

Abstract
Hereditary hemorrhagic telangiectasia (HHT), the most common inherited vascular disorder, is caused by mutations in genes involved in the transforming growth factor beta (TGF-β) signaling pathway (ENG, ACVRL1, and SMAD4). Yet, approximately 15% of individuals with clinical features of HHT do not have mutations in these genes, suggesting that there are undiscovered mutations in other genes for HHT and possibly vascular disorders with overlapping phenotypes. The genetic etiology for 191 unrelated individuals clinically suspected to have HHT was investigated with the use of exome and Sanger sequencing; these individuals had no mutations in ENG, ACVRL1, and SMAD4. Mutations in BMP9 (also known as GDF2) were identified in three unrelated probands. These three individuals had epistaxis and dermal lesions that were described as telangiectases but whose location and appearance resembled lesions described in some individuals with RASA1-related disorders (capillary malformation-arteriovenous malformation syndrome). Analyses of the variant proteins suggested that mutations negatively affect protein processing and/or function, and a bmp9-deficient zebrafish model demonstrated that BMP9 is involved in angiogenesis. These data confirm a genetic cause of a vascular-anomaly syndrome that has phenotypic overlap with HHT.
Main Text
Hereditary hemorrhagic telangiectasia (HHT [MIM 187300 and 600376]) is an autosomal-dominantly inherited vascular-malformation syndrome characterized by telangiectases and arteriovenous malformations (AVMs) and has an incidence of 1 in 10,000 individuals.1 Hallmark features are recurrent epistaxis due to telangiectases of the nasal mucosa; telangiectases on the lips, hands and oral mucosa; solid-organ AVMs, particularly of the lungs, liver, and brain; and a family history of the same. Presentation with three of these criteria is considered diagnostic for HHT.2 The dermal telangiectases are typically pinpoint to pinhead sized, very specifically concentrated on the hands, face and lips, and not diffuse. Telangiectases on the limbs and trunk are not characteristic.
Currently, all known genetic defects that cause HHT are found within the transforming growth factor beta (TGF-β) signaling pathway. Mutations in endoglin (ENG [MIM 131195]), activin A receptor type II-like 1 (ACVRL1, also known as ALK1 [MIM 601284]), and SMAD4 (MIM 600993) cause HHT type 1, HHT type 2, and the combined juvenile polyposis (JP) and HHT (JP-HHT) syndrome, respectively.3–5 Approximately 15% of individuals identified clinically as having HHT currently have no known genetic cause,6 suggesting that there are undiscovered genes associated with HHT.
Dermal telangiectases and cerebral AVMs are also features of capillary-malformation (CM)-AVM syndrome (CM-AVM [MIM 608354]), caused by mutations in RASA1 (MIM 139150).7–10 Recurrent nosebleeds have not been described, and the typical dermal telangiectases generally differ from HHT in location and appearance. The telangiectases seen in CM-AVM include both the small punctate lesions that characterize HHT and the larger telangiectases referred to as CMs. The punctate telangiectases in CM-AVM are more commonly diffuse and tend to cluster in a region of the trunk or limbs (M. Vikkula, personal communication).
We first performed exome sequencing in 38 unrelated individuals reported to have HHT2 (by physicians who had ordered HHT genetic testing) but in whom no mutation had been identified in ENG, ACVRL1 or SMAD4. Informed consent was obtained from all individuals (approved by the institutional review board [00028740] at the University of Utah). Samples were enriched with the use of exome-targeted biotinylated RNA baits (SureSelect 50 Mb) and sequenced with 2× 100 bp paired-end reads (HiSeq2000, Illumina). The Burrows-Wheeler Aligner (0.5.9)11 and the Genome Analysis Toolkit (v.1.6)12 were used for data analysis. To reduce the number of false-positive variant calls, we used raw variant sets as input to a Variant Quality Score Recalibration procedure, and we discarded resulting variants with VQRLOD scores less than −10.0.
We prioritized variants by subjecting them to a two-part classification procedure. First, we computed SIFT,13 PolyPhen-2,14 MutationTaster,15 GERP++,16 PhyloP,17 LRT,18 and SiPhy19 scores for all nonsynonymous variants. We computed scores by consulting a precomputed database of all possible scores distributed with the dbNSFP package, version 2.0.20 We combined these scores into a single value by using a linear weighting scheme, and this value reflected the overall damage to the gene in question. Next, we employed phenotypic ranking by using the VarRanker algorithm (B.O., unpublished data).
Pathogenic missense variants were identified in BMP9 (MIM 605120) in 2 of the 38 samples: c.254C>T (p.Pro85Leu) and c.203G>T (p.Arg68Leu) (Figures S1A and S1B, available online, and Table 1) (RefSeq accession number NM_016204.1). Neither BMP9 variant was present in publically available databases, the affected residues were highly conserved (from humans to zebrafish), and their corresponding mutations were predicted to be damaging by computing algorithms (Table S1).13–15
Table 1.
BMP9 Variants and Case Phenotypes
| Individual | Clinical Description | Nucleotide Change | Protein Change | Presence in Populationa | Family Segregation | Variant Classification |
|---|---|---|---|---|---|---|
| 1 | epistaxis, telangiectasia, first-degree relative reported to have HHT | c.254C>T | p.Pro85Leu | absent | NA | pathogenic |
| 2 | epistaxis, telangiectasia or CM (back, shoulders, mouth, face), AVM (thumb), parent and sibling with history of epistaxis | c.203G>T | p.Arg68Leu | absent | yesb | pathogenic |
| 3 | epistaxis, telangiectasia (80–100 on the left arm and shoulder, 2 on the right palm, 1 possible on the lip); a parent reports frequent epistaxis as a child and teen | c.997C>Tc | p.Arg333Trpc | 0.004 | NA | suspected to be pathogenic |
| 4 | CM and hemihypertrophy | c.−51C>Ac | 5′ UTRc | absent | NA | suspected to be benign |
All nucleotide changes were heterozygous. BMP9 RefSeq accession number NM_016204 was used. “NA” stands for not available.
Presence in population refers to the presence of the variant in 1000 Genomes, dbSNP, or 5,400 control exomes.
Father and sister with epistaxis also carry the mutation.
Identified by Sanger sequencing.
Next, BMP9 exons were Sanger sequenced in an additional 153 unrelated individuals who had been referred for molecular genetic testing as a result of suspicion of HHT by a referring physician, although most (86%) met two of the established diagnostic criteria for HHT and the other 14% met three diagnostic criteria. Another variant (c.997C>T [p.Arg333Trp]) was identified in one individual within this group (Figure S1C and Table 1). Although this variant has been previously reported in dbSNP (rs35129734), it is potentially disease causing because it is very rare (variant frequency of 0.004), the affected residue is strictly conserved among higher vertebrates, and the variant is predicted to be deleterious by SIFT,13 PolyPhen-2,14 and MutationTaster.15
All three individuals identified with a BMP9 mutation had dermal lesions described as telangiectases, yet a detailed review of the location and appearance of their lesions suggested a cutaneous phenotype that differs from classic HHT and has some resemblance to lesions described in individuals with RASA1-related disorders (CM-AVM). Therefore, BMP9 exons were also Sanger sequenced for 60 unrelated individuals who had been clinically suspected of having a mutation in RASA1 by their referring physician but in whom no mutation was identified in this gene. One variant not present in publically available databases was identified in the BMP9 5′ UTR region (c.−51C>A) in one individual (Table 1), who had macrocephaly, extensive CMs, and hemihypertrophy. Given that the individual’s unaffected mother had the same 5′ UTR variant, it is not likely that this variant causes the individual’s phenotype.
Individual 1 (c.254C>T [p.Pro85Leu]) is a 33-year-old female whose onset of epistaxis occurred in early childhood. She reports that her nosebleeds were the most significant during childhood—they occurred two to six times a week—but were still significant enough in her twenties to warrant treatment by nasal cautery. She reported that she currently has approximately three nosebleeds a week but that they are minor in duration relative to how they were during childhood. She had several cutaneous telangiectases on her chest and face. No solid-organ AVMs were known, but she had not been screened for solid-organ AVMs. Her father died suddenly at the age of 45 years, and according to the family, HHT was suspected on the basis of autopsy findings. Unfortunately, this report was not available to confirm details, but it was the suggestion of HHT in her father years ago that led this individual to seek consultation regarding her epistaxis and cutaneous lesions. A sibling had recurrent spontaneous nosebleeds (approximately two per week) and a stroke of unknown etiology at the age of 43 years. No family members were available for examination or variant analysis.
Individual 2 (c.203G>T [p.Arg68Leu]) is a 37-year-old female whose onset of epistaxis occurred at approximately 30 years of age. The frequency of her nosebleeds varied from daily to once every 1–2 weeks. The individual had 15–20 dermal vascular malformations on her face, mouth, upper chest, upper mid back, and hands, and they were first noticed when she was about 30 years old (Figure 1). The individual reported that the larger malformations had faded from a darker to lighter pink over time. An MRI of the brain did not show AVMs. Her father and sister reported infrequent but recurring nosebleeds, but they were not available for clinical examination. DNA was obtained from saliva, and the BMP9 c.203G>T (p.Arg68Leu) variant was identified in both the father and the sister.
Figure 1.
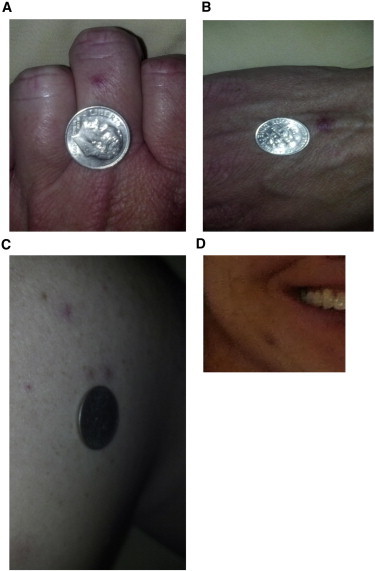
Cutaneous Vascular Lesions in Individual 2
Shown are the middle digit of the left hand (A), the dorsal aspect of the right hand (B), several of approximately 20 lesions on the back and shoulders (C), and a vascular lesion previously treated by laser ablation on the right jawline (D).
Individual 2 had abnormal liver enzymes and portal hypertension. A liver MRI showed findings suggestive of the hepatic vascular findings seen in HHT. In particular, the arterial phase was slightly heterogeneous, and several smaller branches of the hepatic artery were prominent and slightly corkscrew in configuration. Several small focal enhancing nodules were noted on the arterial phase that equilibrated in other phases and were not visible on other sequences, suggesting small focal nodular hyperplasia. However, this individual had also been diagnosed with hepatopulmonary syndrome, and the cause of her hepatic issues is not clear.
Individual 3 (c.997C>T [p.Arg333Trp]) is a 14-year-old male whose onset of epistaxis occurred at approximately 3 years of age. Frequency of epistaxis had ranged from approximately once per week to several times per day. He had 80–100 telangiectases on his left upper extremity from wrist to shoulder, two telangiectases on the palm of his right hand, and one possible telangiectasia on his lower lip. The pathology report of a biopsy of the lesions confirmed them to be telangiectases. No solid-organ involvement (AVM) was known, but he was not screened by the referring physician. Per family history report, his father had nosebleeds of more than once per week in elementary school until his early teens, but none in recent years. DNA was not available from the father for variant analysis.
To characterize the functional effects of the three BMP9 variants identified, we evaluated BMP9 levels and processing into the mature, active form of the protein21 (Figure 2A). Comparison of plasmid-transfected human embryonic kidney (HEK) EBNA cell lysates and supernatants by immunoblot using an antibody specific to mature BMP9 revealed that the processing of the p.Arg68Leu and p.Arg333Trp variants to mature BMP9 was far less efficient than that of the wild-type (WT) BMP9, whereas processing of the p.Pro85Leu variant was less impaired (Figure 2B). This reduction of mature BMP9 in the supernatants was later confirmed with a specific ELISA in HEK cells (Figure 2C), as well as in the hepatoma cell line HepG2 (Figure S2). Importantly, control culture supernatants from HepG2 cells showed only low background levels of BMP9 (<10 pg/ml) (data not shown). Thus, all three variants altered BMP9 processing in vitro. Unfortunately, serum samples were not available for testing BMP9 levels in these individuals.
Figure 2.
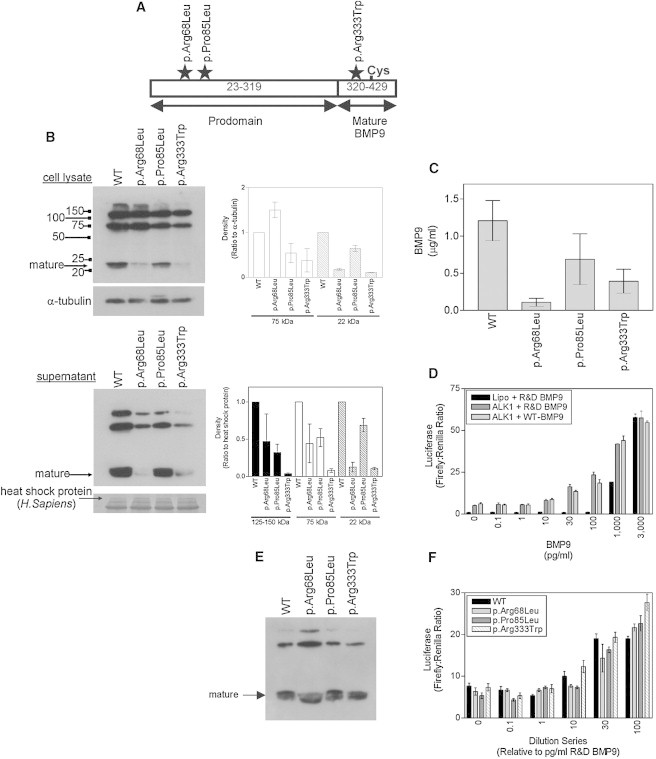
BMP9 Variants Alter Protein Processing and Binding of Ligands to ALK1
(A) BMP9 diagram depicting the alteration locations, prodomain, and mature protein. The BMP9 precursor encodes a signal sequence from amino acids 1–22, a prodomain from amino acids 23–319 (proBMP9), and the mature BMP9 from amino acids 320–429. Secreted BMP9 comprises amino acids 23–429; furin-type proteases cleave its prodomain to generate mature BMP9.21
(B) Plasmids encoding human WT BMP9 or the p.Arg68Leu, p.Pro85Leu, or p.Arg333Trp variants were transfected into HEK EBNA cells. Cell lysates and conditioned media were fractionated with nonreducing SDS-PAGE and immunoblotted for BMP9 with a mouse monoclonal BMP9 antibody (clone #360107, R&D Systems). For cell lysates, equal protein loading was confirmed by subsequent immunoblotting for α-tubulin and densitometry of the bands normalized to the α-tubulin bands. Supernatants were also fractionated and immunoblotted for BMP9. A parallel gel was run and stained with Coomassie blue. For verification, the Coomassie-stained bands were sequenced. WT and variant BMP9 proteins fractionated as three bands corresponding to the mature BMP9 dimer associated with both prodomains (∼125–150 kDa), the mature BMP9 dimer associated with a single prodomain (75 kDa), and mature BMP9 (22 kDa). Mature BMP9 is indicated by an arrow. Densitometry graphs are also shown.
(C) BMP9 levels were determined by a specific ELISA (n = 3). The ELISA was developed with a colorimetric substrate comprising 1 mg/ml 4-nitrophenyl phosphate disodium salt hexahydrate in 1 M diethanolamine (pH 9.8) containing 0.5 mM MgCl2. The assay was developed in the dark at room temperature, and the absorbance was measured at 405 nm. Unknown values were extrapolated from the standard curve with a 4-parameter log curve fit.
(D) C2C12 cells were transfected with human ALK1 and treated with R&D Systems BMP9 (between 0.1 and 3,000 pg/ml) or WT BMP9 diluted to equivalent activities. Cells exposed to Lipofectamine (“Lipo”) alone were treated in parallel with R&D Systems BMP9. All cells were cotransfected with BRE-luciferase and pTK-Renilla. BMP9-driven firefly luciferase responses were normalized to Renilla activity. Data are the mean ± SEM (n = 4 wells) for a representative experiment. The activity of the WT protein was titrated in comparison to commercial BMP9 for the establishment of an activity profile. C2C12 cells transfected with ALK1 became sensitive to commercial BMP9 (mature BMP9, amino acids 320–429) at concentrations as low as 1 pg/ml.
(E) Supernatants from (B) were diluted so that there were equal amounts of mature BMP9 in the media conditioned with WT and variant BMP9. The activity profiles for WT BMP9 were similar to the commercial BMP9 profile, albeit with reduced activity at the lowest concentrations of 1 and 10 pg/ml. For each variant, relative supernatant volumes necessary for achieving equivalent amounts of the mature WT BMP9 were determined.
(F) C2C12 cells were transfected with human ALK1 and treated with WT or variant BMP9. Cells exposed to Lipofectamine alone were treated in parallel with WT BMP9. All cells were cotransfected with BRE-luciferase and pTK-Renilla. BMP9-driven firefly luciferase responses were normalized to Renilla activity. The range over which ALK1 activation occurs is indicated. Data are the mean ± SEM for n = 4 wells from a representative experiment. Top dilutions were prepared at 1 (WT) to 8.3 (p.Arg68Leu) to 1.3 (p.Pro85Leu) to 4 (p.Arg333Trp), and serial dilutions of each BMP9 variant were prepared with the same dilution series as with commercial BMP9. Assays of these BMP9 variants in C2C12 cells transfected with ALK1 revealed that the p.Arg68Leu and p.Pro85Leu variants exhibited less activity than WT BMP9 at 10 pg/ml (p.Arg68Leu: 79.2% ± 10.3% of WT; p.Pro85Leu: 82.2% ± 7.6% of WT) and 30 pg/ml (p.Arg68Leu: 78.9% ± 4.3% of WT; p.Pro85Leu: 78.6% ± 7.2% of WT) equivalent dilutions, but the p.Arg333Trp variant did not (10 pg/ml equivalent: 100.92% ± 11.8% of WT; 30 pg/ml equivalent: 99.6% ± 2.1% of WT).
To assess the relative activities of the WT and variant forms of BMP9, we examined their ability to induce BRE-luciferase activity in C2C12 cells transfected with human ALK1 (Figures 2D–2F). Assays of these BMP9 variants in C2C12 cells transfected with ALK1 revealed that the p.Arg68Leu and p.Pro85Leu variants exhibited less activity than did the WT BMP9 at 10 pg/ml (p.Arg68Leu: 79.2% ± 10.3% of WT; p.Pro85Leu: 82.2% ± 7.6% of WT) and 30 pg/ml (p.Arg68Leu: 78.9% ± 4.3% of WT; p.Pro85Leu: 78.6% ± 7.2% of WT) equivalent dilutions, but the p.Arg333Trp variant did not (10 pg/ml equivalent: 100.92% ± 11.8% of WT; 30 pg/ml equivalent: 99.6% ± 2.1% of WT) (Figure 2F). No luciferase activity was detected in the conditioned medium from HEK EBNA cells transfected with the empty pCEP4 vector (data not shown).
We also tested the biological activity of the BMP9 variants by measuring production of alkaline phosphatase in the ATDC5 mouse chondrogenic cell line. Alkaline phosphatase is a widely accepted marker of the signaling pathway induced by different BMPs and by BMP9 in particular.22 At the same protein concentration (1 ng/ml), the p.Arg68Leu, p.Pro85Leu, and p.Arg333Trp BMP9 variants showed less activity than did the WT protein (Figure 3). Taken together, these experiments demonstrate that the three BMP9 variants affect the biological activity of BMP9.
Figure 3.
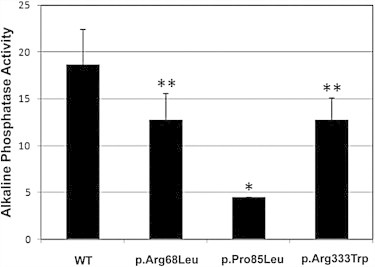
Missense Substitutions in BMP9 Alter Protein Activity
The mouse chondrogenic cell line ATDC5 was incubated with 1 ng/ml of WT BMP9 or the p.Arg68Leu, p.Pro85Leu, or p.Arg333Trp variants produced in HepG2 cells, as shown in Figure S2. Alkaline phosphatase activity was measured in permeabilized ATDC5 cells with the use of p-nitrophenyl phosphate as a soluble substrate. Relative activity units are shown in the vertical axis. ∗p < 0.001; ∗∗p < 0.07.
Next, we performed bmp9 knockdown experiments in zebrafish to identify whether they would exhibit a vascular phenotype similar to HHT. Tg(kdrl:egfp)la116;Tg(gata1.DsRed2)sd2 zebrafish embryos23,24 were injected at the 1- to 4-cell stage with 7 ng bmp9 translation-blocking morpholino (5′-GGAGCAAATGTCCTACGCGCCACAT-3′) or standard control morpholino (GeneTools). Compared to control morphants, bmp9 morphants exhibited small but significant decreases in both anterior-posterior and dorsal-ventral axes, but trunk and tail anatomy were otherwise normal (Figures 4A and 4B). Vascular patterning in bmp9 morphants was relatively normal, although subtle defects were detected in maturation of the caudal vein. Although all control morphants exhibited a dominant ventral-vein return by 2 days postfertilization (dpf) (n = 86/86), in bmp9 morphants the caudal venous plexus failed to resolve and both dorsal and ventral veins continued to carry blood flow (n = 76/91, 83.5%; Figures 4C and 4D). This phenotype persisted as late as 5 dpf, suggesting that it was not the result of developmental delay but instead reflected impaired remodeling. Repeating the knockdown of bmp9 with a splice-blocking morpholino resulted in the same venous remodeling defect (data not shown), supporting a role for BMP9 in angiogenesis. In zebrafish, alk1 mutations result in robust cranial AVMs.25,26 However, we detected no cranial AVMs in bmp9 morphants (n = 0/207), suggesting that other ligands such as BMP10 might compensate for BMP9 in the development of these cranial vessels (B.R., unpublished results).
Figure 4.
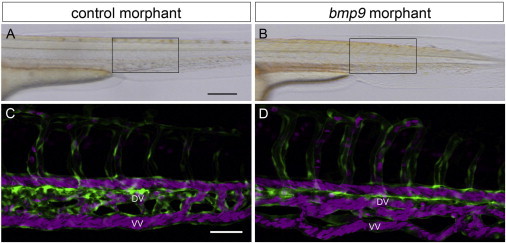
bmp9 Morphant Zebrafish Display Impaired Venous Remodeling
(A and B) Bright-field images of live embryos; boxed areas in (A) and (B) designate regions of focus in (C) and (D), respectively. The scale bar represents 200 μm.
(C and D) Two-photon confocal images of circulation in the caudal vein plexus at 2 dpf. Endothelial cells are green, and red blood cells are magenta. The scale bar represents 50 μm. Abbreviations are as follows: DV, dorsal vein; and VV, ventral vein.
Zebrafish Tg(kdrl:egfp);Tg(gata1:dsRed2) embryos were injected at the 1- to 4-cell stage with 7 ng control morpholino (A and C) or 7 ng bmp9 morpholino (B and D) and imaged at 2 dpf. Embryos were imaged with a Leica TCS SP5 confocal microscope (Leica Microsystems) outfitted with an APO L 20×/1.00 water-immersion objective, standard visible lasers, and Mai Tai DeepSee IR laser (Spectra-Physics, Newport Corp.). EGFP was imaged at 900 nm, and DsRed2 was imaged at 561 nm. Z-series were collected at 2 μm intervals, and two-dimensional projections were generated with MetaMorph 7.7 (Molecular Devices). All images show left anterior lateral views.
Overall, three missense variants in BMP9 were identified in 191 individuals with features of HHT. These three amino acids are conserved from humans through zebrafish, predicted to be damaging, and shown by in vitro studies to alter BMP9 processing to the mature form and BMP9 activity to varying degrees (Figures 2 and 3). Furthermore, knockdown of bmp9 in zebrafish resulted in defects in venous remodeling (Figure 4), supporting a role for BMP9 in angiogenesis. This is consistent with the reported role for BMP signaling during vascular development: blocking BMP9 with a neutralizing antibody in newborn mice significantly increased retinal vascular density.27 Surprisingly, Bmp9-knockout mice did not show any vascular phenotype in retinal vascularization, and no abnormal retinal vascularization was noted in the three individuals with BMP9 variants. Furthermore, BMP9 specifically plays a physiologic role in the control of adult blood-vessel maintenance and angiogenesis by targeting ALK1 on endothelial cells.28
BMP9 exerts its functional effects by binding to specific endothelial cell surface receptors, namely the auxiliary receptor endoglin and the serine-threonine kinase ALK1, both members of the highly conserved TGF-β superfamily. Then, the BMP9-dependent activation of ALK1 leads to the phosphorylation of Smad1, Smad5, and Smad8, and the resulting phospho-Smad proteins associate with Smad4 to form a Smad complex that translocates to the nucleus to regulate gene expression in human microvascular endothelial cells. In addition, ALK1 activation by BMP9 further increases by overexpression of the coreceptor endoglin.28 Furthermore, BMP9 signaling via endoglin promotes a switch from a chemokine-responsive autocrine phenotype to a chemokine-nonresponsive paracrine state that represses endothelial cell migration and promotes vessel maturation.29 These data strongly suggest the involvement of BMP9 in a common TGF-β signaling pathway shared by ALK1, endoglin, and Smad4. Because mutations in ENG, ALK1, and SMAD4 give rise to different types of HHT (HHT1, HHT2, and JP-HHT, respectively), it can be postulated that alterations in BMP9, the upstream component of this signaling route, cause a vascular-malformation syndrome with phenotypic overlap with HHT (Figure 5). Supporting this view, many of the mutations described in ENG involve its orphan domain, a region responsible for binding to BMP9.30–33
Figure 5.
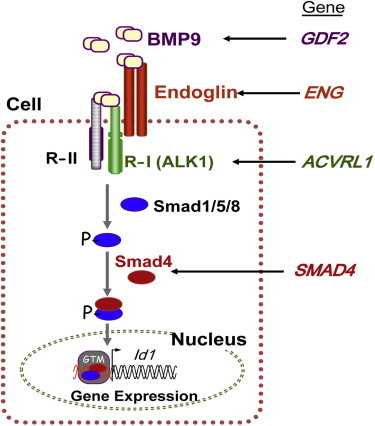
BMP9 and the TGF-β Signaling Pathway
BMP9 binds to specific type I and type II cell-surface receptors (R-I and R-II, respectively) that exhibit serine-threonine kinase activity, as well as to the auxiliary receptor endoglin. Upon ligand binding, R-II transphosphorylates ALK1 (R-I), which then propagates the signal by phosphorylating receptor-regulated (R-Smads) Smad1, Smad5, and Smad8 (“Smad1/5/8”). Once phosphorylated, R-Smads form heteromeric complexes with a cooperating homolog, Smad4, and translocate into the nucleus where they regulate the transcriptional activity of target genes, including Id1. Endoglin, ALK1, and Smad4 are encoded by ENG, ACVRL1 and SMAD4, respectively, whose pathogenic mutations give rise to HHT1, HHT2, and JP-HHT, respectively. BMP9 is encoded by GFDF2, whose pathogenic variants are described here. The following abbreviation is used: GTM, general transcription machinery. This figure was adapted from Figure 2 in Fernández et al.30
We observed that the p.Arg68Leu and p.Arg333Trp variants dramatically affected the degree of processing in the HEK EBNA expression system and thus reduced the amount of mature BMP9 that was being produced inside the expressing cells. Furthermore, the secretion of the mature and proBMP9 forms of the p.Arg68Leu and p.Arg333Trp variants was dramatically reduced. Intriguingly, the p.Pro85Leu variant also exhibited reduced levels of the proBMP9 form, but the processing was less impaired than in the other two variants. In addition to displaying changes in processing, the p.Arg68Leu and p.Pro85Leu BMP9 variants exhibited slightly reduced activity with ALK1. A reduction in activity is consistent with these mutations’ being in a region that might interact with ALK1.21 We did not observe reduced activity of the p.Arg333Trp variant in our C2C12 activity assay, and we observed only a slight reduction in activity of the same variant in ATDC5 cells (Figure 3). This mutation might affect a region that binds to type II or type III receptors, including endoglin. In summary, it is likely that the different BMP9 variants each affect particular aspects of BMP9 processing, secretion, and receptor activity and thus lead to reduced functionality of BMP9.
In our cohort of 191 individuals suspected to have HHT on examination by a referring physician, 3 (1.6%, or 0.24% of the total number of individuals with suspected HHT) of them were found to have a BMP9 variant. These three individuals all had epistaxis and dermal lesions described as telangiectases; however, the lesions were less concentrated on the hands and mouth than in individuals with mutations in ENG, ACVRL1, or SMAD4. In individual 2, the size and appearance of some lesions were more similar to the “atypical CM” seen in CM-AVM. In individual 3, the lesions were more punctate and resembled those in HHT in appearance, but the number and distribution were more typical of telangiectasia described in CM-AVM. The variants were shown to be present in two symptomatic first-degree relatives of one index case. However, no family members were available for examination by our group.
Molecular screening of BMP9 in individuals suspected to have HHT should be considered in individuals with epistaxis and small blanching vascular lesions on the upper body and trunk rather than limited to the hands, face, and mouth (as is typical of HHT). It is of note that the individuals in this study, and many with HHT, have relatively infrequent, mild nosebleeds not routinely reported to physicians during medical history. Because of the small number of individuals presented here and the fact that evaluation for solid-organ AVMs was not routinely performed, little can be concluded about the presence or absence of internal AVMs in this disorder. However, in the one individual in which imaging of solid organs (brain, lungs, and liver) was performed, liver findings similar to those seen in HHT were noted. Interestingly, BMP9 has been reported to be predominantly expressed by the hepatocytes,34 suggesting that the liver vasculature is a potential target of BMP9 deficiency.
Continued molecular analysis and clinical description in families affected by various patterns of CM and/or telangiectases will lead to continued expansion and refinement of hereditary conditions characterized by cutaneous vascular lesions. Both will help guide proper surveillance and possible interventions for this group of individuals.
Acknowledgments
We thank the families and individuals who participated in this study. This work was funded by the Associated Regional and University Pathologists Institute for Clinical and Experimental Pathology, a National Institutes of Health grant R01 (HL079108 to B.L.R.), a British Heart Foundation Programme grant (RG/08/002/24718 to N.W.M), and the Ministerio de Economia y Competitividad of Spain (SAF2010-19222 to C.B.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Richards-Yutz J., Grant K., Chao E.C., Walther S.E., Ganguly A. Update on molecular diagnosis of hereditary hemorrhagic telangiectasia. Hum. Genet. 2010;128:61–77. doi: 10.1007/s00439-010-0825-4. [DOI] [PubMed] [Google Scholar]
- 2.Shovlin C.L., Guttmacher A.E., Buscarini E., Faughnan M.E., Hyland R.H., Westermann C.J., Kjeldsen A.D., Plauchu H. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome) Am. J. Med. Genet. 2000;91:66–67. doi: 10.1002/(sici)1096-8628(20000306)91:1<66::aid-ajmg12>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 3.McDonald M.T., Papenberg K.A., Ghosh S., Glatfelter A.A., Biesecker B.B., Helmbold E.A., Markel D.S., Zolotor A., McKinnon W.C., Vanderstoep J.L. A disease locus for hereditary haemorrhagic telangiectasia maps to chromosome 9q33-34. Nat. Genet. 1994;6:197–204. doi: 10.1038/ng0294-197. [DOI] [PubMed] [Google Scholar]
- 4.Johnson D.W., Berg J.N., Gallione C.J., McAllister K.A., Warner J.P., Helmbold E.A., Markel D.S., Jackson C.E., Porteous M.E., Marchuk D.A. A second locus for hereditary hemorrhagic telangiectasia maps to chromosome 12. Genome Res. 1995;5:21–28. doi: 10.1101/gr.5.1.21. [DOI] [PubMed] [Google Scholar]
- 5.Gallione C.J., Repetto G.M., Legius E., Rustgi A.K., Schelley S.L., Tejpar S., Mitchell G., Drouin E., Westermann C.J., Marchuk D.A. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4) Lancet. 2004;363:852–859. doi: 10.1016/S0140-6736(04)15732-2. [DOI] [PubMed] [Google Scholar]
- 6.Gedge F., McDonald J., Phansalkar A., Chou L.S., Calderon F., Mao R., Lyon E., Bayrak-Toydemir P. Clinical and analytical sensitivities in hereditary hemorrhagic telangiectasia testing and a report of de novo mutations. J. Mol. Diagn. 2007;9:258–265. doi: 10.2353/jmoldx.2007.060117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Revencu N., Boon L.M., Mulliken J.B., Enjolras O., Cordisco M.R., Burrows P.E., Clapuyt P., Hammer F., Dubois J., Baselga E. Parkes Weber syndrome, vein of Galen aneurysmal malformation, and other fast-flow vascular anomalies are caused by RASA1 mutations. Hum. Mutat. 2008;29:959–965. doi: 10.1002/humu.20746. [DOI] [PubMed] [Google Scholar]
- 8.Eerola I., Boon L.M., Mulliken J.B., Burrows P.E., Dompmartin A., Watanabe S., Vanwijck R., Vikkula M. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am. J. Hum. Genet. 2003;73:1240–1249. doi: 10.1086/379793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boon L.M., Mulliken J.B., Vikkula M. RASA1: variable phenotype with capillary and arteriovenous malformations. Curr. Opin. Genet. Dev. 2005;15:265–269. doi: 10.1016/j.gde.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 10.Hershkovitz D., Bercovich D., Sprecher E., Lapidot M. RASA1 mutations may cause hereditary capillary malformations without arteriovenous malformations. Br. J. Dermatol. 2008;158:1035–1040. doi: 10.1111/j.1365-2133.2008.08493.x. [DOI] [PubMed] [Google Scholar]
- 11.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ng P.C., Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwarz J.M., Rödelsperger C., Schuelke M., Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 16.Davydov E.V., Goode D.L., Sirota M., Cooper G.M., Sidow A., Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP++ PLoS Comput. Biol. 2010;6:e1001025. doi: 10.1371/journal.pcbi.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pollard K.S., Hubisz M.J., Rosenbloom K.R., Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;20:110–121. doi: 10.1101/gr.097857.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chun S., Fay J.C. Identification of deleterious mutations within three human genomes. Genome Res. 2009;19:1553–1561. doi: 10.1101/gr.092619.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garber M., Guttman M., Clamp M., Zody M.C., Friedman N., Xie X. Identifying novel constrained elements by exploiting biased substitution patterns. Bioinformatics. 2009;25:i54–i62. doi: 10.1093/bioinformatics/btp190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu X., Jian X., Boerwinkle E. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat. 2011;32:894–899. doi: 10.1002/humu.21517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown M.A., Zhao Q., Baker K.A., Naik C., Chen C., Pukac L., Singh M., Tsareva T., Parice Y., Mahoney A. Crystal structure of BMP-9 and functional interactions with pro-region and receptors. J. Biol. Chem. 2005;280:25111–25118. doi: 10.1074/jbc.M503328200. [DOI] [PubMed] [Google Scholar]
- 22.Miyazono K., Kamiya Y., Morikawa M. Bone morphogenetic protein receptors and signal transduction. J. Biochem. 2010;147:35–51. doi: 10.1093/jb/mvp148. [DOI] [PubMed] [Google Scholar]
- 23.Choi J., Dong L., Ahn J., Dao D., Hammerschmidt M., Chen J.N. FoxH1 negatively modulates flk1 gene expression and vascular formation in zebrafish. Dev. Biol. 2007;304:735–744. doi: 10.1016/j.ydbio.2007.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Traver D., Paw B.H., Poss K.D., Penberthy W.T., Lin S., Zon L.I. Transplantation and in vivo imaging of multilineage engraftment in zebrafish bloodless mutants. Nat. Immunol. 2003;4:1238–1246. doi: 10.1038/ni1007. [DOI] [PubMed] [Google Scholar]
- 25.Corti P., Young S., Chen C.Y., Patrick M.J., Rochon E.R., Pekkan K., Roman B.L. Interaction between alk1 and blood flow in the development of arteriovenous malformations. Development. 2011;138:1573–1582. doi: 10.1242/dev.060467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roman B.L., Pham V.N., Lawson N.D., Kulik M., Childs S., Lekven A.C., Garrity D.M., Moon R.T., Fishman M.C., Lechleider R.J., Weinstein B.M. Disruption of acvrl1 increases endothelial cell number in zebrafish cranial vessels. Development. 2002;129:3009–3019. doi: 10.1242/dev.129.12.3009. [DOI] [PubMed] [Google Scholar]
- 27.Ricard N., Ciais D., Levet S., Subileau M., Mallet C., Zimmers T.A., Lee S.J., Bidart M., Feige J.J., Bailly S. BMP9 and BMP10 are critical for postnatal retinal vascular remodeling. Blood. 2012;119:6162–6171. doi: 10.1182/blood-2012-01-407593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.David L., Mallet C., Mazerbourg S., Feige J.J., Bailly S. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood. 2007;109:1953–1961. doi: 10.1182/blood-2006-07-034124. [DOI] [PubMed] [Google Scholar]
- 29.Young K., Conley B., Romero D., Tweedie E., O’Neill C., Pinz I., Brogan L., Lindner V., Liaw L., Vary C.P. BMP9 regulates endoglin-dependent chemokine responses in endothelial cells. Blood. 2012;120:4263–4273. doi: 10.1182/blood-2012-07-440784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fernández-L A., Sanz-Rodriguez F., Blanco F.J., Bernabéu C., Botella L.M. Hereditary hemorrhagic telangiectasia, a vascular dysplasia affecting the TGF-beta signaling pathway. Clin. Med. Res. 2006;4:66–78. doi: 10.3121/cmr.4.1.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Castonguay R., Werner E.D., Matthews R.G., Presman E., Mulivor A.W., Solban N., Sako D., Pearsall R.S., Underwood K.W., Seehra J. Soluble endoglin specifically binds bone morphogenetic proteins 9 and 10 via its orphan domain, inhibits blood vessel formation, and suppresses tumor growth. J. Biol. Chem. 2011;286:30034–30046. doi: 10.1074/jbc.M111.260133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alt A., Miguel-Romero L., Donderis J., Aristorena M., Blanco F.J., Round A., Rubio V., Bernabeu C., Marina A. Structural and functional insights into endoglin ligand recognition and binding. PLoS ONE. 2012;7:e29948. doi: 10.1371/journal.pone.0029948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Townson S.A., Martinez-Hackert E., Greppi C., Lowden P., Sako D., Liu J., Ucran J.A., Liharska K., Underwood K.W., Seehra J. Specificity and structure of a high affinity activin receptor-like kinase 1 (ALK1) signaling complex. J. Biol. Chem. 2012;287:27313–27325. doi: 10.1074/jbc.M112.377960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bidart M., Ricard N., Levet S., Samson M., Mallet C., David L., Subileau M., Tillet E., Feige J.J., Bailly S. BMP9 is produced by hepatocytes and circulates mainly in an active mature form complexed to its prodomain. Cell. Mol. Life Sci. 2012;69:313–324. doi: 10.1007/s00018-011-0751-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.