel Toomes, and Bernd Wissinger. We thank Ronald Roepmann and Estrada-Cuzcano for protein analysis and Britta Baumann, Marijke N. Zonneveld, and Saskia D. van der Velde-Visser for technical work. We also thank Ramon A.C. van Huet and Lies H. Hoefsloot for patient ascertainment. We thank Francesco Testa and Raffaella Brunetti-Pierri for helpful discussions, Marie-Claude Le Roy for clinical assessment, and Dr Vasiliki Kalatzis for critical reading of the manuscript. We thank the Réseau d’Histologie Expérimentale de Montpellier (facility for immunofluorescence analysis.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Align GVGD, http://agvgd.iarc.fr/index.php
Berkeley Drosophila Genome Project, http://www.fruitfly.org/index.html
ESEfinder, http://rulai.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi?process=home
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Human Splicing Finder, http://www.umd.be/HSF/
NCBI, http://ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PolyPhen2, http://genetics.bwh.harvard.edu/pph2/
Pymol, http://pymol.sourceforge.net
Superlink-online, http://bioinfo.cs.technion.ac.il/superlink-online/
References
- 1.Boon C.J.F., Jeroen Klevering B., Keunen J.E.E., Hoyng C.B., Theelen T. Fundus autofluorescence imaging of retinal dystrophies. Vision Res. 2008;48:2569–2577. doi: 10.1016/j.visres.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 2.Best F. II. Über eine hereditäre Maculaaffektion. Ophthalmologica. 1905;13:199–212. [Google Scholar]
- 3.Boon C.J.F., Klevering B.J., den Hollander A.I., Zonneveld M.N., Theelen T., Cremers F.P.M., Hoyng C.B. Clinical and genetic heterogeneity in multifocal vitelliform dystrophy. Arch. Ophthalmol. 2007;125:1100–1106. doi: 10.1001/archopht.125.8.1100. [DOI] [PubMed] [Google Scholar]
- 4.Mansergh F.C., Kenna P.F., Rudolph G., Meitinger T., Farrar G.J., Kumar-Singh R., Scorer J., Hally A.M., Mynett-Johnson L., Humphries M.M. Evidence for genetic heterogeneity in Best’s vitelliform macular dystrophy. J. Med. Genet. 1995;32:855–858. doi: 10.1136/jmg.32.11.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petrukhin K., Koisti M.J., Bakall B., Li W., Xie G., Marknell T., Sandgren O., Forsman K., Holmgren G., Andreasson S. Identification of the gene responsible for Best macular dystrophy. Nat. Genet. 1998;19:241–247. doi: 10.1038/915. [DOI] [PubMed] [Google Scholar]
- 6.Krämer F., White K., Pauleikhoff D., Gehrig A., Passmore L., Rivera A., Rudolph G., Kellner U., Andrassi M., Lorenz B. Mutations in the VMD2 gene are associated with juvenile-onset vitelliform macular dystrophy (Best disease) and adult vitelliform macular dystrophy but not age-related macular degeneration. Eur. J. Hum. Genet. 2000;8:286–292. doi: 10.1038/sj.ejhg.5200447. [DOI] [PubMed] [Google Scholar]
- 7.Seddon J.M., Afshari M.A., Sharma S., Bernstein P.S., Chong S., Hutchinson A., Petrukhin K., Allikmets R. Assessment of mutations in the Best macular dystrophy (VMD2) gene in patients with adult-onset foveomacular vitelliform dystrophy, age-related maculopathy, and bull’s-eye maculopathy. Ophthalmology. 2001;108:2060–2067. doi: 10.1016/s0161-6420(01)00777-1. [DOI] [PubMed] [Google Scholar]
- 8.Meunier I., Sénéchal A., Dhaenens C.-M., Arndt C., Puech B., Defoort-Dhellemmes S., Manes G., Chazalette D., Mazoir E., Bocquet B., Hamel C.P. Systematic screening of BEST1 and PRPH2 in juvenile and adult vitelliform macular dystrophies: a rationale for molecular analysis. Ophthalmology. 2011;118:1130–1136. doi: 10.1016/j.ophtha.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 9.Sohocki M.M., Sullivan L.S., Mintz-Hittner H.A., Small K., Ferrell R.E., Daiger S.P. Exclusion of atypical vitelliform macular dystrophy from 8q24.3 and from other known macular degenerative loci. Am. J. Hum. Genet. 1997;61:239–241. doi: 10.1016/S0002-9297(07)64299-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hebrard M., Manes G., Bocquet B., Meunier I., Coustes-Chazalette D., Hérald E., Sénéchal A., Bolland-Augé A., Zelenika D., Hamel C.P. Combining gene mapping and phenotype assessment for fast mutation finding in non-consanguineous autosomal recessive retinitis pigmentosa families. Eur. J. Hum. Genet. 2011;19:1256–1263. doi: 10.1038/ejhg.2011.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Catherinot V., Labesse G. ViTO: tool for refinement of protein sequence-structure alignments. Bioinformatics. 2004;20:3694–3696. doi: 10.1093/bioinformatics/bth429. [DOI] [PubMed] [Google Scholar]
- 12.Maeda T., Inoue M., Koshiba S., Yabuki T., Aoki M., Nunokawa E., Seki E., Matsuda T., Motoda Y., Kobayashi A. Solution structure of the SEA domain from the murine homologue of ovarian cancer antigen CA125 (MUC16) J. Biol. Chem. 2004;279:13174–13182. doi: 10.1074/jbc.M309417200. [DOI] [PubMed] [Google Scholar]
- 13.Acharya S., Rayborn M.E., Hollyfield J.G. Characterization of SPACR, a sialoprotein associated with cones and rods present in the interphotoreceptor matrix of the human retina: immunological and lectin binding analysis. Glycobiology. 1998;8:997–1006. doi: 10.1093/glycob/8.10.997. [DOI] [PubMed] [Google Scholar]
- 14.van Lith-Verhoeven J.J.C., Hoyng C.B., van den Helm B., Deutman A.F., Brink H.M.A., Kemperman M.H., de Jong W.H.M., Kremer H., Cremers F.P.M. The benign concentric annular macular dystrophy locus maps to 6p12.3-q16. Invest. Ophthalmol. Vis. Sci. 2004;45:30–35. doi: 10.1167/iovs.03-0392. [DOI] [PubMed] [Google Scholar]
- 15.van den Biesen P.R., Deutman A.F., Pinckers A.J. Evolution of benign concentric annular macular dystrophy. Am. J. Ophthalmol. 1985;100:73–78. doi: 10.1016/s0002-9394(14)74986-4. [DOI] [PubMed] [Google Scholar]
- 16.Deutman A.F. Benign concentric annular macular dystrophy. Am. J. Ophthalmol. 1974;78:384–396. doi: 10.1016/0002-9394(74)90225-6. [DOI] [PubMed] [Google Scholar]
- 17.Coppeto J., Ayazi S. Annular macular dystrophy. Am. J. Ophthalmol. 1982;93:279–284. doi: 10.1016/0002-9394(82)90525-6. [DOI] [PubMed] [Google Scholar]
- 18.Gómez-Faiña P., Alarcón-Valero I., Buil Calvo J.A., Calsina-Prat M., Martín-Moral D., Lillo-Sopena J., Castilla Céspedes M. [Benign concentric annular macular dystrophy] Arch. Soc. Esp. Oftalmol. 2007;82:373–376. doi: 10.4321/s0365-66912007000600011. [DOI] [PubMed] [Google Scholar]
- 19.Martyn L.J., Walker B.A. A kindred showing a disorder of the retinal pigmentary epithelium and choriocapillaris, with characteristic macular changes and autosomal dominant transmission. Birth Defects Orig. Artic. Ser. 1971;7:189–192. [PubMed] [Google Scholar]
- 20.Miyake Y., Shiroyama N., Horiguchi M., Saito A., Yagasaki K. Bull’s-eye maculopathy and negative electroretinogram. Retina. 1989;9:210–215. [PubMed] [Google Scholar]
- 21.Pérez Alvarez M.J., Clement Fernández F. [Benign concentric annular macular dystrophy: two cases] Arch. Soc. Esp. Oftalmol. 2003;78:451–454. [PubMed] [Google Scholar]
- 22.Salinas Alamán A., Sádaba Echarri L.M., Corcóstegui Crespo I., García Layana A. [Benign concentric annular macular dystrophy] Arch. Soc. Esp. Oftalmol. 2005;80:45–48. doi: 10.4321/s0365-66912005000100009. [DOI] [PubMed] [Google Scholar]
- 23.Gehrig A., Felbor U., Kelsell R.E., Hunt D.M., Maumenee I.H., Weber B.H. Assessment of the interphotoreceptor matrix proteoglycan-1 (IMPG1) gene localised to 6q13-q15 in autosomal dominant Stargardt-like disease (ADSTGD), progressive bifocal chorioretinal atrophy (PBCRA), and North Carolina macular dystrophy (MCDR1) J. Med. Genet. 1998;35:641–645. doi: 10.1136/jmg.35.8.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh K.K., Dawson W.W., Krawczak M., Schmidtke J. IMPG1 gene variation in rhesus macular drusen. Vet. Ophthalmol. 2007;10:274–277. doi: 10.1111/j.1463-5224.2007.00549.x. [DOI] [PubMed] [Google Scholar]
- 25.Kuehn M.H., Hageman G.S. Expression and characterization of the IPM 150 gene (IMPG1) product, a novel human photoreceptor cell-associated chondroitin-sulfate proteoglycan. Matrix Biol. 1999;18:509–518. doi: 10.1016/s0945-053x(99)00043-8. [DOI] [PubMed] [Google Scholar]
- 26.Bork P., Patthy L. The SEA module: a new extracellular domain associated with O-glycosylation. Protein Sci. 1995;4:1421–1425. doi: 10.1002/pro.5560040716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akhavan A., Crivelli S.N., Singh M., Lingappa V.R., Muschler J.L. SEA domain proteolysis determines the functional composition of dystroglycan. FASEB J. 2008;22:612–621. doi: 10.1096/fj.07-8354com. [DOI] [PubMed] [Google Scholar]
- 28.Macao B., Johansson D.G.A., Hansson G.C., Härd T. Autoproteolysis coupled to protein folding in the SEA domain of the membrane-bound MUC1 mucin. Nat. Struct. Mol. Biol. 2006;13:71–76. doi: 10.1038/nsmb1035. [DOI] [PubMed] [Google Scholar]
- 29.Acharya S., Foletta V.C., Lee J.W., Rayborn M.E., Rodriguez I.R., Young W.S., 3rd, Hollyfield J.G. SPACRCAN, a novel human interphotoreceptor matrix hyaluronan-binding proteoglycan synthesized by photoreceptors and pinealocytes. J. Biol. Chem. 2000;275:6945–6955. doi: 10.1074/jbc.275.10.6945. [DOI] [PubMed] [Google Scholar]
- 30.Bandah-Rozenfeld D., Collin R.W.J., Banin E., van den Born L.I., Coene K.L.M., Siemiatkowska A.M., Zelinger L., Khan M.I., Lefeber D.J., Erdinest I. Mutations in IMPG2, encoding interphotoreceptor matrix proteoglycan 2, cause autosomal-recessive retinitis pigmentosa. Am. J. Hum. Genet. 2010;87:199–208. doi: 10.1016/j.ajhg.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hollyfield J.G., Varner H.H., Rayborn M.E., Osterfeld A.M. Retinal attachment to the pigment epithelium. Linkage through an extracellular sheath surrounding cone photoreceptors. Retina. 1989;9:59–68. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.