

Abstract
Cancers are caused by the accumulation of genomic alterations. Driver mutations are required for the cancer phenotype, whereas passenger mutations are irrelevant to tumor development and accumulate through DNA replication. A major challenge facing the field of cancer genome sequencing is to identify cancer-associated genes with mutations that drive the cancer phenotype. Here, we describe a powerful and flexible statistical framework for identifying driver genes and driver signaling pathways in cancer genome-sequencing studies. Biological knowledge of the mutational process in tumors is fully integrated into our statistical models and includes such variables as the length of protein-coding regions, transcript isoforms, variation in mutation types, differences in background mutation rates, the redundancy of genetic code, and multiple mutations in one gene. This framework provides several significant features that are not addressed or naively obtained by previous methods. In particular, on the observation of low prevalence of somatic mutations in individual tumors, we propose a heuristic strategy to estimate the mixture proportion of chi-square distribution of likelihood ratio test (LRT) statistics. This provides significantly increased statistical power compared to regular LRT. Through a combination of simulation and analysis of TCGA cancer sequencing study data, we demonstrate high accuracy and sensitivity in our methods. Our statistical methods and several auxiliary bioinformatics tools have been incorporated into a computational tool, DrGaP. The newly developed tool is immediately applicable to cancer genome-sequencing studies and will lead to a more complete identification of altered driver genes and driver signaling pathways in cancer.
Introduction
All cancers arise as a result of changes that have occurred in the DNA sequence of the genome of cancer cells.1 Next-generation sequencing (NGS) technologies have revolutionized cancer genomics research by providing an unbiased and comprehensive method of detecting somatic cancer genome alterations, including nucleotide substitutions, small insertions and deletions, copy-number alterations, and chromosomal rearrangements.2 Recent sequencing experiments have brought success in the identification of several cancer-associated genes that were frequently mutated in tumors, including IDH1 (MIM 147700) and IDH2 (MIM 147650) in gliomas,3 DNMT3A (MIM 602769) in acute myeloid leukemia,4,5 BAP1 (MIM 603089) in metastasizing uveal melanomas6 and malignant pleural mesothelioma (MESOM [MIM 156240]),7 ARID1A (MIM 603024) in ovarian clear cell carcinoma8 and gastric cancer,9 PHF6 (MIM 300414) in T cell acute lymphoblastic leukemia,10 MEN1 (MIM 613733) and DAXX (MIM 603186)/ATRX (MIM 300032) in pancreatic neuroendocrine tumors,11 ARID2 (MIM 609539) in hepatocellular carcinoma,12 MLL2 in diffuse large B cell lymphoma,13 GRIN2A (MIM 138253)14 and GRM3 (MIM 601115)15 in melanoma, and PBRM1 (MIM 606083) in renal carcinoma.16 As genomic sequencing experiments continue to identify large numbers of novel cancer mutations, one big challenge for cancer biologists that remains is to distinguish driver mutations from the larger number of passenger mutations. An impetus for the better identification of driver mutations is the potential therapies targeted against the products of these aberrant genomic alterations.1,2
Driver mutations are required for the cancer phenotype, whereas passenger mutations are irrelevant to tumor development and accumulate through DNA replication. In general, identification of driver genes involves statistical tests of mutated genes followed by experimental validation. The latter involves tailored in vitro and in vivo experiments that collectively form a powerful approach to validate driver genes. However, large-scale functional validation is time consuming and cost prohibitive. Furthermore, such validations are limited because there are no universal functional assays that are suitable for assessing all types of genes and pathways that can be altered in cancers.2 From a statistical point of view, driver genes are defined as those for which the nonsilent mutation rate is significantly higher than a background (or passenger) mutation rate. Silent mutations do not change amino acid residue and generally do not affect protein function and activity and are therefore considered to be passenger mutations. Statistical methods and computational tools are now actively being developed to attempt to assess functional significance of a mutated gene in cancer sequencing studies.17–24 However, applying biological knowledge of the mutational process in tumors into statistical models is not trivial and has not been adequately tested. These biological considerations include length of protein-coding regions (CDS), variation in transcript isoforms, variation in mutation types, differences in background mutation rates, redundancy of genetic code, and number of mutations in one gene. These factors have not yet been fully addressed by current methods in the quest of identifying driver genes.
There is now abundant evidence that alteration of driver genes can be productively organized according to the biochemical pathways and biological processes through which they act.25 Driver mutations can be either common or rare and identification of rare driver mutations may pose a potential challenge. It is possible that mutation in one member of a collection of functionally related genes may result in the same net effect. Furthermore, mutations in certain genes may be observed less frequently if they play functional roles in later stages of tumor development, such as metastasis. As a result, these drivers will appear to be sparsely distributed across a larger number of genes than we expect. Large sample sizes are required to detect these infrequently mutated cancer-associated genes. Alternatively, by analyzing these drivers at the pathway level, the frequency of rare mutations is accumulated and can be detected with sufficient power. Thus, there is an increasing interest in the identification of driver pathways in tumor formation and progression.26
To meet these challenges, we developed a powerful computational tool, DrGaP (driver genes and pathways), for use in cancer genome-sequencing studies (Figure 1). DrGaP incorporates our statistical approaches and several auxiliary bioinformatics tools for better driver gene identification. Biological knowledge of the mutational process is fully integrated into the statistical models and provides several significant improvements and increased power over current methods.
Figure 1.
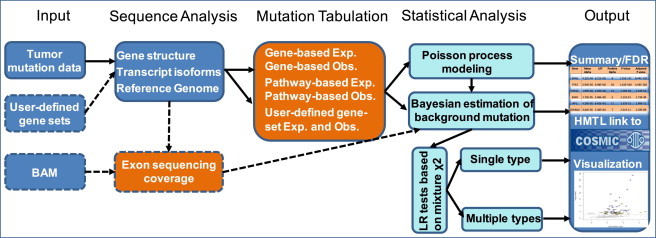
Overview of DrGaP Analysis Pipeline
The core of the pipeline includes input of tumor mutation data, sequencing analysis of somatic mutations, tabulation of somatic variants, significance test of driver genes and pathways, and summary and output of results. Dashed lines indicate optional steps.
Material and Methods
Lessons from Recent Large-Scale Cancer Sequencing Studies
To more accurately model the mutational process in our methods, we first described several significant features learned from recent large-scale cancer genome-sequencing data generated from The Cancer Genome Atlas (TCGA). Specifically, whole-exome sequencing data of 119 lung adenocarcinoma (LUAD) and 127 lung squamous cell carcinoma (LUSC) tumor samples were analyzed. We observed that a total of 7,755 and 11,125 genes were mutated at protein-coding regions (CDS) in LUAD and LUSC, respectively. However, the numbers of somatic mutations vary significantly between tumor histologies and also between individual tumors (Figure S1 available online). The number of silent mutations, which are used for estimating background mutation rates, ranges from zero to several hundred per individual tumor. LUSC was observed to have higher background mutation rates than LUAD, suggesting the necessity of estimating individual background mutation rates in statistical models.
The mutation rate depends not only on the mutated nucleotide base but also on the neighboring sequences. For example, somatic variants occur predominantly at G/C base pairs with the most prevalent changes being G/C to A/T and G/C to T/A in tobacco exposure-related tumors17,27,28 (Figure S2). Furthermore, mutations at G/C show differential rates between CpG and non-CpG sites because of deamination of cytosine at CpG dinucleotides.29 To reduce the risk of bias, 11 different mutation types are considered in our statistical models (Table S1).
We also commonly observed that some tumors do not have any silent mutations in specific mutation types (Figure S3), which can lead to a problem in estimating background mutations. Therefore, Bayesian methods are favored to estimate the distribution of background mutation rates in individual tumors.30 Based on the analysis of silent mutations from large-scale TCGA cancer sequencing data, we found that a prior beta distribution of background mutation fits the real data better than the uniform distribution that has been commonly used in previous studies17,18,22–24 (Figure S4).
Because of the nature of NGS, some CDS cannot be captured in library preparation or by sequencing because of the existence of regions with high GC content. Somatic mutations are called only when both tumor and matched normal tissue simultaneously have sufficient sequence coverage that is generally defined to be at least 8× for identifying mutations in whole-exome studies.31 We estimated that, on average, less than 85% of CDS in the genome have sequence coverage ≥8× in both tumor and matched normal samples by analyzing TCGA lung cancer sequencing data (Figure S5A and Tables S2 and S3). Furthermore, the proportion of CDS with sufficient coverage varies substantially among genes within the same sample (Figure S5B). Statistical models accounting for sequence coverage may increase sensitivity of identifying driver mutations. Below, we will describe how biological information is integrated into our statistical models (Table 1).
Table 1.
Biological Information Is Integrated into Statistical Models
| Biological Knowledge | Statistical Interpretation |
|---|---|
| transcript isoforms | sum aggregate of CDS from multiple isoforms of the same gene |
| variation in mutation types | consider 11 different mutation types |
| background mutation rates | beta prior of which is background rate of mutation type j in individual i |
| differences in background mutation rates | estimate separate mutation rates for each individual tumor |
| redundancy of the genetic code | define and as the number of base pairs in CDS of gene k that can give rise to nonsilent and silent mutations |
| multiple mutations in one gene | addressed by the Poisson process |
| sequencing coverage | is the proportion of CDS with a minimum eight sequence coverage in both a tumor and its matched normal DNA from individual i |
| CDS size | where L is length of CDS for gene k |
Poisson Process
To better identify driver mutations, we introduced a Poisson process to model the random nature of somatic mutations. Suppose we have I tumor samples to analyze and J types of mutations (Table S1) across the tumor samples. For each sample, K genes are analyzed. For each type j we will calculate the number of base pairs in CDS of gene k that can give rise to nonsilent and silent mutations. These counts are denoted by and , respectively. Furthermore, suppose that for sample i, the number of nonsilent and silent mutations in the screened CDS of type j is a Poisson process with rate and , respectively, where , is the background mutation rate in type j from sample i, and is the driver effect and can be interpreted as the increased rate of mutation due to the extent of the “driver” property of gene k of type j. Suppose that and are the number of nonsilent and silent mutations actually observed in gene k with type j from sample i. The probability of observing a given set of mutations of type j in gene k from sample i follows the Poisson distribution for nonsilent and silent mutations:
| (Equation 1) |
| (Equation 2) |
Both and can be potentially further adjusted by the sequence coverage of CDS, to increase sensitivity of identifying driver mutations, where is defined as the proportion of CDS with sufficient sequence coverage in both a tumor and its matched normal DNA at gene k in individual i (Figure S5). The nonsilent mutations can be further classified into five different functional types, i.e., missense, splicing, nonsense, in-frame, and out-of-frame indel.
Log-Likelihood
For nonsilent mutations, the log-likelihood of observed nonsilent mutations can be expressed
| (Equation 3) |
For silent mutations, the log-likelihood is
| (Equation 4) |
Maximum Likelihood Estimation
We can obtain the maximum likelihood estimation (MLE) of from Equation 4:
| (Equation 5) |
Note that in real data, some can be 0 if the sample i has no mutations of type j (Figure S3). Therefore, Bayesian methods will be used to estimate the distribution of in individual tumors, which borrow information from all the samples for estimating each individual and therefore give more smooth estimates.30 Based on the observation from the large-scale TCGA data, a prior beta distribution of will be used, which is more appropriate than the uniform distribution that has been commonly used in previous studies17,18,22–24 (Figure S4). Because it is the conjugate prior of the binomial distribution, the posterior distribution is still a beta distribution. We can estimate and . and are the moment estimation of the parameters and , where is the sample mean and is the sample variance. Then, we can obtain the MLE of by substituting into Equation 3, which is the root of the following equation:
| (Equation 6) |
It is subject to the constraint . If the root of Equation 6 is negative or , then will be 0.
Likelihood Ratio Test
Significance of the driver mutation rate for type j in gene k can be tested by the likelihood ratio test (LRT) under null hypothesis. If we want to test each single mutation type of separately, it will be a one-side test and the parameter space of is . LRT will be performed under H0: :
| (Equation 7) |
However, based on our empirical observation (Figures S1 and S3), some may be too small to observe any mutations in samples. This results in a larger probability of zero estimation of than expected under the above Poisson models and may lead to incorrect type I error. To remedy this problem, we will correct the mixture proportion of asymptotic distribution by a factor :
| (Equation 8) |
We may estimate the parameter by using simulations (Appendix A). Similarly, if we want to test whether any mutation type has increased rate of mutation resulting from the extent of the “driver” property of gene k, the distribution of the statistic of LRT will be a more complicated mixture of chi-square distributions. The null hypothesis H0 is ; the alternative hypothesis H1 is . We can express the statistic of LRT in gene k as
| (Equation 9) |
Throughout the manuscript we will use the term LRT-S when multiple types of mutations are summed into a single type in the likelihood ratio tests (i.e., and in Equation 9); LRT-M when multiple types of mutations are jointly considered in likelihood ratio tests but without correction of the mixture proportion of chi-square distribution (i.e., and ); and LRT-C when multiple types of mutations are jointly considered in likelihood ratio tests with correction of the mixture proportion of chi-square distribution by estimating ε (i.e., and ). LRT-S, LRT-M, and LRT-C represent three different likelihood ratio test statistics and their asymptotic distributions are a mixture of chi-square distribution. These statistics may have different statistical power for identify driver genes. The Benjamini-Hochberg method will be used to control false discovery rate (FDR) in all statistical tests.32
Pathway Approach
The above Poisson models (Equations 1 and 2) for a single gene can be easily extended to analyze a pathway or gene set by treating multiple genes within a pathway as a “big” gene. In brief, somatic mutations within a pathway are counted and classified into silent, missense, nonsense, splicing mutations, in-frame, and out-of-frame indels; each type of mutation except indels are further classified into one of nine nucleotide types based on the base of interest and its flanking bases and location of CpG islands (Table S1). Similarly, the expectations of base pairs that can give rise to different types of mutations are summed: and where gene k belongs to pathway p and is the proportion of CDS with at least 8× in both a tumor and its matched normal DNA. LRT will be performed to examine the significance of a pathway. Currently, we have collected 880 gene sets, including 186 KEGG, 217 BioCarta, and 400 Reactome pathways and multiple user-specified gene sets such as Chromatin remodeling, HMTs histone methylation reader, HATs HDACs, and DNMTs and Methyl-CpG binding.
Simulation
We evaluated our DrGaP statistical approaches through simulations under a range of scenarios comparable to recent tumor mutation data generated by the TCGA sequencing projects. Two different strategies of simulations were performed. One is to generate somatic mutations from probability models. In brief, background mutation rate η was first sampled from a beta distribution, i.e., , which we observed in TCGA data sets (Figure S4). Then, we generated silent mutations by Poisson distribution with parameter where M is sampled from and is the number of base pairs that can give rise to silent mutations and η is the background mutation rate. Driver nonsilent mutations were generated in a similar way (i.e., ). To evaluate the effects of multiple types of mutations on statistical power, we considered that there are 1, 5, and 11 types of driver mutations occurring in tumor samples. In each simulation, we generated 100 tumor samples with 10,000 genes and 11 different mutation types. Each simulation was replicated 100 times; the power is defined as the proportion of driver genes identified by a statistical test with a significance level of 0.05.
Another simulation strategy is to generate mutation data by directly sampling somatic mutations in TCGA data sets, including LUAD, LUSC,33 high-grade serous ovarian cancer (HGS-OvCa),34 and colorectal carcinoma (CRC)35 (Table S4). In each data set, we randomly assigned observed somatic mutations into CDS and their splicing sites (within 2 bp of an exon/intron boundary) across the genome. To maximally match simulated data to real observed mutation data, the mutation types (Table S1) remained unchanged during sampling. For example, if an observed somatic mutation in CDS is A→G, we randomly sample a base position corresponding to base A in CDS across the genome and change A to G at that position. Then, we determine whether the new mutation A→G is a silent or nonsilent mutation according to the genetic code. We applied the same sampling rule to mutations occurring in CpG sites. After the mutation reshuffling, the mutations become evenly distributed across the genome. Finally, we chose 100 simulated tumor samples from each data set and made 300 driver genes by adding 2–5 nonsilent mutations to these 300 selected genes.
With these simulation data, we also compared our DrGaP with several previous methods, including Bernoulli,24 Binomial-S and Binomial-M,17,18,22,23 Poisson,19 and TRAB.21 In order to test driver mutations, these statistical models need to specify background mutation rates. However, estimation of background mutation rates is not explicitly described in most of these methods. For a fair comparison, we used the same method as our DrGaP to estimate individual background mutation rates, , for the Bernoulli method.24 Averaging over individual tumors, we obtained background mutation rates of different mutation types that were subsequently used for multiple-parameter Binomial model (Binomial-M)17,18,22,23 and Greenman’s Poisson model.19 Similarly, we estimated the overall background mutation rate in the sample and used it for single-parameter Binomial model (Binomial-S).17,18,22,23 The TRAB method is a Bayesian method that models the occurrence of tumor mutation as the Poisson-Gamma distribution.21 We implemented its R code with the default parameter setting in our simulations.
Software
Several bioinformatics tools, together with the newly proposed statistical approaches, are integrated into an open-source software called DrGaP (Figure 1). Other auxiliary bioinformatics tools were also developed including (1) estimating depth of coverage of CDS in paired samples from sequence alignment files (i.e., BAM), (2) determining the sum aggregate of CDS from multiple isoforms, (3) analyzing sequences in CDS, and (4) mutation tabulation.
Results
Simulation Studies
We first evaluated type I error and parameter estimation under the null hypothesis . Estimators of α and η are unbiased under the null hypothesis. p values from statistical tests that either consider a single type of mutation (i.e., LRT-S) or jointly consider multiple type of mutation without correcting LRT statistics (i.e., LRT-M) are not uniformly distributed [0, 1]. The LRT-M test is also conservative. We thus proposed to estimate the mixture proportion of chi-square distribution, ε. The corrected LRT (i.e., LRT-C) has uniformly distributed p values and approximately correct type I error under the null hypothesis (Figure 2). When at least one of the mutation types has an increased rate of occurrence in tumor samples (i.e., under the alternative hypothesis), estimators of α and η are also unbiased (Figure S6). As expected, the statistical power for detecting mutations increases when the mutations rate α increases. LRT-C performs consistently better than the other two likelihood ratio statistics, LRT-S and LRT-M, in all of scenarios. The increased power of LRT-C over LRT-M becomes more apparent when there are multiple types of driver mutations that occur in tumor samples (Figure 3). The performance of LRT-S is improved when multiple types of driver mutations occur in tumor samples. This is because as increased types of mutations occur, the bias in estimating mutation rates is reduced when treating all mutations as a single type in LRT-S. We observed that lower background mutation often leads to a larger estimate of ε and thus has a higher impact on likelihood ratio statistics (Figure 4). Under these circumstances, statistical tests correcting the mixture proportion of chi-square distribution (i.e., LRT-C) show a greater advantage over those without correction (e.g., LRT-M). We will present results only from LRT-C for our methods below.
Figure 2.
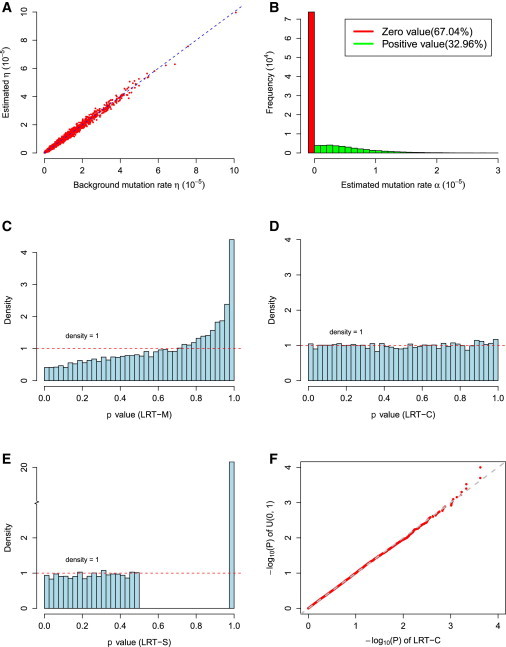
Parameter Estimation and Type I Error in DrGaP
(A and B) Parameter estimation of η and α.
(C–E) Distribution of p values from different likelihood ratio statistics: LRT-M, LRT-C, and LRT-S.
(F) Q-Q plot of uniform distribution [0, 1] and p values from LRT-C under null hypothesis.
LRT-M refers to LRT test jointly considering multiple types of mutation without estimating ε; LRT-C refers to LRT test jointly considering multiple types of mutations and correcting mixture proportion of chi-square distribution by estimating ε. LRT-S refers to LRT test considering a single type of mutation. That is, multiple types of mutations are summed into a single type in LRT-S.
Figure 3.
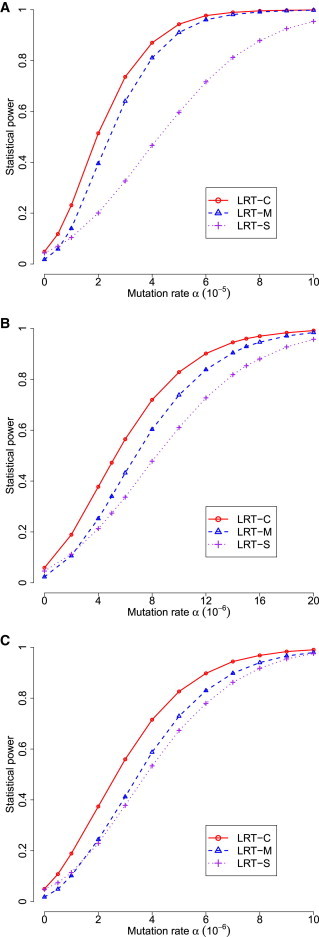
Statistical Power of Three Statistics in DrGaP
(A) Only one type of driver mutation occurred in tumors.
(B) Five of 11 types of driver mutations occurred in tumors.
(C) All 11 types of driver mutations occurred in tumors.
For comparison, the total driver mutation rates are equal among the three scenarios; the mutation rates of individual types are also equal in the latter two scenarios.
Figure 4.
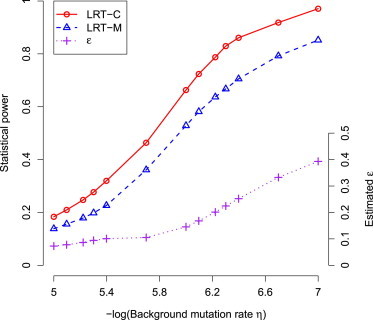
Impact of Background Mutation Rate on Likelihood Ratio Statistics
Lower background mutation (η) often leads to a larger estimate of the mixture proportion of chi-square distribution (ε).
We then compared our DrGaP approach with several previous methods that use either Bernoulli,24 Binomial,17,18,22,23 Poisson,19 or Poisson-Gamma21 related statistical models to detect driver mutations. TRAB is a Bayesian approach and computes a posteriori probability of a driver mutation, whereas the other methods give a p value, i.e., a probability that none of driver mutations is true. We therefore used receiver operating characteristic (ROC) curves to evaluate the sensitivity and specificity of these statistical methods for detecting driver mutations. First, we evaluated their performance under various numbers of driver mutation types occurring in tumor samples (Figure 5). Our method generally has higher sensitivity and specificity than the other methods in different scenarios. It shows a greater advantage over the other methods when multiple types of driver mutations occur in tumors. The performance of Bernoulli, Binomial-S, and TRAB methods is comparable, whereas the performance of Binomial-M depends on the number of mutation types and often works better when fewer types of driver mutation occur in tumors. Second, we evaluated their performance in data simulated from TCGA cancer sequencing studies (LUAD, LUSC, CRC, and HGS-OvCa) that show a wide spectrum of somatic mutations in tumors (Figure S7). Similarly, DrGaP consistently outperforms the other methods in four different TCGA data sets. It works relatively better in LUAD and LUSC than in nonhypermutated CRC and HGS-OvCa data sets. Tumors in LUAD and LUSC often have higher frequencies of somatic mutations and a large variability in background mutation rates.
Figure 5.
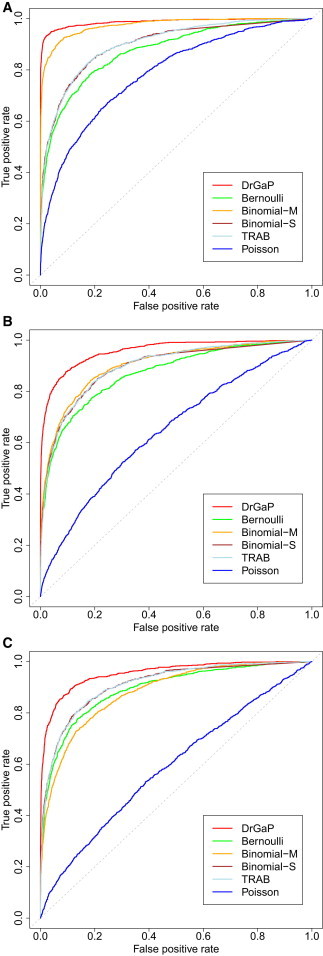
ROC Plots of Sensitivity and Specificity of Six Statistical Methods under Different Tumor Mutation Patterns
(A) Only one type of driver mutation occurred in tumors.
(B) Five of 11 types of driver mutations occurred in tumors.
(C) All 11 types of driver mutations occurred in tumors.
Although multiple methods exist to detect a single driver gene, few methods to detect a whole driver pathway are available. Our method is flexible and is also applicable for identifying driver pathways and gene sets. We therefore compared our method with PathScan for identifying driver pathways. PathScan is a tool for testing whether a pathway is significantly mutated in tumors. It is based on a Bernoulli distribution and its statistic is the number of genes that are mutated in the pathway.37 Simulation studies shows that DrGaP also has increased power over PathScan. The advantage of DrGaP over PathScan is more evident when fewer driver genes are mutated in the pathway (Figure S8).
Comparison of the Results from the Study of Ding et al.17
We applied our DrGaP methods to several cancer sequencing studies (Table S4). As before, in each study, somatic mutations are classified into silent, missense, nonsense, splicing mutations, in-frame, and out-of-frame indels. Each type of mutation, except indels, is further classified into nine nucleotide types based on the base of interest and its flanking bases and location of CpG islands (Table S1). These mutation data were then input into our DrGaP analysis pipeline (Figure 1) for identifying driver genes and pathways.
Ding et al.17 sequenced coding exons and splice donor/acceptor sites (dinucleotides in the 5′/3′ ends of introns) of 623 genes in 188 LUAD samples and identified 1,013 nonsilent mutations. They also selected a subset of 250 genes to identify 108 silent mutations for measuring a background mutation rate. Ding et al.17 identified 22 driver genes with 5% FDR. Recently, Youn and Simon also applied their approach to the same data set and identified 28 genes with 5% FDR.24 A total of 20 genes overlapped between Ding’s and Youn’s methods, and together they identified 30 mutated driver genes. However, our DrGaP method identified 59 driver genes at 5% FDR and identified 29 out of the 30 total genes by Ding and/or Youn’s methods17,24 (Figure 6). The single gene that was missed by our method reached marginal significance (FDR = 6%) and was the least significant in the combined Ding and Youn list (Table S5).
Figure 6.
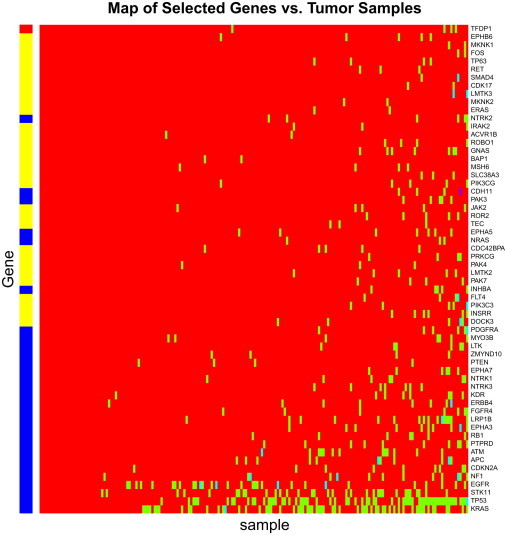
Driver Mutated Genes in the Study Ding et al.17
Tumor samples with or without mutations in genes are labeled green or red, respectively. The red/blue/yellow banner across the left side of the map shows the difference between genes selected by DrGaP and those selected by the other methods (Ding et al.17 and Youn and Simon24). The genes covered by the red bar are identified by the Ding/Youn methods but are missed by DrGaP and those covered by the yellow bar are the additional genes found by DrGaP. The genes covered by the blue bar are those which both DrGaP and Ding/Youn’s methods find significant.
In addition to its high reproducibility, DrGaP identified an additional 30 driver genes: INSRR (MIM 147671), DOCK3 (MIM 603123), PAK4 (MIM 605451), PIK3C3 (MIM 602609), FLT4 (MIM 136352), PAK7 (MIM 608038), LMTK2 (MIM 610989), TEC (MIM 600583), PRKCG (MIM 176980), CDC42BPA (MIM 603412), SLC38A3 (MIM 604437), JAK2 (MIM 147796), BAP1, PIK3CG (MIM 601232), ROR2 (MIM 602337), MSH6 (MIM 600678), ERAS (MIM 300437), ROBO1 (MIM 602430), MKNK2 (MIM 605069), CDK17 (MIM 603440), ACVR1B (MIM 601300), LMTK3, MKNK1 (MIM 606724), RET (MIM 164761), SMAD4 (MIM 600993), IRAK2 (MIM 603304), GNAS (MIM 139320), TP63 (MIM 603273), FOS (MIM 164810), and GATA1 (MIM 305371). Most have been suggested to play important roles in tumorigenesis in a broad range of published studies. For example, genetic variation in TP63 was recently found to contribute to the susceptibility of lung adenocarcinoma in two large lung cancer genome-wide association studies.38,39 RET (also called ret proto-oncogene) is a receptor tyrosine kinase that is one of the cell surface molecules that transduce signals for cell growth and differentiation. Germline gain-of-function mutations are known to predispose to multiple endocrine neoplasia type 2 (MEN2), characterized by medullary thyroid cancer, pheochromocytoma, and hyperparathyroidism.40 More recently, a novel fusion gene between KIF5B (MIM 602809) and RET, as generated by a pericentric inversion in chromosome 10, was identified in lung adenocarcinomas.41–44 Jak2 is a protein tyrosine kinase involved in a specific subset of cytokine receptor signaling pathways and has been implicated in a variety of cancers including lung and ovary.45,46 We believe our method provides higher accuracy and sensitivity than other methods for detecting driver mutated genes.
DrGaP is applicable not only to identify driver genes but also to identify driver pathway or any gene sets. To illustrate its utility, we applied DrGaP to the data of Ding et al.17 to find significantly mutated KEGG pathways (Table S6). In their original pathway analysis, Ding and her colleagues used two statistical methods. One is a binomial test that examines whether nonsilent mutation rates are higher than background mutation rates in a pathway; the other is a Fisher’s exact test that examines whether the number of gene mutations occurring in a pathway is proportionally higher than in the rest of the genome. Recently, Wendl et al.37 also applied their PathScan to the same data set to resolve the inconsistencies between binomial and Fisher’s tests from the study of Ding et al.17 DrGaP yielded largely consistent results with PathScan, but the p values from DrGaP tend to be smaller than those from PathScan. However, there are a few exceptions including Jak-STAT and TGF-β signaling pathways. Fisher’s tests gave FDR values of 0.0006 and 0.03 for these two pathways whereas binomial tests did not reach significance. PathScan concluded that none of these pathways are significant. DrGaP found these inconclusive pathways to actually be significant. Indeed, mutations in the JAKs are often found in myeloproliferative disorders (MPDs) and leukemia, and the constitutive phosphorylation of STATs is a common occurrence in many hematological and solid tumors.47 Alterations in TGF-β signaling are linked to a variety of human diseases, including cancer and inflammation. Disruption of TGF-β homeostasis occurs in several human cancers.48
Lung Adenocarcinoma and Squamous Cell Cancers
We also applied DrGaP to the analysis of whole-exome sequencing data of 119 LUAD and 127 LUSC tumor samples from TCGA. We found that 7,755 and 11,125 genes were mutated in CDS in LUAD and LUSC, respectively. Each individual tumor carried a median of 105 and 181 nonsilent mutations in LUAD and LUSC, respectively (Figure S1). Such large numbers of mutations per tumor are also observed in other tobacco-exposure related tumors (e.g., larynx, oral cavity, esophagus, and bladder cancers).49–51 Thus, it is critically important to apply statistical approaches to narrow down a much smaller and more relevant list for subsequent functional validation.
Our method identified 110 driver mutated genes in LUAD tumor samples at 5% FDR, accounting for approximately 0.5% of genes in the genome. Each individual LUAD tumor carries a median of 6 driver mutations. In LUSC tumor samples, a total of 260 driver genes were identified with a median of 16 driver genes per tumor (Figure 7). A total of 36 genes are commonly mutated in both LUAD and LUSC tumors, including CDH10 (MIM 604555), CSMD3 (MIM 608399), GRM1 (MIM 604473), KEAP1 (MIM 606016), LRP1B (MIM 608766), NAV3 (MIM 611629), NF1 (MIM 613113), and TP53 (MIM 191170), to name a few (Tables S7 and S8). In addition, we identified multiple driver pathways significantly mutated in LUAD, including focal adhesion, MAPK, tight junction, apoptosis, and cell cycle pathways (Table S9). In LUSC, multiple cellular pathways including TGF-β, Hedgehog, mTOR, Jak-STAT, and Wnt signaling pathways were significantly mutated in tumors (Table S10). Interestingly, chromatin remodeling and histone methylation pathways were significantly mutated in both LUAD and LUSC tumors.
Figure 7.
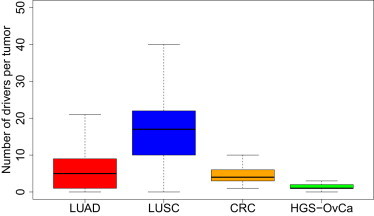
Numbers of Driver Mutated Genes per Tumor in TCGA Data Sets
LUAD, LUSC, nonhypermutated CRC, and HGS-OvCa. Genes with <5% FDR from DrGaP are presented in the above studies.
Colorectal Cancer
We further applied DrGaP to the driver gene analysis of two additional TCGA data sets (CRC and HGS-OvCa) with low prevalence of somatic mutations. In CRC, we analyzed 194 nonhypermutated tumor samples with a median of 58 nonsilent mutations per tumor.35 DrGaP identified a total of 44 driver genes with 0.05 FDR (Table S11). Each nonhypermutated CRC tumor carries a median of 4 driver mutations (Figure 7). TCGA reported 17 significantly mutated genes defined by FDR less than 10% and/or manual curation,35 15 of which were also identified by DrGaP. Two genes missed by DrGaP are MLK4 (MIM 614793) and EDNRB (MIM 131244). The q value of EDNRB exceeds 5% in the TCGA study; there are 6 missense and 3 silent mutations detected in EDNRB in nonhypermutated CRC tumors. DrGaP found an additional 29 driver genes, of which SMAD2 (MIM 601366), LRP1B (MIM 608766), BRAF (MIM 164757), TNFRSF10C (MIM 603613), ARID1A (MIM 603024), LIFR (MIM 151443), ERBB4 (MIM 600543), SLITRK1 (MIM 609678), ATM (MIM 607585), TGIF1 (MIM 602630), and CASP14 (MIM 605848) are particularly interesting candidates. For example, LIFR is a key tumor suppressor and mediates the action of the leukemia inhibitory factor. Its activation has been reported in the CRC and many other cancers.52,53
Ovarian Carcinoma
In HGS-OvCa, we analyzed 316 tumor samples with a median of 40 nonsilent mutations per tumor.34 We identified a total of 29 driver genes with 0.05 FDR and a median of 1 driver mutations per tumor (Table S12). TCGA reported 9 significantly mutated genes, 7 of which were also identified by DrGaP. Two genes that were not detected by DrGaP are FAT3 (MIM 612483) and GABRA6 (MIM 137143). Although these two genes were claimed to be significantly mutated genes, their likelihood ratio FDR and convolution FDR were reported 0.09 and 0.02 for FAT3 and 0.09 and 0.12 for GABRA6 in the TCGA study. Our DrGaP yielded FDR of 0.09 for FAT3 and 0.11 for GABRA6. In addition, DrGaP identified an additional 22 driver genes in HGS-OvCa, of which HIST1H1C (MIM 142710), CREBBP (MIM 600140), RB1CC1 (MIM 606837), BAI3 (MIM 602684), DUSP19 (MIM 611437), GNAS (MIM 139320), CDC27 (MIM 116946), and EFEMP1 (MIM 601548) are particularly interesting candidates. For example, RB1 (MIM 614041) signaling is the most significant pathways altered in HGS-OvCa. RB1CC1 is RB1-inducible coiled-coil 1, which enhances the RB1 pathway through transcriptional activation of RB1, CDKN1A (MIM 116899), and CDKN2A (MIM 600160).54
Discussion
A major challenge facing the field of cancer genome sequencing is to identify cancer-associated genes with mutations that drive the cancer phenotype. In this paper, we described a powerful and flexible statistical framework for identifying driver genes and pathways in cancer genome-sequencing data. Our methods provide several novel features that were either naively obtained or were unattainable by previous methodologies.
First, biological knowledge of the mutational process in tumors is fully integrated into our statistical models (Table 1). Second, multiple types of mutations are considered to reduce the risk of bias in estimating mutation rates in our statistical methods (Table S1 and Figure S2). This increases statistical power compared with a single-type mutation test in which all types of mutations are summed into one. Third, we incorporate the mixture proportion ε of chi-square distribution in our LRT statistics. We observed that some tumor samples have a very low background mutation rate η (Figure S3), leading to one-side LRT statistic as zero to be overrepresented in the standard mixture chi-square distribution under null hypothesis. Thus, the appropriate mixture chi-square distribution of the LRT statistics is under H0. Our simulation shows that considering parameter ε in the LRT statistic (i.e., LRT-C) corrects type I error and increases statistical power. Interestingly, the parameter ε was estimated up to 0.4 in lung cancer sequencing studies, suggesting the large impact of parameter ε on likelihood ratio statistics. We expect that our method will be of greater advantage for analyzing tumors with low prevalence of somatic mutations, such as in cancers of the hematological system. Fourth, more appropriate and informative Bayesian prior of background mutation rates, , was used in our statistical methods (Figure S4). After analyzing large-scale TCGA data, we found that a prior beta distribution of background mutation rates fits the real data better than uniform distribution commonly used in previous studies.17,18,22–24 Fifth, sequence coverage at CDS varies across studies, individual tumors, and genes in NGS experiments (Figure S5). In our methods, CDS sizes are adjusted for sequence coverage in order to increase sensitivity of identifying driver mutations. Finally, although multiple methods exist to detect a single driver gene, few methods exist to detect a whole driver pathway. Our methods can do both, because they are implemented in the same statistical framework. Because of the innovative features described above, we believe our proposed methodology increases power and sensitivity for identifying driver genes and driver pathways, compared to current methods. It should be also noted that cancers with copy-number alterations may cause allelic imbalance and thus potentially affect inference of somatic mutations. However, identification of driver mutations in our methods is focused on observed (or already detected) somatic mutations and its estimation of mutation rate per se won’t be affected by copy-number alterations.
All cancers are as a result of somatically acquired changes in the DNA of cancer cells. However, how many driver mutations does it take to make a tumor? Not all detected somatic abnormalities present in a cancer genome are required for the development of the cancer. Indeed, most of them have made no contribution at all.1 On the basis of age-incidence statistics, it has been suggested that common adult epithelial cancers such as breast, colorectal, lung, and prostate require 5–7 rate-limiting events, possibly equating to drivers, whereas cancers of the hematological system may require fewer.55 We applied our tool DrGaP to the analysis of four TCGA data sets: LUAD, LUSC, nonhypermutated CRC, and HGS-OvCa. DrGaP not only recaptured a large majority of driver genes previously reported by TCGA studies,33–35 but it also identified a much longer list of additional candidate genes whose mutations potentially drive cancer phenotypes. Most of them have been suggested to play important roles in neoplasm initiation and progression. Numbers of somatically mutated driver genes vary among individual tumors and cancer types. We observed a median of 6 driver mutations in LUAD, 16 in LUSC, 4 in nonhypermutated CRC, and 1 in HGS-OvCa (Figure 7). Interestingly, different tumors usually carry different sets of driver mutations. These data demonstrated the extreme complexity and heterogeneity of tumor cells and have important implications in targeted cancer therapy.
Large number of driver mutations involved in lung tumors, especially in LUSC, may be also attributed to potent carcinogen from life-long tobacco exposure. Tobacco exposure causes a large number of somatic mutations on the genome. This increases the chance that mutations conferring small cell growth advantage (i.e., small effect) are selected in lung microenvironment. Many mutations with small effects can be accumulated in a specific group of cells over time and collectively lead to tumor initiation and progression in lung. In tumors such as HGS-OvCa with a low prevalence of somatic mutations, fewer driver mutations (but with large effects) are expected in each individual tumor.
In the past few decades, a number of agents have been developed to target pathways that are deregulated in cancer. However, even when there is a well-known target and a highly specific drug, increased survival is generally very limited.56 For example, Gefitinib, an EGFR-tyrosine-kinase inhibitor, is a standard first-line treatment for patients with advanced non-small-cell lung cancer whose tumors have activating EGFR (MIM 131550) mutations. Although Gefitinib is one of the most specific drugs targeting EGFR-activating mutations, it prolongs life by only a median of 5-month progression-free survival compared with platinum-based doublet chemotherapy.57,58 One of the reasons why current target therapy does not work is that multiple driver mutations shape the tumorigenic process. Combination therapy targeting multiple driver mutations and pathways simultaneously has the potential to dramatically increase survival.
In summary, we have developed a powerful and flexible statistical framework for identifying driver genes and pathways in cancer genome-sequencing studies. Our statistical approaches and several auxiliary bioinformatics tools have been incorporated into a computational tool, DrGaP, for cancer genomics research. This newly developed tool is immediately applicable to cancer sequencing studies. We believe that DrGaP provides significantly improved accuracy and sensitivity and can be used to identify a more complete array of driver genes and pathways altered in cancers.
Acknowledgments
We thank TCGA for providing access to lung cancer exome-sequencing data. We thank Haris Vikis for reading and commenting the manuscript, Liping Li for creating webpage for the software DrGaP and pairdoc, and two anonymous reviewers for useful comments. This work has been support in part by National Institutes of Health grants (1R21AG040777, 5R01CA134433, and 5R01CA134682), start-up from Advancing a Healthier Wisconsin Fund (FP00001701 and FP00001703), and National Natural Science Foundation of China (No 11271346).
Contributor Information
Pengyuan Liu, Email: pliu@mcw.edu.
Yan Lu, Email: ylu@mcw.edu.
Appendix A
The LRT statistics in Equation 8 follow a mixture of and where is a positive factor, . This correction is necessary because some tumors have too low background mutation rate η to observe any occurrences of a certain type of mutations in samples. The parameter increases as the η decreases. To prove this fact, we start with a simple example that considers only one single gene with one single mutation type. Let and be the length of CDS that can give rise to silent and nonsilent mutations. Suppose there are tumor samples with background mutation rates . Denote . We can calculate the probability that there are no mutations occurring across all tumor samples under the null hypothesis (i.e., no driver mutations):
This probability reaches nearly 1 when become very small. The probability of zero LRT statistics will be larger than 0.5, because we can observe only positive discrete number of mutations , and no mutations across all samples that result in zero LRT statistics.
In the above example, can be estimated by . However, in reality we should consider all different mutation types across all different genes for estimating. Therefore, we propose a simulation method to estimate . In brief, we randomly generate somatic mutation data under the null hypothesis. That is, we set all of the driver mutation rates . We simulate the occurrence of somatic mutations by the Poisson process with , which is estimated by Equation 5. Then, we calculate the LRT statistics for each gene and each mutation type . The parameter can be estimated by . Note that the positive , we can also estimate by the mean of : .
Supplemental Data
Supplemental Data include 8 figures and 12 tables and can be found with this article online at http://www.cell.com/AJHG/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
HUGO Gene Nomenclature Committee, http://www.genenames.org/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Pairdoc, http://code.google.com/p/pairdoc/
The Cancer Genome Atlas, http://cancergenome.nih.gov/
TRAB, http://bcb.dfci.harvard.edu/∼gp/software/trab/index.html
References
- 1.Stratton M.R., Campbell P.J., Futreal P.A. The cancer genome. Nature. 2009;458:719–724. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meyerson M., Gabriel S., Getz G. Advances in understanding cancer genomes through second-generation sequencing. Nat. Rev. Genet. 2010;11:685–696. doi: 10.1038/nrg2841. [DOI] [PubMed] [Google Scholar]
- 3.Yan H., Parsons D.W., Jin G., McLendon R., Rasheed B.A., Yuan W., Kos I., Batinic-Haberle I., Jones S., Riggins G.J. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ley T.J., Ding L., Walter M.J., McLellan M.D., Lamprecht T., Larson D.E., Kandoth C., Payton J.E., Baty J., Welch J. DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 2010;363:2424–2433. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yan X.J., Xu J., Gu Z.H., Pan C.M., Lu G., Shen Y., Shi J.Y., Zhu Y.M., Tang L., Zhang X.W. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat. Genet. 2011;43:309–315. doi: 10.1038/ng.788. [DOI] [PubMed] [Google Scholar]
- 6.Harbour J.W., Onken M.D., Roberson E.D., Duan S., Cao L., Worley L.A., Council M.L., Matatall K.A., Helms C., Bowcock A.M. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010;330:1410–1413. doi: 10.1126/science.1194472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bott M., Brevet M., Taylor B.S., Shimizu S., Ito T., Wang L., Creaney J., Lake R.A., Zakowski M.F., Reva B. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat. Genet. 2011;43:668–672. doi: 10.1038/ng.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones S., Wang T.L., Shih IeM., Mao T.L., Nakayama K., Roden R., Glas R., Slamon D., Diaz L.A., Jr., Vogelstein B. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330:228–231. doi: 10.1126/science.1196333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang K., Kan J., Yuen S.T., Shi S.T., Chu K.M., Law S., Chan T.L., Kan Z., Chan A.S., Tsui W.Y. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat. Genet. 2011;43:1219–1223. doi: 10.1038/ng.982. [DOI] [PubMed] [Google Scholar]
- 10.Van Vlierberghe P., Palomero T., Khiabanian H., Van der Meulen J., Castillo M., Van Roy N., De Moerloose B., Philippé J., González-García S., Toribio M.L. PHF6 mutations in T-cell acute lymphoblastic leukemia. Nat. Genet. 2010;42:338–342. doi: 10.1038/ng.542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiao Y., Shi C., Edil B.H., de Wilde R.F., Klimstra D.S., Maitra A., Schulick R.D., Tang L.H., Wolfgang C.L., Choti M.A. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331:1199–1203. doi: 10.1126/science.1200609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li M., Zhao H., Zhang X., Wood L.D., Anders R.A., Choti M.A., Pawlik T.M., Daniel H.D., Kannangai R., Offerhaus G.J. Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nat. Genet. 2011;43:828–829. doi: 10.1038/ng.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pasqualucci L., Trifonov V., Fabbri G., Ma J., Rossi D., Chiarenza A., Wells V.A., Grunn A., Messina M., Elliot O. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat. Genet. 2011;43:830–837. doi: 10.1038/ng.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wei X., Walia V., Lin J.C., Teer J.K., Prickett T.D., Gartner J., Davis S., Stemke-Hale K., Davies M.A., Gershenwald J.E., NISC Comparative Sequencing Program Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nat. Genet. 2011;43:442–446. doi: 10.1038/ng.810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prickett T.D., Wei X., Cardenas-Navia I., Teer J.K., Lin J.C., Walia V., Gartner J., Jiang J., Cherukuri P.F., Molinolo A. Exon capture analysis of G protein-coupled receptors identifies activating mutations in GRM3 in melanoma. Nat. Genet. 2011;43:1119–1126. doi: 10.1038/ng.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Varela I., Tarpey P., Raine K., Huang D., Ong C.K., Stephens P., Davies H., Jones D., Lin M.L., Teague J. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011;469:539–542. doi: 10.1038/nature09639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ding L., Getz G., Wheeler D.A., Mardis E.R., McLellan M.D., Cibulskis K., Sougnez C., Greulich H., Muzny D.M., Morgan M.B. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Getz G., Höfling H., Mesirov J.P., Golub T.R., Meyerson M., Tibshirani R., Lander E.S. Comment on “The consensus coding sequences of human breast and colorectal cancers”. Science. 2007;317:1500. doi: 10.1126/science.1138764. [DOI] [PubMed] [Google Scholar]
- 19.Greenman C., Wooster R., Futreal P.A., Stratton M.R., Easton D.F. Statistical analysis of pathogenicity of somatic mutations in cancer. Genetics. 2006;173:2187–2198. doi: 10.1534/genetics.105.044677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin J., Gan C.M., Zhang X., Jones S., Sjöblom T., Wood L.D., Parsons D.W., Papadopoulos N., Kinzler K.W., Vogelstein B. A multidimensional analysis of genes mutated in breast and colorectal cancers. Genome Res. 2007;17:1304–1318. doi: 10.1101/gr.6431107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parmigiani G., Chen S. TRAB: testing whether mutation frequencies are above an unknown background. Stat. Appl. Genet. Mol. Biol. 2008;7:e11. doi: 10.2202/1544-6115.1277. [DOI] [PubMed] [Google Scholar]
- 22.Sjöblom T., Jones S., Wood L.D., Parsons D.W., Lin J., Barber T.D., Mandelker D., Leary R.J., Ptak J., Silliman N. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 23.Wood L.D., Parsons D.W., Jones S., Lin J., Sjöblom T., Leary R.J., Shen D., Boca S.M., Barber T., Ptak J. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 24.Youn A., Simon R. Identifying cancer driver genes in tumor genome sequencing studies. Bioinformatics. 2011;27:175–181. doi: 10.1093/bioinformatics/btq630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parsons D.W., Li M., Zhang X., Jones S., Leary R.J., Lin J.C., Boca S.M., Carter H., Samayoa J., Bettegowda C. The genetic landscape of the childhood cancer medulloblastoma. Science. 2011;331:435–439. doi: 10.1126/science.1198056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vogelstein B., Kinzler K.W. Cancer genes and the pathways they control. Nat. Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 27.Lee W., Jiang Z., Liu J., Haverty P.M., Guan Y., Stinson J., Yue P., Zhang Y., Pant K.P., Bhatt D. The mutation spectrum revealed by paired genome sequences from a lung cancer patient. Nature. 2010;465:473–477. doi: 10.1038/nature09004. [DOI] [PubMed] [Google Scholar]
- 28.Pleasance E.D., Stephens P.J., O’Meara S., McBride D.J., Meynert A., Jones D., Lin M.L., Beare D., Lau K.W., Greenman C. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2010;463:184–190. doi: 10.1038/nature08629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chapman M.A., Lawrence M.S., Keats J.J., Cibulskis K., Sougnez C., Schinzel A.C., Harview C.L., Brunet J.-P., Ahmann G.J., Adli M. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467–472. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Casella G. An introduction to empirical Bayes data analysis. Am. Stat. 1985;39:83–97. [Google Scholar]
- 31.Ng S.B., Turner E.H., Robertson P.D., Flygare S.D., Bigham A.W., Lee C., Shaffer T., Wong M., Bhattacharjee A., Eichler E.E. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc., B. 1995;57:289–300. [Google Scholar]
- 33.Cancer Genome Atlas Research Network Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cancer Genome Atlas Network Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wendl M.C., Wallis J.W., Lin L., Kandoth C., Mardis E.R., Wilson R.K., Ding L. PathScan: a tool for discerning mutational significance in groups of putative cancer genes. Bioinformatics. 2011;27:1595–1602. doi: 10.1093/bioinformatics/btr193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu Z., Wu C., Shi Y., Guo H., Zhao X., Yin Z., Yang L., Dai J., Hu L., Tan W. A genome-wide association study identifies two new lung cancer susceptibility loci at 13q12.12 and 22q12.2 in Han Chinese. Nat. Genet. 2011;43:792–796. doi: 10.1038/ng.875. [DOI] [PubMed] [Google Scholar]
- 39.Miki D., Kubo M., Takahashi A., Yoon K.A., Kim J., Lee G.K., Zo J.I., Lee J.S., Hosono N., Morizono T. Variation in TP63 is associated with lung adenocarcinoma susceptibility in Japanese and Korean populations. Nat. Genet. 2010;42:893–896. doi: 10.1038/ng.667. [DOI] [PubMed] [Google Scholar]
- 40.Zbuk K.M., Eng C. Cancer phenomics: RET and PTEN as illustrative models. Nat. Rev. Cancer. 2007;7:35–45. doi: 10.1038/nrc2037. [DOI] [PubMed] [Google Scholar]
- 41.Ju Y.S., Lee W.C., Shin J.Y., Lee S., Bleazard T., Won J.K., Kim Y.T., Kim J.I., Kang J.H., Seo J.S. A transforming KIF5B and RET gene fusion in lung adenocarcinoma revealed from whole-genome and transcriptome sequencing. Genome Res. 2012;22:436–445. doi: 10.1101/gr.133645.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kohno T., Ichikawa H., Totoki Y., Yasuda K., Hiramoto M., Nammo T., Sakamoto H., Tsuta K., Furuta K., Shimada Y. KIF5B-RET fusions in lung adenocarcinoma. Nat. Med. 2012;18:375–377. doi: 10.1038/nm.2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lipson D., Capelletti M., Yelensky R., Otto G., Parker A., Jarosz M., Curran J.A., Balasubramanian S., Bloom T., Brennan K.W. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat. Med. 2012;18:382–384. doi: 10.1038/nm.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takeuchi K., Soda M., Togashi Y., Suzuki R., Sakata S., Hatano S., Asaka R., Hamanaka W., Ninomiya H., Uehara H. RET, ROS1 and ALK fusions in lung cancer. Nat. Med. 2012;18:378–381. doi: 10.1038/nm.2658. [DOI] [PubMed] [Google Scholar]
- 45.Colomiere M., Ward A.C., Riley C., Trenerry M.K., Cameron-Smith D., Findlay J., Ackland L., Ahmed N. Cross talk of signals between EGFR and IL-6R through JAK2/STAT3 mediate epithelial-mesenchymal transition in ovarian carcinomas. Br. J. Cancer. 2009;100:134–144. doi: 10.1038/sj.bjc.6604794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Looyenga B.D., Hutchings D., Cherni I., Kingsley C., Weiss G.J., Mackeigan J.P. STAT3 is activated by JAK2 independent of key oncogenic driver mutations in non-small cell lung carcinoma. PLoS ONE. 2012;7:e30820. doi: 10.1371/journal.pone.0030820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Costa-Pereira A.P., Bonito N.A., Seckl M.J. Dysregulation of janus kinases and signal transducers and activators of transcription in cancer. Am. J. Cancer Res. 2011;1:806–816. [PMC free article] [PubMed] [Google Scholar]
- 48.Jeon H.S., Jen J. TGF-beta signaling and the role of inhibitory Smads in non-small cell lung cancer. J. Thorac. Oncol. 2010;5:417–419. doi: 10.1097/JTO.0b013e3181ce3afd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gui Y., Guo G., Huang Y., Hu X., Tang A., Gao S., Wu R., Chen C., Li X., Zhou L. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat. Genet. 2011;43:875–878. doi: 10.1038/ng.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Agrawal N., Frederick M.J., Pickering C.R., Bettegowda C., Chang K., Li R.J., Fakhry C., Xie T.X., Zhang J., Wang J. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333:1154–1157. doi: 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stransky N., Egloff A.M., Tward A.D., Kostic A.D., Cibulskis K., Sivachenko A., Kryukov G.V., Lawrence M.S., Sougnez C., McKenna A. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cho Y.G., Chang X., Park I.S., Yamashita K., Shao C., Ha P.K., Pai S.I., Sidransky D., Kim M.S. Promoter methylation of leukemia inhibitory factor receptor gene in colorectal carcinoma. Int. J. Oncol. 2011;39:337–344. doi: 10.3892/ijo.2011.1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Iorns E., Ward T.M., Dean S., Jegg A., Thomas D., Murugaesu N., Sims D., Mitsopoulos C., Fenwick K., Kozarewa I. Whole genome in vivo RNAi screening identifies the leukemia inhibitory factor receptor as a novel breast tumor suppressor. Breast Cancer Res. Treat. 2012;135:79–91. doi: 10.1007/s10549-012-2068-7. [DOI] [PubMed] [Google Scholar]
- 54.Chano T., Ikebuchi K., Ochi Y., Tameno H., Tomita Y., Jin Y., Inaji H., Ishitobi M., Teramoto K., Nishimura I. RB1CC1 activates RB1 pathway and inhibits proliferation and cologenic survival in human cancer. PLoS ONE. 2010;5:e11404. doi: 10.1371/journal.pone.0011404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miller D.G. On the nature of susceptibility to cancer. The presidential address. Cancer. 1980;46:1307–1318. doi: 10.1002/1097-0142(19800915)46:6<1307::aid-cncr2820460602>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 56.Marty M., Cognetti F., Maraninchi D., Snyder R., Mauriac L., Tubiana-Hulin M., Chan S., Grimes D., Antón A., Lluch A. Randomized phase II trial of the efficacy and safety of trastuzumab combined with docetaxel in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer administered as first-line treatment: the M77001 study group. J. Clin. Oncol. 2005;23:4265–4274. doi: 10.1200/JCO.2005.04.173. [DOI] [PubMed] [Google Scholar]
- 57.Maemondo M., Inoue A., Kobayashi K., Sugawara S., Oizumi S., Isobe H., Gemma A., Harada M., Yoshizawa H., Kinoshita I., North-East Japan Study Group Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 2010;362:2380–2388. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 58.Zhang L., Ma S., Song X., Han B., Cheng Y., Huang C., Yang S., Liu X., Liu Y., Lu S., INFORM investigators Gefitinib versus placebo as maintenance therapy in patients with locally advanced or metastatic non-small-cell lung cancer (INFORM; C-TONG 0804): a multicentre, double-blind randomised phase 3 trial. Lancet Oncol. 2012;13:466–475. doi: 10.1016/S1470-2045(12)70117-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.