

Abstract
Epileptic encephalopathies are genetically heterogeneous severe disorders in which epileptic activity contributes to neurological deterioration. We studied two unrelated children presenting with a distinctive early-onset epileptic encephalopathy characterized by refractory epilepsy and absent developmental milestones, as well as thick and short corpus callosum and persistent cavum septum pellucidum on brain MRI. Using whole-exome sequencing, we identified biallelic mutations in seizure threshold 2 (SZT2) in both affected children. The causative mutations include a homozygous nonsense mutation and a nonsense mutation together with an exonic splice-site mutation in a compound-heterozygous state. The latter mutation leads to exon skipping and premature termination of translation, as shown by RT-PCR in blood RNA of the affected boy. Thus, all three mutations are predicted to result in nonsense-mediated mRNA decay and/or premature protein truncation and thereby loss of SZT2 function. Although the molecular role of the peroxisomal protein SZT2 in neuronal excitability and brain development remains to be defined, Szt2 has been shown to influence seizure threshold and epileptogenesis in mice, consistent with our findings in humans. We conclude that mutations in SZT2 cause a severe type of autosomal-recessive infantile encephalopathy with intractable seizures and distinct neuroradiological anomalies.
Main Text
Children presenting with neonatal or infantile encephalopathies featuring epilepsy often follow a devastating course leading to premature death or to a poor neurological outcome. The developing brain seems to be particularly prone and susceptible to seizure activity. The impairment of active processes of myelination, synaptogenesis, neuron migration, and apoptosis occurring in the neonatal and infantile periods have been implicated in both the initiation and the propagation of seizures, as well as in the often devastating results of seizure activity.1,2 The motor, sensory, and cognitive development of a child with epilepsy is often impaired by ongoing epileptic activity.3 Thus, in those conditions featuring frequent seizures and/or prominent epileptiform abnormalities on electroencephalography (EEG), epilepsy itself might contribute to cognitive impairment, and these disorders are classified as epileptic encephalopathies.
Early-onset epileptic encephalopathies (EOEEs) are highly heterogeneous, and when the most common acquired, malformative, and metabolic etiologies are ruled out, the possibility of a genetic cause must be addressed. Specific syndromes are defined on the basis of seizure type, age of onset, and the results of ancillary tests, and this has proved useful for guiding genetic studies.4,5 However, the genetic origins and underlying molecular processes of EOEEs have been described in only a few well-defined syndromes,5,6 such as Ohtahara syndrome (MIM 308350 and 612164). Ohtahara syndrome, one of the most severe and earliest forms of epilepsy, is characterized by a suppression burst pattern on EEG and can be caused by heterozygous de novo mutations in STXBP1 (MIM 602926).7,8 Mutations in more than ten additional genes cause early infantile epileptic encephalopathy in a broad sense,6 but because of the lack of specific clinical, electrophysiologic, and neuroradiologic signs, many affected children have no specific diagnosis. Contemporary tools for genetic analysis, such as microarray-based comparative genomic hybridization (array CGH) and whole-exome sequencing, have greatly enhanced the ability to identify pathogenic copy-number variations and mutations even in rare disease phenotypes.9,10 Understanding the genetic basis of EOEEs will enable more precise diagnosis, genetic counseling, and classification of these diseases and might eventually assist in the development of targeted treatments.
We aimed at the identification of causative alleles in genetically unresolved forms of EOEE. The study was approved by the ethics committee of the University of Ulm and the Helsinki committee of the Rabin Medical Center in accordance with a protocol approved by the National Committee for Genetic Studies, Israeli Ministry of Health. Written informed consent to participate in the study was obtained from the affected individuals’ parents.
We studied two unrelated individuals with unexplained EOEE and remarkable clinical and radiological similarities. Clinical features are summarized in Table S1, available online. Individual 1 (II:6, Figure 1A), a 10-year-old girl, is the sixth child of healthy nonconsanguineous parents who are both of Iraqi Jewish descent. One of her brothers (individual II:5) was probably affected by the same condition; he died at age 3 years from respiratory infection, and no DNA was available for genetic studies. Individual 2 (II:1, Figure 1B), a 9-year-old boy, is the first child of healthy nonconsanguineous Spanish parents. Both unrelated children had common facial dysmorphic features (Figure 1C), severe developmental delay with hypotonia in infancy, decreased tendon reflexes, and absence of developmental milestones; there was no microcephaly. Seizures began at the age of 4 years in individual 1 and at the age of 2 months in her affected brother and in individual 2. Seizures in individual 1 were characterized by loss of consciousness, drooling, and perioral cyanosis and were at times followed by tonic-clonic generalization. Initially described as focal, seizures in individual 2 affected either side of the body with frequent tonic generalization. The current seizure pattern includes multiple tonic seizures per day and atypical absences. Seizures have proved highly refractory to multiple antiepileptic drug combinations in both children. EEG of individual 1 was normal prior to epilepsy onset. After the onset of epilepsy, EEG showed an abnormal background trace and prominent epileptiform abnormalities in both children (Figure S1), but no suppression burst pattern. In individual 1, EEG at the age of 4 years showed slowed 4–5 Hz background activity with isolated spike waves and sharp waves in the right frontopolar and right frontocentral areas, as well as focal 12–14 and 6 Hz epileptic activity with secondary generalization, corresponding to clinical seizures. In individual 2, EEG at the age of 8 years revealed altered background activity and no topographic differentiation and multifocal spikes in either hemisphere. Brain MRI showed a short and thick corpus callosum and persistent cavum septum pellucidum in both children, as well as in the deceased brother of individual 1 (Figure 2). Electromyography, nerve conduction velocities, array CGH, and metabolic investigations in blood, urine, and cerebrospinal fluid all gave normal results.
Figure 1.

Pedigrees and Pictures of Individuals with EOEE
(A and B) Pedigrees of family 1 (A) and family 2 (B). Individuals affected or likely affected by EOEE are represented by filled black and gray symbols, respectively. The index individuals are depicted by arrows. The plus sign (+) denotes the reference sequence, and “mut” indicates the respective mutation. Red “mut” represents the c.73C>T (p.Arg25∗) nonsense mutation, blue “mut” represents the c.1496G>T exonic splice-site mutation, and green “mut” represents the c.2092C>T (p.Gln698∗) nonsense mutation.
(C) Individual 1 at the age of 10 years (left) and individual 2 at the age of 9 years (right). Common facial dysmorphic features include a high forehead, downslanting palpebral fissures, ptosis, and arched and laterally extended eyebrows.
Figure 2.
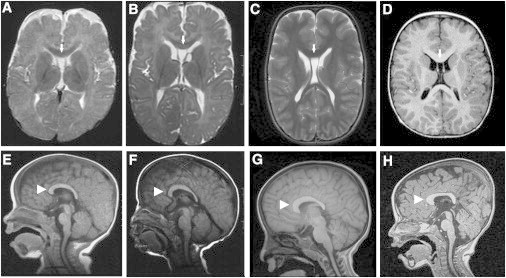
Brain MRI Findings
Persistent cavum septum pellucidum (arrows) and thick and short corpus callosum (arrowheads) in the three affected children.
(A and E) Individual II-5 at the age of 6 months.
(B, C, F and G) Individual II-6 (individual 1) at the ages of 7 months (B and F) and 4 years and 8 months (C and G).
(D and H) Individual 2 at the age of 2 years.
To identify the underlying genetic cause of the putatively autosomal-recessive encephalopathy, we sequenced the exomes of unrelated individuals 1 and 2. For the exome sequence analysis, we used the SeqCap EZ Human Exome Library v.2.0 and v.3.0 enrichment kits in individuals 1 and 2, respectively, and an Illumina HiSeq2000 sequencer with a paired-end 100 bp protocol. Sequence reads were aligned to the human reference genome (hg19 assembly, UCSC Genome Browser). Mean coverage was 84× and 111× for the exome of individuals 1 and 2, respectively, and approximately 95% of the target sequences were covered at least 10× in both exomes. Data analysis was performed with a combination of available tools and custom scripts (details are available on request) and, in the case of individual 2, with the recently established VARBANK exome pipeline from the Cologne Center for Genomics (H.T. and P.N., unpublished data). We focused on private and rare (minor allele frequency < 0.01) presumptively damaging variants.
Neither of the two exomes displayed an apparent pathogenic allele in any of the 15 OMIM-referenced genes mutated in early infantile epileptic encephalopathy. Because the parents of individual 1 are both of Iraqi Jewish origin, we assumed that a disease-causing mutation would be present in the homozygous state. In individual 1, SNP-array-based genotyping with an Affymetrix Human Mapping 50K Xba 240 array and HomozygosityMapper11 identified five homozygosity regions > 2 Mb (on chromosomes 1, 3, 4, 10, and 12; Table S2); collectively, they span 20 Mb and contain >200 genes, none of which is known to be implicated in EOEEs. Four genes mapping to these regions of homozygosity contained a rare homozygous variant (Tables S3 and S4). Only one of these genes, SZT2, also harbored two distinct rare variants in individual 2, consistent with compound-heterozygous mutations. SZT2 was the only gene in which rare or unique biallelic damaging mutations were observed in the exomes of both affected individuals.
We confirmed the three SZT2 (RefSeq accession number NM_015284.3) mutations by Sanger sequencing on an ABI3730 DNA Analyzer after PCR amplification of the exons of interest. Individual 1 was homozygous for a c.73C>T transition in exon 2. This mutation leads to a premature stop codon (p.Arg25∗) (Figure 3A) and is predicted to result in a truncation of SZT2 after 24 of 3,375 amino acids and/or in nonsense-mediated mRNA decay (NMD). Alternatively, Met50, the next downstream in-frame methionine, might be used as a translation initiation codon, leading to an N-terminally-shortened protein. The c.73C>T nonsense mutation cosegregated with the disease in the family (Figure 1A) and was absent from 143 Iraqi Jewish control subjects. Individual 2 was compound heterozygous for a c.2092C>T (p.Gln698∗) nonsense mutation inherited from the mother and a paternally inherited c.1496G>T mutation (Figures 1B and 3B) that was predicted to lead to the amino acid substitution p.Ser499Ile. The latter mutation affects the guanine at the last position of exon 10 (Figure 3B) and is predicted by the NNSPLICE and VARBANK-MaxEntScan splice prediction tools to significantly weaken the exon 10 splice donor site (NNSPLICE: splice score 0.88 for wild-type versus 0.11 for the mutation; VARBANK-MaxEntScan: splice score 7.7 for wild-type versus 1.36 for the mutation). For the splicing analysis, total RNA from peripheral white blood cells of individual 2 and a control was extracted from fresh EDTA blood by a standard trizol protocol. RT-PCR was performed with the QIAGEN OneStep RT-PCR kit with primers located in SZT2 exons 9 and 11. Using direct sequencing of RT-PCR products amplified from RNA of individual 2, we observed skipping of exon 10 (Figure 3C), consistent with the bioinformatic prediction. This out-of-frame exon-skipping event is predicted to lead to a frameshift followed by a premature termination codon (p.Gly412Alafs∗86). Upon RT-PCR, we detected the two mutations identified in individual 2 in a heterozygous state with largely similar intensity (Figure 3C and data not shown), suggesting that—at least in blood—these mutations induce no or incomplete NMD and are predicted to produce truncated proteins. None of the three mutations identified in the two children are present in the >6,000 European American and African American individuals included in the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project Exome Variant Server.
Figure 3.
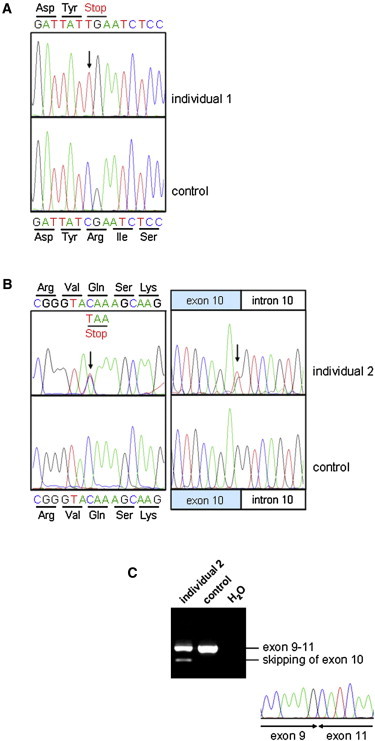
SZT2 Mutations
(A) Homozygous nonsense mutation c.73C>T (p.Arg25∗) detected in individual 1.
(B) Compound-heterozygous mutation c.2092C>T (p.Gln698∗) (left) and c.1496G>T (right) identified in individual 2.
(C) RT-PCR on total RNA extracted from the blood of individual 2 and a control shows that the c.1496G>T exonic splice-site mutation leads to the skipping of exon 10.
We have identified mutations in SZT2 as the cause of EOEE in two unrelated children. The disease is characterized by severe developmental delay, refractory epilepsy, and a thick corpus callosum and persistent cavum septum pellucidum. Recent advances in molecular genetics are leading to an expanding list of genes that are mutated in EOEEs.6 Ideally, genetic testing should be guided by the phenotype, but this is not possible for many EOEEs that do not show distinctive clinical or radiological features in addition to epilepsy and developmental delay. Thus, the causative mutation remains unknown for most children with allegedly genetic epileptic encephalopathy, which poses additional difficulties for discussing prognosis and genetic counseling. In contrast to the features of other genetic EOEEs, a thick corpus callosum and persistent cavum septum pellucidum appear as clues that should raise the suspicion of SZT2 mutation.
Agenesis or dysgenesis of the corpus callosum is a relatively frequent finding in neurodevelopmental disorders. It has been described in several EOEE-associated Mendelian syndromes, namely Aicardi syndrome (MIM 304050) and the encephalopathy associated with mutations in aristaless-related homeobox (ARX [MIM 300382 and 308350]).12 Thick corpus callosum has been described in Cohen syndrome (MIM 216550), a disorder featuring microcephaly, ocular abnormalities, and cognitive impairment,13 and in interstitial 6q deletions.14 In most of the cases, a relatively large corpus callosum has been associated with additional structural brain abnormalities.15 Another neurodevelopmental syndrome with partial clinical overlap with the EOEE we describe is the genetically unresolved syndrome of megalencephaly, mega corpus callosum, and complete lack of motor development; this syndrome might be associated with infantile spasms and persistent cavum septum pellucidum.16,17
Whether SZT2 participates in the processes of cellular proliferation, axonal growth, or glial patterning occurring at the midline during development remains to be investigated, and thus how mutations in SZT2 might influence callosal and/or cortical morphogenesis is unknown. However, it is conceivable that defective axonal pruning might have led to the thick corpus callosum. The finding of a persistent cavum pellucidum has been associated with epilepsy,18 but proof of causality is lacking and its frequency in the general population is presently unknown.
Use of the term epileptic encephalopathy presupposes that epilepsy itself contributes to encephalopathy. However, it is often difficult to weigh the role of epilepsy in newborns or infants against the pre-existing severe neurological dysfunction, given that neither seizures nor electroencephalographic abnormalities substantially modify a poor outcome that is mainly explained by the underlying etiology. The existence of severe developmental delay antedating the onset of epilepsy and EEG abnormalities in the affected girl suggests that mutations in SZT2 also impair brain maturation and that this impairment occurs independently of the seizures.
The 71-exon gene SZT2 (seizure threshold 2, previously known as TIGR, C1orf84, and KIAA0467) encodes a large protein of unknown function. It is widely expressed and has high expression in the CNS in humans and mice, predominantly in the parietal and frontal cortices, hippocampus, cerebellum, and dorsal root ganglia.19,20 SZT2 contains predicted functional domains, namely a superoxide dismutase motif and a PTS1 peroxisomal targeting signal, and was shown to colocalize with catalase at the peroxisome.19 In that study, a suppression-subtractive-hybridization procedure was used for identifying upregulation of SZT2 (called TIGR by the authors for “transcript increased in glutamate resistance”) as protective against oxidative glutamate toxicity and H2O2-induced oxidative stress in a neuronal cell model. Thus, oxidative glutamate toxicity and excitotoxicity are plausible mechanisms underlying epileptogenesis caused by SZT2 mutations.
Szt2 has been shown to influence seizure threshold and epileptogenesis in mice. Szt2m1Frk-homozygous mouse mutants are susceptible to induced seizures, although they do not display spontaneous seizures.20 The Szt2m1Frk allele contains a splice donor mutation after exon 32, predicting transcriptional read-through, a translational frameshift, and a premature stop. Male mutant mice treated daily with a tetanic, high-frequency electrical stimulus developed seizures significantly earlier than did wild-type controls. Most of the mice that were homozygous for another mutation, Szt2Gt(XH662)By (a gene-trap mutation in exon 21), died before birth; surviving mutant mice had a low seizure threshold.20 Thus, in mice, truncating mutations in Szt2 can confer low seizure thresholds and embryonic lethality, phenotypes consistent with the pharmacoresistant seizures and developmental arrest in the human disease we describe. Unexpectedly, Szt2 mRNA levels were upregulated rather than diminished in both mutant mouse models.20 Although the underlying pathomechanism remains to be elucidated, the effect of the mutations at the protein level might well be loss of function, as is supposedly the case in the human disease.
Our study defines an infantile epileptic syndrome and highlights an important role for SZT2 in epileptogenesis. Severe developmental delay antedating the onset of epilepsy suggests that SZT2 might also play a role in human brain development. The identification of additional persons with biallelic SZT2 mutations will shed further light on the clinical spectrum of this emerging disorder.
Acknowledgments
We thank the families for participating in this study and M. van der Knaap for reviewing MRI images. We thank G.J. Halpern for her help with editing the manuscript. The study was supported by the Adler Chair in Pediatric Cardiology at Tel Aviv University, the Israeli Ministry of Health Chief Scientist Foundation (grant 3-4963 to L.B.-V.), the Israeli Science Foundation (grant 558/09 to L.B.-V.), the German Federal Ministry of Education and Research (NGFNplus-EMINet to P.N. and C.K.), the EuroEPINOMICS program (Deutsche Forschungsgemeinschaft [DFG] grant NU50/8-1 to P.N.), the Ministerio de Ciencia y Competitividad at Instituto de Salud Carlos III (grant PI12/01005 to A.M.), the Agència de Gestió d’Ajuts Universitaris i de Recerca (grant SGR 2009/0078 to A.M.), and the DFG (to G.B.). D.D. and A.H. are the owners of Toldot Genetics Ltd.
Contributor Information
Lina Basel-Vanagaite, Email: basel@post.tau.ac.il.
Guntram Borck, Email: guntram.borck@uni-ulm.de.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Berkeley Drosophila Genome Project NNSplice 0.9, http://www.fruitfly.org/seq_tools/splice.html
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Holmes G.L., Ben-Ari Y. The neurobiology and consequences of epilepsy in the developing brain. Pediatr. Res. 2001;49:320–325. doi: 10.1203/00006450-200103000-00004. [DOI] [PubMed] [Google Scholar]
- 2.Rho J.M. Basic science behind the catastrophic epilepsies. Epilepsia. 2004;45(Suppl 5):5–11. doi: 10.1111/j.0013-9580.2004.05001.x. [DOI] [PubMed] [Google Scholar]
- 3.Ben-Ari Y., Holmes G.L. Effects of seizures on developmental processes in the immature brain. Lancet Neurol. 2006;5:1055–1063. doi: 10.1016/S1474-4422(06)70626-3. [DOI] [PubMed] [Google Scholar]
- 4.Zupanc M.L. Clinical evaluation and diagnosis of severe epilepsy syndromes of early childhood. J. Child Neurol. 2009;24(Suppl):6S–14S. doi: 10.1177/0883073809338151. [DOI] [PubMed] [Google Scholar]
- 5.Noh G.J., Jane Tavyev Asher Y., Graham J.M., Jr. Clinical review of genetic epileptic encephalopathies. Eur. J. Med. Genet. 2012;55:281–298. doi: 10.1016/j.ejmg.2011.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tavyev Asher Y.J., Scaglia F. Molecular bases and clinical spectrum of early infantile epileptic encephalopathies. Eur. J. Med. Genet. 2012;55:299–306. doi: 10.1016/j.ejmg.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 7.Saitsu H., Kato M., Mizuguchi T., Hamada K., Osaka H., Tohyama J., Uruno K., Kumada S., Nishiyama K., Nishimura A. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat. Genet. 2008;40:782–788. doi: 10.1038/ng.150. [DOI] [PubMed] [Google Scholar]
- 8.Pavone P., Spalice A., Polizzi A., Parisi P., Ruggieri M. Ohtahara syndrome with emphasis on recent genetic discovery. Brain Dev. 2012;34:459–468. doi: 10.1016/j.braindev.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 9.Bamshad M.J., Ng S.B., Bigham A.W., Tabor H.K., Emond M.J., Nickerson D.A., Shendure J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011;12:745–755. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- 10.Mefford H.C., Yendle S.C., Hsu C., Cook J., Geraghty E., McMahon J.M., Eeg-Olofsson O., Sadleir L.G., Gill D., Ben-Zeev B. Rare copy number variants are an important cause of epileptic encephalopathies. Ann. Neurol. 2011;70:974–985. doi: 10.1002/ana.22645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seelow D., Schuelke M., Hildebrandt F., Nürnberg P. HomozygosityMapper—an interactive approach to homozygosity mapping. Nucleic Acids Res. 2009;37(Web Server issue):W593–W599. doi: 10.1093/nar/gkp369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paul L.K., Brown W.S., Adolphs R., Tyszka J.M., Richards L.J., Mukherjee P., Sherr E.H. Agenesis of the corpus callosum: genetic, developmental and functional aspects of connectivity. Nat. Rev. Neurosci. 2007;8:287–299. doi: 10.1038/nrn2107. [DOI] [PubMed] [Google Scholar]
- 13.Kivitie-Kallio S., Autti T., Salonen O., Norio R. MRI of the brain in the Cohen syndrome: a relatively large corpus callosum in patients with mental retardation and microcephaly. Neuropediatrics. 1998;29:298–301. doi: 10.1055/s-2007-973581. [DOI] [PubMed] [Google Scholar]
- 14.Rosenfeld J.A., Amrom D., Andermann E., Andermann F., Veilleux M., Curry C., Fisher J., Deputy S., Aylsworth A.S., Powell C.M. Genotype-phenotype correlation in interstitial 6q deletions: a report of 12 new cases. Neurogenetics. 2012;13:31–47. doi: 10.1007/s10048-011-0306-5. [DOI] [PubMed] [Google Scholar]
- 15.Lerman-Sagie T., Ben-Sira L., Achiron R., Schreiber L., Hermann G., Lev D., Kidron D., Malinger G. Thick fetal corpus callosum: an ominous sign? Ultrasound Obstet. Gynecol. 2009;34:55–61. doi: 10.1002/uog.6356. [DOI] [PubMed] [Google Scholar]
- 16.Göhlich-Ratmann G., Baethmann M., Lorenz P., Gärtner J., Goebel H.H., Engelbrecht V., Christen H.J., Lenard H.G., Voit T. Megalencephaly, mega corpus callosum, and complete lack of motor development: a previously undescribed syndrome. Am. J. Med. Genet. 1998;79:161–167. doi: 10.1002/(sici)1096-8628(19980923)79:3<161::aid-ajmg2>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 17.Dagli A.I., Stalker H.J., Williams C.A. A patient with the syndrome of megalencephaly, mega corpus callosum and complete lack of motor development. Am. J. Med. Genet. A. 2008;146A:204–207. doi: 10.1002/ajmg.a.32079. [DOI] [PubMed] [Google Scholar]
- 18.Iannetti P., Papetti L., Nicita F., Castronovo A., Ursitti F., Parisi P., Spalice A., Verrotti A. Developmental anomalies of the medial septal area: possible implication for limbic epileptogenesis. Childs Nerv. Syst. 2011;27:765–770. doi: 10.1007/s00381-010-1322-8. [DOI] [PubMed] [Google Scholar]
- 19.Toutzaris D., Lewerenz J., Albrecht P., Jensen L.T., Letz J., Geerts A., Golz S., Methner A. A novel giant peroxisomal superoxide dismutase motif-containing protein. Free Radic. Biol. Med. 2010;48:811–820. doi: 10.1016/j.freeradbiomed.2009.12.023. [DOI] [PubMed] [Google Scholar]
- 20.Frankel W.N., Yang Y., Mahaffey C.L., Beyer B.J., O’Brien T.P. Szt2, a novel gene for seizure threshold in mice. Genes Brain Behav. 2009;8:568–576. doi: 10.1111/j.1601-183X.2009.00509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.