

Abstract
Orofaciodigital syndrome (OFD) is a recognized clinical entity with core defining features in the mouth, face, and digits, in addition to various other features that have been proposed to define distinct subtypes. The three genes linked to OFD—OFD1, TMEM216, and TCTN3—play a role in ciliary biology, a finding consistent with the clinical overlap between OFD and other ciliopathies. Most autosomal-recessive cases of OFD, however, remain undefined genetically. In two multiplex consanguineous Arab families affected by OFD, we identified a tight linkage interval in chromosomal region 1q32.1. Exome sequencing revealed a different homozygous variant in DDX59 in each of the two families, and at least one of the two variants was accompanied by marked reduction in the level of DDX59. DDX59 encodes a relatively uncharacterized member of the DEAD-box-containing RNA helicase family of proteins, which are known to play a critical role in all aspects of RNA metabolism. We show that Ddx59 is highly enriched in its expression in the developing murine palate and limb buds. At the cellular level, we show that DDX59 is localized dynamically to the nucleus and the cytoplasm. Consistent with the absence of DDX59 representation in ciliome databases and our demonstration of its lack of ciliary localization, ciliogenesis appears to be intact in mutant fibroblasts but ciliary signaling appears to be impaired. Our data strongly implicate this RNA helicase family member in the pathogenesis of OFD, although the causal mechanism remains unclear.
Main Text
Orofaciodigital syndrome (OFD [MIM 311200]) is a well-recognized clinical entity that combines characteristic manifestations in the mouth, face, and digits; these include cleft lip and palate, lobulated tongue, multiple oral frenulae, and polydactyly.1 In addition to these core features, which have been described in the original report by Mohr in 1941 and subsequently by Claussen in 1946 and Papillon-Leage and Psaume in 1954, several subsequent reports have added various skeletal, ocular, and neurological manifestations and proposed various subtypes (OFD1–OFD13).2–4 However, many of the subtypes were isolated reports, and there is significant controversy about the justification for the extensive “splitting” in the OFD nomenclature.5
As in many clinically heterogeneous conditions, molecular analysis offered helpful insight into the nosology of OFD. The identification of OFD1 (MIM 300170) as the first gene to be linked to OFD clearly defined OFD1 as a distinct subtype that is X-linked and lethal in males.6 Subsequently, mutations in TMEM216 (MIM 613277) were observed in individuals with OFD6 (Varadi syndrome [MIM 277170]), in which individuals have the additional presence of cerebellar anomalies.7 The finding of OFD1 and TMEM216 mutations in individuals with such classic ciliopthies as Joubert syndrome and Meckel-Gruber syndrome confirms that at least a subset of OFD can be categorized under the increasingly inclusive label of ciliopathy.7 In support of this notion, TCTN3 (MIM 613847), which is mutated in a subset of individuals with OFD, is also mutated in individuals with Meckel-Gruber-like phenotype.8 However, many OFD individuals remain molecularly undiagnosed, highlighting the need for further research into the genetics of this genetically heterogeneous condition.
In this study, we enrolled two multiplex Arab families in which all affected individuals have the core features of cleft palate, lobulated tongue, and polydactyly, in addition to variable expressivity of other clinical phenotypes (Figure 1 and Table 1). After obtaining written informed consent (King Faisal Specialist Hospital and Research Center institutional review board 2080006), blood samples in EDTA tubes were collected from all relevant family members for DNA extraction. Because both pedigrees are consistent with autosomal-recessive inheritance and because the parents in each family share a common set of ancestors, we hypothesized that OFD in each family is caused by a homozygous mutation that resides within an autozygous interval such that autozygosity mapping would reveal the disease locus. Therefore, we proceeded with autozygosity mapping essentially as described before.9 In brief, we performed genome-wide SNP genotyping on the Axiom platform according to the manufacturer’s protocol (Affymetrix) and subsequently carried out autozygome determination of each individual by using autoSNPa.10 In family 1, the two available cousins were found to share only two autozygous intervals corresponding to chr1: 192,263,777–203,181,377 and chr15: 42,273,528–45,568,618. On the other hand, affected members of family 2 were found to share only one interval corresponding to chr1: 197,301,687–204,123,671. Thus, the two families share one minimal autozygous interval corresponding to chr1: 197,262,220–201,811,027 (Figure S1, available online). Reassuringly, linkage analysis combining both families revealed a single peak with a LOD score of 5.8 (Figure 2). No known ciliopathy-linked gene was identified after the 26 genes within the interval were examined.
Figure 1.
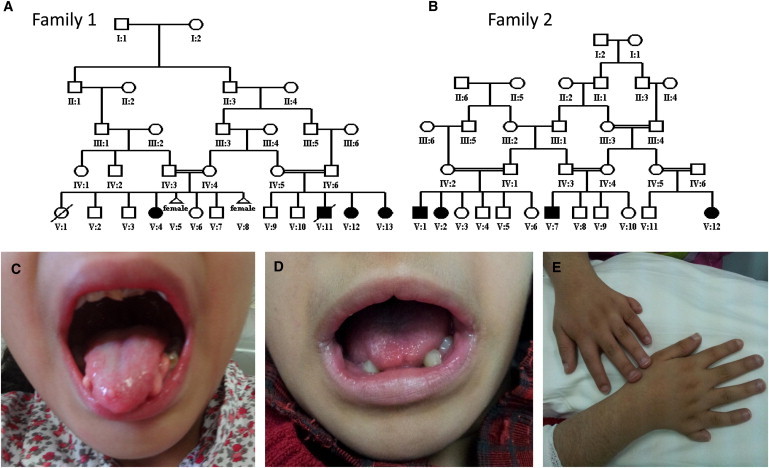
Identification of Two Multiplex Consanguineous Families Affected by OFD
(A and B) Pedigrees of families 1 (A) and 2 (B).
(C–E) Representative clinical images of lobulated tongue (C and D) and polydactyly (E). Please note the subtle midline lip defect in (D).
Table 1.
Clinical Characteristics of the Affected Members in Both Study Families
| Family | Individual | Palate | Tongue | Face | Digits | Heart | Cognitive | Others |
|---|---|---|---|---|---|---|---|---|
| 1 | V:4 | cleft palate | lobulation | frontal bossing, hypertelorism | polydactyly | TOF | ID | scoliosis, fused kidneys, ACC |
| 1 | V:13 | cleft palate | lobulation | frontal bossing, hypertelorism | polydactyly | VSD | ID | - |
| 2 | V:1 | bifid uvula | lobulation | frontal bossing, hypertelorism | polydactyly | - | ID | Hirschsprung disease |
| 2 | V:2 | bifid uvula | lobulation | frontal bossing, hypertelorism | polydactyly | - | ID | - |
| 2 | V:7 | bifid uvula | lobulation | frontal bossing, hypertelorism | polydactyly | - | ID | - |
| 2 | V:12 | bifid uvula | lobulation | frontal bossing, hypertelorism | polydactyly | - | ID | - |
Abbreviations are as follows: TOF, tetralogy of Fallot; ID, intellectual disability; ACC, agenesis of corpus callosum; and VSD, ventricular septal defect.
Figure 2.
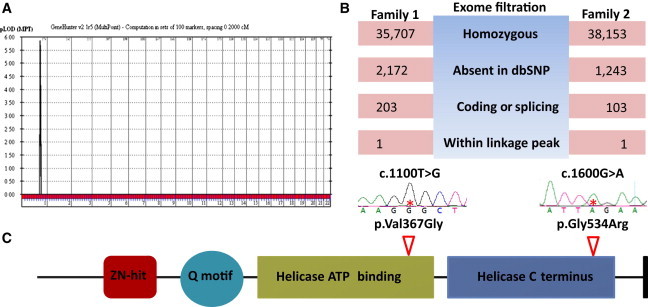
Identification of an OFD Locus in Chromosomal Region 1q32.1 and DDX59 Mutations Therein
(A) Linkage analysis combining the two families confirms the critical autozygosity interval in (A) with a LOD score of 5.8.
(B) Filtration strategy used for analyzing the exome results of families 1 and 2. Note that a single variant affecting DDX59 survived filtration in each family.
(C) Schematic of DDX59. The locations of the two alterations are indicated along with the sequence chromatograms.
The haplotype within the linkage interval was different between the two families, consistent with their different tribal origin in Arabia. Thus, we exome sequenced one affected individual from each family and filtered the resulting variants as follows: homozygous → absent in dbSNP132 → coding or splicing → within the linkage interval (Figure 2). Each of the two individuals was found to have one variant in DDX59 as the only variant that survived filtration: c.1100T>G (p.Val367Gly) in family 1 and c.1600G>A (p.Gly534Arg) in family 2 (RefSeq accession number NM_001031725.4) (Figure 2). Both variants survived the test of segregation with the disease within their respective pedigrees, are absent in 300 in-house ethnically matched exomes and 200 ethnically matched controls by direct sequencing, and are highly predicted to be pathogenic by PolyPhen-2 and SIFT (0.999 and 0.00 for p.Val367Gly and 1.00 and 0.00 for p.Gly534Arg, respectively). DDX59 encodes a member of a large family of DEAD-box-containing RNA helicases. The missense change p.Val367Gly affects a highly conserved ATP-binding domain, whereas p.Gly534Arg affects a highly conserved helicase domain, and both residues are conserved down to rice and Arabidopsis and in other human DDX proteins (Figure S2). In addition, immunoblot analysis of fibroblasts from one individual with p.Val367Gly revealed marked reduction in protein abundance, suggesting a quantitative defect in addition to the proposed qualitative defect (Figure S3). The above data highly suggest that the two variants are causally linked to OFD in the two families.
DEAD-box-containing RNA helicases are involved in virtually all aspects of RNA metabolism, including splicing, nuclear export, and ribosomal biogenesis, although the exact mechanism remains unclear, especially because it has been shown that these proteins can only unwind short segments of double-stranded RNA.11,12 It has also been shown that these helicases can displace proteins in RNP complexes, and some have selective subcellular localization that is thought to be relevant to their function, such as mitochondrial RNA metabolism.13–16 Targeted deletion of some of these genes in yeast was shown to be lethal but was rescued by the overexpression of other members, suggesting some level of redundancy.17 In humans, a bioinformatics search for all potential DEAD-box-containing RNA helicases revealed the presence of more than 40, but the function of many of these members, including DDX59, remains unknown.11
In order to gain insight into a potential mechanistic link between DDX59 mutations and OFD, we first analyzed its developmental expression pattern and the cellular localization of the protein it encodes. Using whole-mount in situ hybridization of the developing mouse palate, we showed that Ddx59 is highly enriched in the lips, palatal shelves (secondary palate), and developing limb buds (Figure 3). At the cellular level, immunofluorescence experiments using DDX59 antibody showed a granular cytoplasmic and nuclear localization, the latter of which was dynamic. This pattern was maintained even in fibroblasts from affected individual family 1_V:4 (Figure 4). The dynamic nuclear localization and the cytoplasmic localization of DDX59 suggest a diverse role in RNA biology and that this role might provide a mechanistic link between its deficiency and OFD, although the nature of this link will require further investigation.
Figure 3.
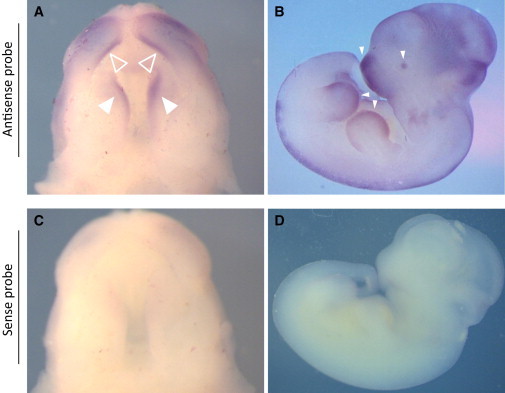
Ddx59 Is Expressed in the Developing Palate and Limb Buds of Mouse Embryos
(A and C) Whole-mount in situ hybridization on embryonic day (E) 13.5 palates shows a strong domain on expression of Ddx59 in the secondary palate (solid triangles) and lip (empty triangles) (A), but not in the sense control shown in (C).
(B and D) Expression in an E11.5 embryo is seen in the developing snout region, eye, and limb buds (solid triangles) (B), but not in the sense control shown in (D). The riboprobe corresponds to nucleotides c.942 to c.∗18 in the 3′ UTR (RefSeq NM_026500.3).
Figure 4.
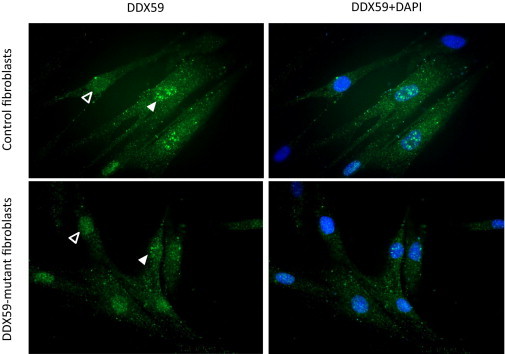
DDX59 Is Localized to the Nucleus and Cytoplasm
Granular pattern of both cytoplasmic and nuclear localization of DDX59 (Abcam ab69521) is observed in control and affected individual fibroblasts (family 1_V:4) with no clear difference. Note the dynamic nature of the granular nuclear localization pattern, indicated by its variable presence (solid triangles) and absence (empty triangles).
Given that the three genes linked to OFD to date appear to play a role in ciliary biology, we asked whether cells from affected individuals have any ciliary defect. Using primary fibroblasts from a skin biopsy of individual family 1_V:4, we conducted a stress-induced ciliogenesis assay as described before.18 After 72 hr of serum starvation, staining with acetylated alpha-tubulin antibody revealed normal ciliogenesis in nearly all control fibroblasts (Figure 5). The affected individual also appeared to have normally ciliated fibroblasts (Figure 5), suggesting that altered DDX59 is compatible with normal ciliogenesis. Indeed, DDX59 is not listed in the publically available ciliome databases, suggesting that it is not abundantly localized to the cilia. In order to investigate whether the normal-looking cilia in fibroblasts from individuals with DDX59 mutations are functionally normal, we set out to test SHH (sonic hedgehog) signaling by using GLI1 expression as a readout, a standard test of ciliary signaling.19 Upon stimulating fibroblasts with SAG (smoothened agonist), fibroblasts from individual family 1_V:4 showed significantly less SHH signaling than did control cells (p < 0.0017) (Figure 5). Finally, we fully sequenced DDX59 in four simplex OFD individuals without OFD1 mutations but did not identify any pathogenic mutations, suggesting that the contribution of DDX59 mutations to the overall mutation rate in OFD is not high, although larger cohorts will be needed for assessing this more accurately.
Figure 5.
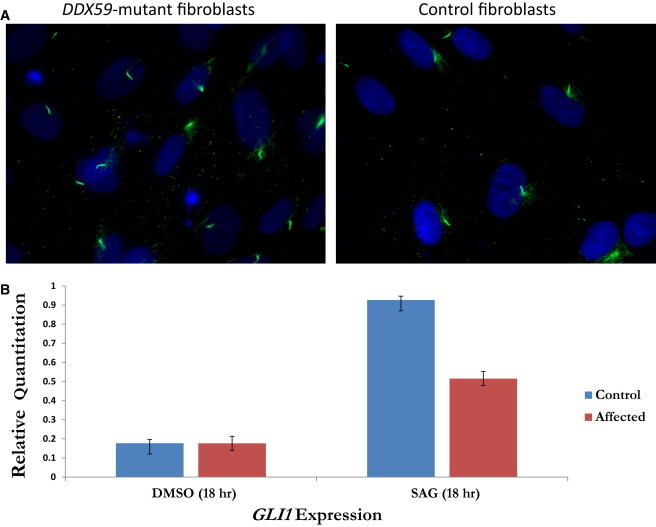
DDX59-Related OFD Is Not Associated with Ciliogenesis Defect but Is Associated with Impaired SHH Signaling
(A) Compared to control fibroblasts, fibroblasts from family 1_V:4 show normal ciliogenesis at 100× magnification.
(B) GLI1 expression is markedly lower in fibroblasts from family 1_V:4 than in control fibroblasts in response to SAG stimulation.
Taken together, our data show that mutations in DDX59 cause an autosomal-recessive form of OFD and probably converge with other causes of OFD on impaired ciliary signaling. It will be of interest in the future to investigate the overall contribution of DDX59 mutations to all recessive forms of OFD, as well as the exact mechanism through which defects in this RNA helicase causes OFD.
Acknowledgments
We thank the families for their enthusiastic participation. We also thank the Genotyping and Sequencing Core Facilities at King Faisal Specialist Hospital and Research Center for their technical help. This work was supported by King Abdulaziz City for Science and Technology grant 09-MED941-20 (to F.S.A.) and a Dubai Harvard Foundation for Medical Research Collaborative Research Grant (to F.S.A.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
MAFFT version 7, http://mafft.cbrc.jp/alignment/software/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
PolyPhen-2, www.genetics.bwh.harvard.edu/pph2/
SIFT, www.sift.jcvi.org/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Baraitser M. The orofaciodigital (OFD) syndromes. J. Med. Genet. 1986;23:116–119. doi: 10.1136/jmg.23.2.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Claussen O. Et arvelig syndrom omfattende tungemisdannelse og polydaktyli. Nord. Med. Tidskr. 1946;30:1147–1151. [Google Scholar]
- 3.Mohr O.L. A hereditary sublethal syndrome in man. Nor Vidensk Akad Oslo I Mat Naturv Klasse. 1941;14:3–18. [Google Scholar]
- 4.Papillon-Leage P.J., Psaume J. Dysmorphie des freins buccaux. Actual. Odontostomatol. (Paris) 1954;8:7–26. [PubMed] [Google Scholar]
- 5.Gurrieri F., Franco B., Toriello H., Neri G. Oral-facial-digital syndromes: review and diagnostic guidelines. Am. J. Med. Genet. A. 2007;143A:3314–3323. doi: 10.1002/ajmg.a.32032. [DOI] [PubMed] [Google Scholar]
- 6.Ferrante M.I., Giorgio G., Feather S.A., Bulfone A., Wright V., Ghiani M., Selicorni A., Gammaro L., Scolari F., Woolf A.S. Identification of the gene for oral-facial-digital type I syndrome. Am. J. Hum. Genet. 2001;68:569–576. doi: 10.1086/318802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Valente E.M., Logan C.V., Mougou-Zerelli S., Lee J.H., Silhavy J.L., Brancati F., Iannicelli M., Travaglini L., Romani S., Illi B. Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat. Genet. 2010;42:619–625. doi: 10.1038/ng.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomas S., Legendre M., Saunier S., Bessières B., Alby C., Bonnière M., Toutain A., Loeuillet L., Szymanska K., Jossic F. TCTN3 mutations cause Mohr-Majewski syndrome. Am. J. Hum. Genet. 2012;91:372–378. doi: 10.1016/j.ajhg.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alkuraya F.S. Discovery of rare homozygous mutations from studies of consanguineous pedigrees. Curr. Protoc. Hum. Genet. 2012;Chapter 6:12. doi: 10.1002/0471142905.hg0612s75. [DOI] [PubMed] [Google Scholar]
- 10.Carr I.M., Flintoff K.J., Taylor G.R., Markham A.F., Bonthron D.T. Interactive visual analysis of SNP data for rapid autozygosity mapping in consanguineous families. Hum. Mutat. 2006;27:1041–1046. doi: 10.1002/humu.20383. [DOI] [PubMed] [Google Scholar]
- 11.Linder P. Dead-box proteins: a family affair—active and passive players in RNP-remodeling. Nucleic Acids Res. 2006;34:4168–4180. doi: 10.1093/nar/gkl468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abdelhaleem M., Maltais L., Wain H. The human DDX and DHX gene families of putative RNA helicases. Genomics. 2003;81:618–622. doi: 10.1016/s0888-7543(03)00049-1. [DOI] [PubMed] [Google Scholar]
- 13.Schmidt U., Lehmann K., Stahl U. A novel mitochondrial DEAD box protein (Mrh4) required for maintenance of mtDNA in Saccharomyces cerevisiae. FEMS Yeast Res. 2002;2:267–276. doi: 10.1016/S1567-1356(02)00109-5. [DOI] [PubMed] [Google Scholar]
- 14.Séraphin B., Simon M., Boulet A., Faye G. Mitochondrial splicing requires a protein from a novel helicase family. Nature. 1989;337:84–87. doi: 10.1038/337084a0. [DOI] [PubMed] [Google Scholar]
- 15.Jankowsky E., Fairman M.E. Duplex unwinding and RNP remodeling with RNA helicases. Methods Mol. Biol. 2008;488:343–355. doi: 10.1007/978-1-60327-475-3_22. [DOI] [PubMed] [Google Scholar]
- 16.Jankowsky E., Bowers H. Remodeling of ribonucleoprotein complexes with DExH/D RNA helicases. Nucleic Acids Res. 2006;34:4181–4188. doi: 10.1093/nar/gkl410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jamieson D.J., Beggs J.D. A suppressor of yeast spp81/ded1 mutations encodes a very similar putative ATP-dependent RNA helicase. Mol. Microbiol. 1991;5:805–812. doi: 10.1111/j.1365-2958.1991.tb00753.x. [DOI] [PubMed] [Google Scholar]
- 18.Shaheen R., Faqeih E., Shamseldin H.E., Noche R.R., Sunker A., Alshammari M.J., Al-Sheddi T., Adly N., Al-Dosari M.S., Megason S.G. POC1A truncation mutation causes a ciliopathy in humans characterized by primordial dwarfism. Am. J. Hum. Genet. 2012;91:330–336. doi: 10.1016/j.ajhg.2012.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caspary T., Larkins C.E., Anderson K.V. The graded response to Sonic Hedgehog depends on cilia architecture. Dev. Cell. 2007;12:767–778. doi: 10.1016/j.devcel.2007.03.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.