

Abstract
BAP31 is one of the most abundant endoplasmic reticulum (ER) membrane proteins. It is a chaperone protein involved in several pathways, including ER-associated degradation, export of ER proteins to the Golgi apparatus, and programmed cell death. BAP31 is encoded by BCAP31, located in human Xq28 and highly expressed in neurons. We identified loss-of-function mutations in BCAP31 in seven individuals from three families. These persons suffered from motor and intellectual disabilities, dystonia, sensorineural deafness, and white-matter changes, which together define an X-linked syndrome. In the primary fibroblasts of affected individuals, we found that BCAP31 deficiency altered ER morphology and caused a disorganization of the Golgi apparatus in a significant proportion of cells. Contrary to what has been described with transient-RNA-interference experiments, we demonstrate that constitutive BCAP31 deficiency does not activate the unfolded protein response or cell-death effectors. Rather, our data demonstrate that the lack of BAP31 disturbs ER metabolism and impacts the Golgi apparatus, highlighting an important role for BAP31 in ER-to-Golgi crosstalk. These findings provide a molecular basis for a Mendelian syndrome and link intracellular protein trafficking to severe congenital brain dysfunction and deafness.
Main Text
In the search for mutations causing X-linked intellectual disability (XLID), we used next-generation sequencing technologies to sequence the X chromosome exome in 11 unrelated male individuals suffering from severe intellectual disability. These individuals were selected because they were severely affected, and either they had a clinical presentation that did not correspond to a known syndrome or molecular tests failed to detect a mutation in known genetic causes of XLID. Most of them displayed additional clinical signs involving the CNS, such as deafness or congenital microcephaly. In order to sequence the X chromosome exome, we captured exonic sequences with the Agilent SureSelect Human Exome V3 Kit and performed sequencing with the Illumina Hiseq pipeline v.1.5 at the Centre National de Génotypage (Evry), the French national genotyping center. Sequences were mapped to the human reference genome (hg19 assembly of the UCSC Genome Browser) with the commercial software CLCBIO Genomics Workbench v.4.9. We also performed SNP detection with CLCBIO and used an additional filter to select the variants located on the human X chromosome. We selected variants with a minimum quality of 30 and a minimum average quality of surrounding bases of 20. All SNPs were filtered against dbSNP132. We discarded variants for which we had fewer than ten reads and those located outside coding sequences or farther than ±5 nucleotides from an exon. Possible disease-causing sequence variants were analyzed by several bioinformatics tools, such as UMD-HTS,1 MutationTaster, the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project Exome Variant Server, and the Human Gene Mutation Database (Table S1, available online).
In one of the affected individuals (III.2, family 1), who also presented with sensorineural deafness and dystonia, we identified a c.194−2A>G mutation in exon 4 of BCAP31 (MIM 300398), encoding B-cell-receptor-associated protein 31 (BAP31), in Xq28 (Figure 1). Amplification and sequencing of BCAP31 transcripts in the primary fibroblasts of the affected individual revealed that the c.194−2A>G mutation (RefSeq accession number NM_001139441.1) activates a cryptic splice site located at positions −42 and −43 relative to exon 4 of BCAP31 and results in a transcript that is predicted to encode a truncated BAP31 (p.Ile64fs∗25) (Figures S1A and S1B). We measured the amount of BCAP31 transcript in the fibroblasts of individual F1-III.2 and found it to be 7% of the wild-type level, suggesting that the mutant transcript is a substrate for nonsense-mediated mRNA decay (data not shown). There is no antibody available to detect the N-terminal region of BAP31, so we therefore do not know whether a truncated peptide could be produced by the BAP31-deficient cells in this family. Nonetheless, we showed that the c.194−2A>G mutation leads to the absence of normal BAP31 in the fibroblasts of affected persons (Figure S1C).
Figure 1.
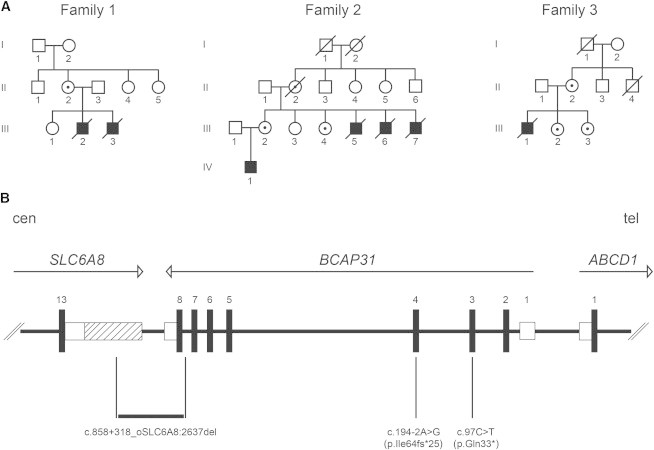
Pedigrees and Identified Mutations
(A) Pedigrees of the three families show the seven affected individuals and female carriers of a BCAP31 mutation.
(B) Schematic representation of the human chromosome Xq28 region containing BCAP31 and flanking SLC6A8 and ABCD1. Exon numbers are provided, and arrows indicate the transcription orientation (cen, centromere; tel, telomere). UTRs are represented by open boxes, and the alternative 3′ UTR of SLC6A8 is hatched. The three mutations are indicated below the genes. The Xq28 deletion is numbered according to HGVS nomenclature: RefSeq NM_005629.3 for SLC6A8 and NM_001139441 for BCAP31.
BAP31 is a chaperone and is one of the most abundant endoplasmic reticulum (ER) membrane proteins.2 This ubiquitous 31 kDa protein has a role in the export of secreted proteins from the ER,3,4 the recognition of abnormally folded proteins, and their targeting to the ER-associated-degradation (ERAD) pathway.5,6 BAP31 also serves as a cargo receptor for the export of transmembrane proteins.5 It is highly abundant in neurons.7
We screened BCAP31 for mutations in 29 male probands presenting with severe intellectual disability, dystonia, and sensorineural deafness, a phenotype similar to that of affected males in family 1. Two additional BCAP31 mutations were identified. The first mutation was identified in a family with four affected males, and the second was found in a family with a single affected male (families 2 and 3, respectively, Figure 1A). We found a deletion of BCAP31 exon 8 in family 2 and a truncating c.97C>T (p.Gln33∗) mutation in exon 3 in family 3 (Figure 1B and Figure S1A). The three mutations were inherited through carrier females who do not have intellectual disability. Clinical cerebral MRI and sample collection from affected individuals were performed at the Centre de Référence Maladies Rares “Leucodystrophie” (LeukoFrance network, LeucoEpimar, Leukobiobank) with the ethical agreement of the Comité de Protection des Personnes Sud-Est VI (no. AU788), the Commission National de l’Informatique et des Libertés (no. 1406552), and the Agence Française de Sécurité Sanitaire des Produits de Santé (no. B90298-60), and signed informed consent was obtained from the affected individuals or their legal guardians.
In order to precisely characterize the deletion present in family 2, we performed a series of PCR amplifications by using primers located 5′ and 3′ to the deleted exon in BCAP31 to amplify the deletion junction fragment from the DNA of affected individuals in that family (Figure S1D). Sequencing of the junction fragment revealed that the deletion spans 5,344 bp and results in the loss of exon 8 of BCAP31 and 248 bp of the 3′ UTR of the neighboring SLC6A8 (MIM 300036), encoding a creatine transporter (Figure 1B). SLC6A8 mutations cause cerebral creatine deficiency syndrome (MIM 300352), and affected males have intellectual disability. To assess the potential involvement of the 3′ UTR deletion of SLC6A8 in the phenotype of the affected individuals in family 2, we measured the amount of SLC6A8 mRNA in their fibroblasts and found that it was reduced by 54% (Figure S1E). However, the creatine peaks on proton magnetic resonance spectroscopy were found to be normal in the only living male individual in this family (IV.1, data not shown). The expression of an SLC6A8 transcript (Ensembl reference sequence ENST00000597046.1) with a shorter 3′ UTR that is not deleted in family 2 (Figure 1B) might account for this observation and for the fact that SLC6A8 transcripts were present at 46% of the wild-type levels in the fibroblasts of the affected individuals. We conclude that SLC6A8 is not involved in the phenotype of the affected individuals in family 2. In addition, the similarity of the clinical presentation among the three families (two of which were devoid of any SCL6A8 anomaly) further supports this conclusion. Indeed, comparison of the clinical phenotypes in the three affected families, comprising a total of seven affected individuals, defines an XLID syndrome consisting of severe motor and intellectual disabilities, early dystonia, sensorineural deafness, and hypomyelinating white-matter changes associated with failure to thrive, microcephaly, and facial dysmorphism (Figure 2 and Table 1). Death occurred during the first years of life, suddenly, or, in four cases, during a febrile episode. Progressive aggravation of the phenotype with cerebral atrophy was observed in two individuals, who died at the end of adolescence. The only surviving person improved clearly after 3 years of age and remained stable at the last examination at age 13. If one considers the clinically homogeneous cohort composed of family 1 (coming from the exome sequencing project) and the 29 families screened in a second phase because they had a similar phenotype, mutations in BCAP31 were found in 10% (3/30). This relatively high detection rate suggests that BCAP31 mutations might not be rare in males suffering from deafness, dystonia, and white-matter changes.
Figure 2.
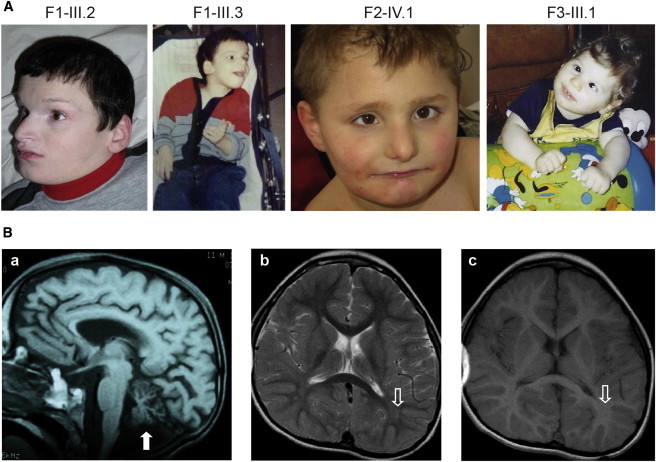
Phenotype of the Affected Individuals
(A) Facial dysmorphic features observed in the different affected members of the three families. Note the dystonic posture of the upper limbs of individuals F1-III.3 and F3-III.1.
(B) Sagittal cerebral MRI sequence of person F1-III.2 at 11 months of age shows the atrophy of the cerebellar vermis (white arrow) (a). Axial cerebral MRI of individual F2-IV.1 at 6.5 years of age shows a mild abnormal hyperintense signal on the T2 sequence in the parieto-occipital white matter (b) and a normal hyperintense signal on the T1 sequence (c) (open white arrows), suggesting poor myelination.
Table 1.
Summary of the Clinical and MRI Features Identified in the Affected Individuals
|
Family 1 |
Family 2 |
Family 3 |
|||||
|---|---|---|---|---|---|---|---|
| III.2 | III.3 | III.5 | III.6 | III.7 | IV.1 | III.1 | |
| Age at last exam | 22 years | 11 years | NA | 10 months | 12 months | 13 years | 3 years |
| Age of onset | congenital | congenital | congenital | 6 weeks | 6 weeks | congenital | congenital |
| Age at death | 24 years | 13 years | 7 months (sudden) | 1 year (fever) | 2 years (sudden) | living | 3 years (fever) |
| Deafness (onset) | + (1 year) | + (1 year) | NA | + (1 year) | + (1 year) | + (normal at 6 weeks, 70 db at 7 months, 0 db at 3 years) | + (5 months; 20 db at 8 months) |
| Facial dysmorphism | ++ | + | NA | + | + | + | +/− |
| Best Acquisitions | |||||||
| Motor (acquired at) | none | none | none | none | head (12 months) | head (6 months), sitting (5 years), autonomous wheelchair (8 years) | head (1 year; lost at 1.5 years) |
| Cognitive | none | none | none | none | none | simple sign language and scribbling | none |
| Neurological Signs | |||||||
| Ocular | congenital strabismus | congenital strabismus, abnormal eye movements | NA | optic atrophy | congenital strabismus | congenital strabismus, optic atrophy | congenital strabismus |
| Dystonia (onset) | ++ | ++ | NA | +++ | +++ | ++ (10 months) | +++ (6 months) |
| Seizures (onset) | + (4 years) | + | NA | none | none | + (8 years) | none |
| Other | pyramidal signs, quadriplegia, unexplained episodic fever, aggressiveness | pyramidal signs, quadriplegia, unexplained episodic fever | NA | pyramidal signs, quadriplegia | pyramidal signs, quadriplegia | pyramidal signs, paraplegia, unexplained episodic fever, hyperactivity | pyramidal signs, quadriplegia, unexplained episodic fever |
| Growth | |||||||
| Failure to thrive | +++ (IUGD, weight −7 SDs, height −8 SDs) | +++ (IUGD, weight −6 SDs, height −5 SDs) | NA | + | + | +++ (IUGD, weight and height −3 SDs at 0–3 years and −2 SDs at >3 years) | + (normal at birth, weight and height −2 SDs at >1 year) |
| Microcephaly | +++ (−5 SDs) | +++ (−5 SDs) | NA | NA | NA | + (−3 SDs at 2 months, −2 SDs at 2 years) | ++ (−2 SDs at 0–6 months, −3 SDs at >6 months) |
| MRI | |||||||
| White matter (performed at) | NP | periventricular hypomyelination (11 years) | NP | NP | NP | periventricular hypomyelination (5.6 years) | diffuse hypomyelination (2.5 years) |
| Atrophy | NP | cerebral cortex, cerebellum | NP | NP | NP | 0 | corpus callosum, frontal lobe, white matter |
Abbreviations are as follows: +++, severe; ++, moderate; +, mild; +/−, very mild; IUGD, intrauterine growth delay; NA, not available; and NP, not performed.
Approximately 30% of all proteins in eukaryotic cells are targeted to the ER to be folded and to receive posttranslational modifications.8 During translation, the nascent polypeptide enters the reticulum via the translocon, a pore-forming complex. Several translocon-associated proteins ensure that polypeptides entering the ER fold properly. If incorrect folding is detected, for example in proteins containing missense substitutions, a mechanism of retrograde transport operating from the ER lumen to the cytoplasm targets the protein for degradation by the proteasome after polyubiquitinylation.9 If the cell cannot maintain an acceptable level of misfolded proteins, it will launch apoptosis programs and die.10 The three mutations identified in BCAP31 are probable loss-of-function alleles that abolish the production of normal BAP31 (Figure S1C). Given the role and cellular distribution of BAP31, we next examined ER morphology in the fibroblasts of the affected individuals. We labeled calreticulin (CALR), a soluble protein chaperone located in the ER lumen, where it enables the correct folding of nascent proteins. CALR labeling revealed a swollen aspect in all BAP31-deficient fibroblasts (Figure 3A). To exclude a possible CALR-specific observation, we repeated these experiments by using heat shock 70 kDa protein 5 (HSPA5, also known as glucose-regulated protein 78 [GRP78]), another abundant ER chaperone (Figure 3A), and we confirmed that the size of the mutant fibroblasts was increased (Figure 3B).
Figure 3.
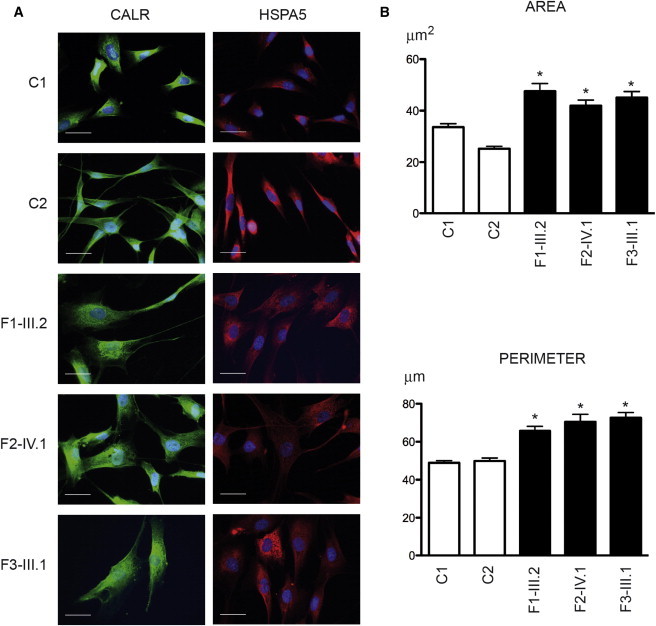
BAP31-Deficient Fibroblasts Have Abnormal Morphology
(A) Immunohistochemistry performed on fibroblasts of two control individuals (C1 and C2), individual III.2 from family 1 (F1-III.2), individual IV.1 from family 2 (F2-IV.1), and individual III.1 from family 3 (F3-III.1) with an antibody against CALR (1:200, Stressgen ADI-SPA-600-F) (left panels) or HSPA5 (1:200, Abcam ab151269) (right panels). These images reveal the abnormal size and shape of BAP31-deficient fibroblasts compared with control fibroblasts. Cells were stained with DAPI for visualization of nuclear DNA. Scale bars represent 25 μm.
(B) Quantification of the area and perimeter of control and affected individuals’ fibroblasts immunolabeled with CALR and shown in (A) demonstrates the statistically different values (∗p < 0.0001, Student’s t test) between the fibroblasts of the two control individuals and the three affected individuals. For each fibroblast, a region of interest delineated with ImageJ software defined the cell boundaries. The program measured the area and the perimeter expressed in square micrometers and micrometers, respectively. We measured the morphometric parameters of randomly selected fibroblasts (104 cells were analyzed for C1, 103 for C2, 80 for individual F1-III.2, 82 for individual F2-IV.1, and 60 for individual F3-III.1). Error bars represent the SEM.
We reasoned that, in the absence of BAP31, ERAD dysfunction might lead to the accumulation of misfolded proteins in the ER. We used the ΔF508 (c.1521_1523delCTT [RefSeq NM_000492.3]) altered version of cystic fibrosis transmembrane conductance regulator (CFTR), the most frequent cause of cystic fibrosis (MIM 219700), as an ERAD substrate whose degradation involves BAP31.11,12 Transfection with pEGFP-ΔF508CFTR was performed, and fibroblasts were grown for 24 hr after transfection. BCAP31 mutations appeared to lead to the presence of increased amounts of EGFP-ΔF508CFTR (Figure S2), as would be expected from reduced ERAD activity. However, the difference between affected and control fibroblasts was not statistically significant when the fluorescence was quantified by image analysis (Figure S2A) or flow cytometry (Figure S2B). These findings suggest that the BCAP31 mutations identified in the three families do not lead to a massive accumulation of misfolded proteins within the ER, contrary to what was found via transient RNAi experiments.11,12 Most likely, BAP31-independent pathways are able to cope with the misfolded proteins present and thus avoid an accumulation of unwanted proteins in the ER.
Next, we tested whether the unfolded protein response (UPR) was activated in BAP31-deficient fibroblasts. We measured the transcript levels of three genes whose expression is known to be increased upon UPR activation:10 DDIT3 (MIM 126337), HSPA5 (MIM 138120), and XBP1 (MIM 194355). Unexpectedly, we found that the UPR was not activated in the affected individuals’ fibroblasts, although these cells were able to activate their UPR when treated with ER-stress inducer thapsigargin (Figure S3A). Accordingly, levels of HSPA5 and XBP1 were unchanged in BAP31-deficient fibroblasts (Figure S3B). DDIT3 levels could not be tested because of the poor specificity of the DDIT3 antibody in immunoblot experiments.
In addition to UPR and ERAD, BAP31 was recently shown to have a role in cell death via an interaction with the protein product of FIS1.13 We also investigated this aspect to determine whether our human BAP31-deficient fibroblasts were launching apoptosis programs. We found no evidence of cell death in the fibroblasts lacking BAP31. The mitochondrial-respiratory-chain activities in the muscle of person F3-III.1 were normal (data not shown), and the mitochondrial network displayed a normal morphology in all affected fibroblasts (Figure S4A). In addition, immunoblot analysis failed to detect cleaved caspase-3, a mediator of apoptotic cell death, in BAP31-deficient fibroblasts (Figure S4B). From these data, we conclude that the phenotype of the individuals carrying a BCAP31 mutation was not caused by exacerbated cell death.
BAP31 was shown to serve as a cargo receptor for the export of transmembrane proteins.5 Given that the ER was abnormal in BAP31-deficient fibroblasts, we wondered whether the Golgi apparatus morphology was preserved. To visualize the Golgi apparatus in affected fibroblasts, we used an antibody raised against GOLGA2, a membrane protein involved in the maintenance of the Golgi apparatus structure.14 We found that the Golgi apparatus was disorganized in up to 29.5% of BAP31-deficient fibroblasts (Figures 4A and 4B). To better observe the cellular ultrastructure, we studied these cells by using electron microscopy and confirmed that a significant proportion of BAP31-deficient fibroblasts had an unusually large ER and abnormal Golgi morphology. Strikingly, we found that the mutant fibroblasts contained large cytoplasmic vesicles partially filled with electron-dense inclusions (Figure 4C). These vesicles possibly originated from the disorganization of the Golgi apparatus, given that they were frequently observed in close vicinity to residual Golgi-like structures.
Figure 4.
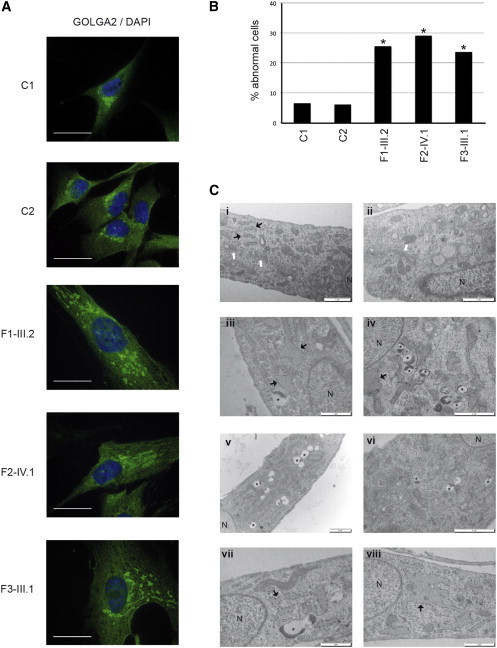
The Golgi Apparatus Is Disorganized in a Large Proportion of BAP31-Deficient Fibroblasts
(A) Representative images show the abnormal structure of the Golgi apparatus in BAP31-deficient fibroblasts. The typical bean-shaped perinuclear Golgi structure observed in control fibroblasts was lost in a large proportion of BAP31-deficient fibroblasts, where it was replaced by small patches of GOLGA2-positive structures scattered in the cytoplasm. Immunohistochemistry was performed on fibroblasts from two control individuals (C1 and C2), individual III.2 from family 1 (F1-III.2), individual IV.1 from family 2 (F2-IV.1), and individual III.1 from family 3 (F3-III.1) with an antibody specific to GOLGA2 (green) (1:200, Sigma G7295). The fibroblasts were stained with DAPI for visualization of nuclear DNA. Scale bars represent 25 μm.
(B) Percentage of fibroblasts showing a severely perturbed Golgi apparatus morphology in control samples and in the fibroblasts of the three affected individuals (∗p < 0.01, Chi-square test; 100 cells were counted in each of two experiments for each condition).
(C) Representative electron-microscopy images reveal the ultrastructure of control fibroblasts (i and ii) and BAP31-deficient fibroblasts in individuals F1-III.2 (iii and iv), F2-IV.1 (v and vi), and F3-III.1 (vii and viii). The cells were washed, fixed in 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4), postfixed in 1% osmium tetroxide, scraped, and centrifuged, and the pellet was dehydrated in graded alcohol solutions and embedded in SPURR low-viscosity medium. Ultrathin sections (50–60 nm) were counterstained with uranyl acetate and lead citrate before observation with a JEOL JEM1400 electron microscope at 80 kV. Images were obtained with a MegaView III camera (Soft Imaging System). The affected individuals’ fibroblasts contained an enlarged ER (black arrows) and numerous vesicles containing electron-dense material (asterisks) and lost the normal structure of their Golgi apparatus (white arrows in control fibroblasts). N = nucleus. Scale bars represent 2 μm (iv, v, and vi) or 1 μm (i, ii, iii, vii, and viii).
Taken together, our results support the notion that BAP31 has a role in ER-to-Golgi exchanges and that this function cannot be compensated in human cells. In human BAP31-deficient fibroblasts, misfolded proteins do not massively accumulate and there is no activation of either the UPR or cell-death programs. A disorganized Golgi apparatus is predicted to have major consequences for the production of plasma membrane or secreted proteins. The seven affected individuals’ phenotype, combining intellectual disability, dystonia, and sensorineural deafness, demonstrates that this is particularly deleterious for the CNS. In the brain of the BAP31-deficient individuals, we observed white-matter changes reminiscent of those observed in hypomyelinating leukodystrophies,15 confirming the key role of ER protein trafficking in the myelination process.16 BCAP31 is flanked by two genes: SLC6A8 on its centromeric side and ABCD1 (MIM 300371) on its telomeric side. Large deletions involving BCAP31 and ABCD117,18 or SCL6A8 and BCAP31 have been reported.19 Deletions involving ABCD1 and BCAP31 have been reported in four cases and have been given the acronym CADDS (contiguous ABCD1 DXS1357E deletion syndrome [MIM 300475]). In one case, however, the deletion extended over seven genes in Xq28,18 thereby complicating the interpretation of the clinical findings. The affected persons suffered from neonatal hypotonia, failure to thrive, cholestatic liver disease, and white-matter abnormalities (ranging from mild hypomyelination to neuronal heterotopias) not described in adrenoleukodystrophy (MIM 300100). Seizures were inconstant. The four individuals described with CADDS died before the first year of age. Facial dysmorphism was described in a single case. They all had sensorineural deafness. Deletions of SLC6A8 and BCAP31 cause dystonia, profound intellectual disability, mild liver dysfunction, sensorineural deafness, and myelination delay.19 These clinical signs, also found in our series of seven BAP31-deficient individuals, do not belong to the clinical picture of individuals presenting with a dysfunction of either SLC6A8 or ABCD1; these latter individuals display, respectively, cerebral creatine deficiency syndrome or adrenoleukodystrophy. It is very likely that the liver dysfunction reported in persons carrying large deletions involving BCAP31 and ABCD1 or SCL6A8 and BCAP31 can be attributed to BCAP31 loss. Four affected individuals in our families harboring mutations in BCAP31 demonstrated transitory episodes of a 2- to 3-fold increase of liver transaminase levels during banal febrile illnesses. However, in contrast to previous reports,17,18 liver ultrasound did not detect any abnormalities in the affected individuals we describe.
The identification of loss-of-function mutations in BCAP31 provides the molecular basis of a Mendelian syndrome and links ER-to-Golgi exchange to severe congenital brain dysfunction and deafness.
Acknowledgments
We thank Pascale Fanen and Frédéric Becq for the gift of the ΔF508CFTR plasmids. We thank Caroline Lacoste and the Centre de Ressources Biologiques of Hôpital d’Enfants de la Timone (Karine Bertaux, Cécile Mouradian, and Andrée Robaglia-Schlupp) for assistance with the cell lines used in this study. We thank Joël Courageot and Alexandre Altié from the Service Commun de Microscopie Electronique of the Faculté de Médecine de Marseille and Eleonore Eymard Pierre (Cytogénétique, Centre Hospitalier Universitaire de Clermont Ferrand, France) for providing affected-individual samples from the LeukoFrance biobank. This work was supported by funding from INSERM and Aix-Marseille Université to L.V. and from the European Association against Leukodystrophy (ELA) Foundation (ELA 2009-00714) to O.B.T. and was supported in part by funding from Agence Nationale de la Recherche Labex project “Medical Genomics” to M.L.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Human Gene Mutation Database, http://www.hgmd.cf.ac.uk/ac/index.php
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Mutation Taster, http://www.mutationtaster.org
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS
Universal Mutation Database High Throughput Sequencing, http://www.umd-hts.eu/
References
- 1.Frédéric M.Y., Lalande M., Boileau C., Hamroun D., Claustres M., Béroud C., Collod-Béroud G. UMD-predictor, a new prediction tool for nucleotide substitution pathogenicity — application to four genes: FBN1, FBN2, TGFBR1, and TGFBR2. Hum. Mutat. 2009;30:952–959. doi: 10.1002/humu.20970. [DOI] [PubMed] [Google Scholar]
- 2.Bell A.W., Ward M.A., Blackstock W.P., Freeman H.N., Choudhary J.S., Lewis A.P., Chotai D., Fazel A., Gushue J.N., Paiement J. Proteomics characterization of abundant Golgi membrane proteins. J. Biol. Chem. 2001;276:5152–5165. doi: 10.1074/jbc.M006143200. [DOI] [PubMed] [Google Scholar]
- 3.Annaert W.G., Becker B., Kistner U., Reth M., Jahn R. Export of cellubrevin from the endoplasmic reticulum is controlled by BAP31. J. Cell Biol. 1997;139:1397–1410. doi: 10.1083/jcb.139.6.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paquet M.E., Cohen-Doyle M., Shore G.C., Williams D.B. Bap29/31 influences the intracellular traffic of MHC class I molecules. J. Immunol. 2004;172:7548–7555. doi: 10.4049/jimmunol.172.12.7548. [DOI] [PubMed] [Google Scholar]
- 5.Wakana Y., Takai S., Nakajima K., Tani K., Yamamoto A., Watson P., Stephens D.J., Hauri H.P., Tagaya M. Bap31 is an itinerant protein that moves between the peripheral endoplasmic reticulum (ER) and a juxtanuclear compartment related to ER-associated Degradation. Mol. Biol. Cell. 2008;19:1825–1836. doi: 10.1091/mbc.E07-08-0781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Geiger R., Andritschke D., Friebe S., Herzog F., Luisoni S., Heger T., Helenius A. BAP31 and BiP are essential for dislocation of SV40 from the endoplasmic reticulum to the cytosol. Nat. Cell Biol. 2011;13:1305–1314. doi: 10.1038/ncb2339. [DOI] [PubMed] [Google Scholar]
- 7.Manley H.A., Lennon V.A. Endoplasmic reticulum membrane-sorting protein of lymphocytes (BAP31) is highly expressed in neurons and discrete endocrine cells. J. Histochem. Cytochem. 2001;49:1235–1243. doi: 10.1177/002215540104901005. [DOI] [PubMed] [Google Scholar]
- 8.Brodsky J.L., Skach W.R. Protein folding and quality control in the endoplasmic reticulum: Recent lessons from yeast and mammalian cell systems. Curr. Opin. Cell Biol. 2011;23:464–475. doi: 10.1016/j.ceb.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith M.H., Ploegh H.L., Weissman J.S. Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science. 2011;334:1086–1090. doi: 10.1126/science.1209235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 11.Lambert G., Becker B., Schreiber R., Boucherot A., Reth M., Kunzelmann K. Control of cystic fibrosis transmembrane conductance regulator expression by BAP31. J. Biol. Chem. 2001;276:20340–20345. doi: 10.1074/jbc.M011209200. [DOI] [PubMed] [Google Scholar]
- 12.Wang B., Heath-Engel H., Zhang D., Nguyen N., Thomas D.Y., Hanrahan J.W., Shore G.C. BAP31 interacts with Sec61 translocons and promotes retrotranslocation of CFTRDeltaF508 via the derlin-1 complex. Cell. 2008;133:1080–1092. doi: 10.1016/j.cell.2008.04.042. [DOI] [PubMed] [Google Scholar]
- 13.Iwasawa R., Mahul-Mellier A.L., Datler C., Pazarentzos E., Grimm S. Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J. 2011;30:556–568. doi: 10.1038/emboj.2010.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakamura N. Emerging new roles of GM130, a cis-Golgi matrix protein, in higher order cell functions. J. Pharmacol. Sci. 2010;112:255–264. doi: 10.1254/jphs.09r03cr. [DOI] [PubMed] [Google Scholar]
- 15.Schiffmann R., van der Knaap M.S. Invited article: an MRI-based approach to the diagnosis of white matter disorders. Neurology. 2009;72:750–759. doi: 10.1212/01.wnl.0000343049.00540.c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roboti P., Swanton E., High S. Differences in endoplasmic-reticulum quality control determine the cellular response to disease-associated mutants of proteolipid protein. J. Cell Sci. 2009;122:3942–3953. doi: 10.1242/jcs.055160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Corzo D., Gibson W., Johnson K., Mitchell G., LePage G., Cox G.F., Casey R., Zeiss C., Tyson H., Cutting G.R. Contiguous deletion of the X-linked adrenoleukodystrophy gene (ABCD1) and DXS1357E: a novel neonatal phenotype similar to peroxisomal biogenesis disorders. Am. J. Hum. Genet. 2002;70:1520–1531. doi: 10.1086/340849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iwasa M., Yamagata T., Mizuguchi M., Itoh M., Matsumoto A., Hironaka M., Honda A., Momoi M.Y., Shimozawa N. Contiguous ABCD1 DXS1357E deletion syndrome: report of an autopsy case. Neuropathology. 2013;33:292–298. doi: 10.1111/j.1440-1789.2012.01348.x. [DOI] [PubMed] [Google Scholar]
- 19.Osaka H., Takagi A., Tsuyusaki Y., Wada T., Iai M., Yamashita S., Shimbo H., Saitsu H., Salomons G.S., Jakobs C. Contiguous deletion of SLC6A8 and BAP31 in a patient with severe dystonia and sensorineural deafness. Mol. Genet. Metab. 2012;106:43–47. doi: 10.1016/j.ymgme.2012.02.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.