

Abstract
The (R)-BINOL•SnCl4-catalyzed formal (3 + 2) cycloaddition between 3-substituted indoles and benzyl 2-trifluoroacetamidoacrylate is a direct, enantioselective method to prepare pyrroloindolines from simple starting materials. However, under the originally disclosed conditions, the pyrroloindolines are formed as mixtures of diastereomers, typically in the range of 3:1 to 5:1 favoring the exo-product. The poor diastereoselectivity detracts from the synthetic utility of the reaction. We report here that use of methyl 2-trifluoroacetamidoacrylate in conjunction with (R)-3,3'-dichloro-BINOL•SnCl4 provides the corresponding pyrroloindolines with improved diastereoselectivity (typically ≥10:1). Guided by mechanistic studies, a one-flask synthesis of enantioenriched indolines by in situ reduction of a persistent iminium ion is also described.

Keywords: pyrroloindoline, (3 + 2) cycloaddition, enantioselective catalysis
1. Introduction
Nitrogen-containing heterocycles, such as indolines1 and pyrroloindolines,2 are prevalent in a variety of natural products that exhibit an array of promising biological activities (see Figure 1 for representative examples).3 Many of these compounds possess all-carbon quaternary stereocenters at C3 of the indoline or pyrroloindoline, and the synthetic challenge presented by this structural feature has driven innovative research aimed at the discovery of new methods to stereoselectively prepare such heterocycles. These methods include several stereoselective transformations starting from tryptophan,4 as well as catalytic asymmetric reactions using both organo-5 and transition metal catalysts.6
Figure 1.

Indoline and pyrroloindoline containing natural products.
In 2010, our group reported an enantioselective synthesis of pyrroloindolines (8) via an (R)-BINOL•SnCl4-catalyzed formal (3 + 2) cycloaddition between 3-substituted indoles (5) and benzyl 2-trifluoroacetamidoacrylate (6) (Scheme 1).7 Good yields, moderate exo/endo diastereoselectivity, and high enantioselectivities were obtained for a variety of indole substrates. Unexpectedly, studies aimed at epimerizing C2 revealed that exo-8 and endo-8 possess the opposite absolute configurations at the bridgehead carbons.
Scheme 1.
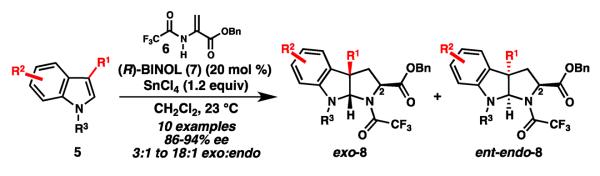
(R)-BINOL•SnCl4-catalyzed pyrroloindoline formation.
Although further mechanistic studies are required, this observation led us to propose a cooperative Lewis acid/Lewis acid-assisted Brønsted acid (LBA) mechanism, shown in Scheme 2. Activation of acrylate 6 by coordination to SnCl4 would result in reversible conjugate addition by the indole to generate a racemic mixture of Sn-enolates 9 and ent-9. Highly face-selective protonation of 9/ent-9 by an (R)-BINOL•SnCl4 complex (11) would serve to resolve the two enantiomers into diastereomers in a rate- and stereoselectivity-determining step. In this scenario, the ee of the two pyrroloindoline products would reflect the facial selectivity of the protonation step, while the dr would reflect the difference in protonation rates for the two chiral enolates 9 and ent-9, due to matching and mismatching effects with chiral complex 11.8 The stoichiometric proton source required to turn over complex 12 could potentially be the N-H of trifluoroacetamide exo-13/ent-endo-13, which upon coordination to SnCl4 would be rendered sufficiently acidic.
Scheme 2.

Proposed mechanism for pyrroloindoline formation.
The proposed mechanism is consistent with Yamamoto's prior reports that (R)-BINOL•SnCl4(11) behaves as a Lewis acid-assisted Brønsted acid (LBA), which is capable of catalyzing the enantioselective protonation of silyl enolates.9 However, the pyrroloindoline formation described above constitutes the first example of a tandem conjugate addition/asymmetric protonation process catalyzed by 11. Based on our mechanistic proposal for pyrroloindoline formation, we recently identified similar conditions for the synthesis of unnatural tryptophan derivatives (18) from 2-substituted indoles (15) and methyl 2-acetamidoacrylate (16), in which the sole stereogenic center is set during an asymmetric protonation step (Scheme 3).10 This tandem Friedel–Crafts conjugate addition/asymmetric protonation reaction provides direct access to a variety of tryptophan derivatives (18) without the need for pre-functionalization of the indole substrate.
Scheme 3.

17•SnCl4-catalyzed conjugate addition/asymmetric protonation.
Returning to the enantioselective pyrroloindoline synthesis shown in Scheme 1, the fact that exo-8 and ent-endo-8 are of opposite enantiomeric series presents a practical challenge for synthetic applications. Specifically, in order to preclude erosion of ee during subsequent synthetic steps, it is imperative to separate the two diastereomers. Unfortunately, depending on the indole substitution pattern, separation of the diastereomers can be tedious using standard silica gel chromatography. In addition, the modest diastereoselectivity results in lower isolated yields of the pure exo-diastereomer. In an effort to identify conditions more amenable to application in total synthesis endeavors, we sought to re-optimize the reaction parameters for the formal (3 + 2) cycloaddition with the objective of improving the diastereoselectivity while maintaining synthetically useful enantioselectivity. Herein, we report the successful realization of this objective, as well as mechanism-guided studies that resulted in the development of a one-flask reduction process for the direct synthesis of related indoline derivatives.
2. Results and Discussion
The optimization studies disclosed in our original report determined that the enantio- and diastereoselectivity of pyrroloindoline formation was highly dependent on the substitution of the 2-amidoacrylate: the highest enantioselectivity was obtained using benzyl 2-trifluoroacetamidoacrylate (6), while the highest diastereoselectivity was attained using methyl 2-trifluoroacetamidoacrylate (20) (Scheme 4). In the latter case, the ee's of the two diastereomers were only modestly reduced. Thus, we returned to the use of acrylate 20 in the cycloaddition, and sought to improve the ee and dr by optimizing the catalyst structure.
Scheme 4.

Dependence of diastereoselectivity on acrylate substitution.
A screen of (R)-BINOL derivatives revealed that several catalysts containing substitution at the 3- and 3'-positions provided an improvement in both dr and ee. Hypothesizing that catalyst selectivity might correlate to the pKa of the BINOL O-H protons, several 6,6'-derivatives were also prepared in order to isolate the electronic and steric effects; however, no linear correlation was observed. Ultimately, (R)-3,3'-Cl2-BINOL (25) was identified as the catalyst that provided the optimal combination of ee, dr, and overall yield.
With these newly optimized conditions in hand, a survey of indole substrates was conducted (Table 2). As previously observed, indoles bearing electron-donating substituents on the aryl backbone provide the highest yields; however, both electron-rich and electron-poor substrates provide high ee's. More sterically-hindered indoles are less reactive and require 1.6 equivalents of SnCl4, but nevertheless furnish the desired pyrroloindoline in good yield with high selectivity.
Table 2.
Substrate scope of pyrroloindoline formation.

|
Determined by 1H NMR analysis of crude reaction mixture. Values in parentheses are dr obtained using acrylate 6 and catalyst 7.
Determined by SFC using chiral stationary phase. Values in parentheses are ee obtained using acrylate 6 and catalyst 7.
Isolated yield of exo-diastereomer.
1.6 equiv SnCl4 was employed.
Having identified conditions that provide the pyrroloindoline products with improved diastereoselectivity, we sought to obtain further mechanistic insight by monitoring the reaction using in situ 1H NMR spectroscopy. For this purpose, we returned to the reaction between 1,3-dimethylindole (5a) and benzyl 2-trifluoroacetamidoacrylate (6) because it is homogeneous over the course of the reaction. Figure 2 shows a sample of 1H NMR spectra taken over the first 9 hours of the reaction, and reveals several interesting aspects of this transformation. Notably, upon addition of SnCl4 and (R)-BINOL (7) to a mixture of 5a and 6, the indole proton resonances broaden significantly (Figure 2, t = 0). This broadening is also observed in the absence of acrylate 6; however, SnCl4 alone does not alter the 1H NMR spectrum of 5a. It is possible that this broadening is due to a rapid, dynamic proton exchange process promoted by 7•SnCl4, which is consistent with the finding that deuteurated 5a undergoes rapid D-H exchange under the reaction conditions. Interestingly, the chemical shifts of the acrylate remain unchanged, indicating that there is no significant accumulation of an acrylate–SnCl4 complex. Over the course of the reaction, resonances corresponding to an indole–acrylate adduct grow in; however, these peaks do not correspond to the pyrroloindoline product.
Figure 2.

In situ monitoring of the formal (3 + 2) cycloaddition using 1H NMR.
We hypothesized that in the presence of a strong Lewis acid, such as SnCl4, coordination of the amide might favor the ring-opened iminium ions exo-14/ent-endo-14. To test this hypothesis, we resubjected diastereomerically pure exo-8a to SnCl4 (1.2 equiv) and varying equivalents of (R)-BINOL (7) (Figure 3). The NMR spectra of the mixtures were dependent on the concentration of 7. In the presence of 20 mol % 7, the 1H NMR spectrum closely resembles that of the indole-acrylate adduct observed in the 1H NMR experiment. Notably, this species exhibits a resonance between 9 and 10 ppm (depending on concentration of 7, Figure 3, a and b), which we assign to the indolinium proton (Hb). In addition, the N-methyl group (Ha) in this species is shifted downfield relative to the pyrroloindoline (4.0 ppm versus 3.1 and 2.9 ppm for the two rotamers of the exo diastereomer), and is consistent with literature data for other iminium ions.11 This structural assignment is further supported by 2D 1H–13C NMR correlation data. In the presence of SnCl4 alone, the pyrroloindoline peaks broaden, likely due to dynamic interconversion between the ring-opened and -closed forms (Figure 3, c). The fact that addition of 1.2 equivalents of 7 resolves this mixture into one species suggests that 7•SnCl4, an LBA,9a might preferentially stabilize the open structure. Importantly, following aqueous work-up, pyrroloindoline exo-8a is cleanly reisolated with no indication of epimerization or racemization.
Figure 3.

NMR spectra of (a) pyrroloindoline exo-8a, SnCl4 (1.2 equiv), (R)-BINOL (7) (1.2 equiv) (b) pyrroloindoline exo-8a, SnCl4 (1.2 equiv), (R)-BINOL (7) (0.2 equiv) (c) pyrroloindoine exo-8a, SnCl4 (1.2 equiv) (d) pyrroloindoline exo-8a.
The progress of the reaction can be quantified by integrating the vinyl protons of the acrylate relative to 1,4-diethylbenzene as an internal standard. However, further kinetic analysis of the (R)-BINOL-catalyzed reaction has been complicated by several factors, including the racemic background reaction (particularly at higher SnCl4:(R)-BINOL ratios) and product inhibition.
Given that iminium ions exo-14/ent-endo-14 are observed under the reaction conditions, we wondered if it would be possible to trap these intermediates with an external hydride source. The resulting product would be an indoline-containing amino acid derivative bearing an all-carbon quaternary stereocenter at the C3-position (Scheme 5). Commonly employed strategies for the enantioselective generation of indolines include asymmetric hydrogenation of prochiral indoles,12 reduction of oxindoles,13 C-H activation of substituted anilines,14 and kinetic resolution of racemic mixtures of indolines.15 However, these methods are often limited in scope or require multiple steps.
Scheme 5.

Intercepting an iminium ion intermediate to yield indolines.
To assess the feasibility of our proposed transformation, the formal (3 + 2) cycloaddition was performed under the previously optimized conditions, but in the presence of a reductant (Table 3). Whereas weaker reductants such as triethylsilane and sodium triacetoxyborohydride proved ineffective, we were pleased to find that use of Hantzsch ester 29 did provide the indoline product, albeit in low yield (entry 3). Alternatively, use of sodium borohydride furnishes 28a in good yield, 15:1 dr, and 92% ee (entry 5). The more soluble reducing agent lithium borohydride provided a lower yield of the desired product along with a greater amount of byproducts. The limited solubility of NaBH4 and LiBH4 in methylene chloride likely contributes to the compatibility of all the reagents, allowing the reaction to be carried out in one pot.
Table 3.
Screen of reducing agents.

| entry | Reductant | yield (%) |
|---|---|---|
| 1 | Et3SiH | 0 |
| 2 | NaBH(OAc)3 | 0 |
| 3 | Hantzsch ester 29 | 3 |
| 4 | LiBH4 | 85 |
| 5 | NaBH4 | 93a |
product isolated in 15:1 dr and 92% ee.

Having identified an optimal reducing agent, a survey of indole substrates was conducted (Table 4). Indoles with either electron-donating or -withdrawing substituents are good substrates for the reaction. At the 3-position, n-butyl and phenylethyl groups are tolerated, but reactivity decreases with increasing steric bulk, and 1.6 equivalents of SnCl4 are required to achieve good reactivity. Whereas cleavage of the protecting group was observed when TBS-protected tryptophol was employed, use of the TIPS-protected trpytophol furnished 28k in good yield. The indole nitrogen can also be protected with an allyl group, making this method more useful when an N-Me functionality is not desired in the product.
Table 4.
In situ reduction for the synthesis of indolines.

|
Determined by 1H NMR analysis of crude reaction mixture.
Determined by SFC using chiral stationary phase.
Isolated yield of exo-diastereomer.
1.6 equiv SnCl4 was employed.
The reduced products 28 are formed with the same diastereomeric and enantiomeric ratios as the corresponding pyrroloindolines, suggesting that the reduction does not affect the selectivities of the other steps. If a methylene chloride solution of pyrroloindoline exo-8a and SnCl4 is re-exposed to sodium borohydride, the pyrroloindoline is reduced to indoline 28a in quantitative yield.
3. Conclusion
Building on our initial report of a formal (3 + 2) cycloaddition between 3-substituted indoles and 2-amidoacrylates to yield enantioenriched pyrroloindolines, we have further optimized the catalyst structure to significantly improve the diastereoselectivity of this reaction while maintaining comparable yields and ee's. Subsequent NMR studies have provided a more thorough understanding of this reaction, revealing that the initial product of the reaction is an indolinium ion. Based on these findings, a method has been developed to trap this intermediate with sodium borohydride to yield enantioenriched indoline products with a quaternary stereocenter at C3. The study and development of additional transformations that proceed by cooperative Lewis acid–LBA mechanisms are the subject of ongoing research in our laboratory.
4. Experimental Section
4.1. General
Unless otherwise stated, reactions were performed under a nitrogen atmosphere using freshly dried solvents. Tetrahydrofuran (THF), methylene chloride (CH2Cl2), and toluene were dried by passing through activated alumina columns. Deuterated methylene chloride (CD2Cl2) was dried by passing through a plug of activated alumina in a glovebox. Dimethylformamide (DMF) was dried over activated molecular sieves, dichloroethane (DCE) was distilled over calcium hydride. All other commercially obtained reagents were used as received unless specifically indicated. All reactions were monitored by thin-layer chromatography using EMD/Merck silica gel 60 F254 pre-coated plates (0.25 mm). Flash column chromatography was performed either as described by Still et al. (Still, W. C., Kahn, M.; Mitra, A. J. Org. Chem. 1978, 43, 2923–2925.) using silica gel (partical size 0.032–0.063) purchased from Silicycle or using pre-packaged RediSep®Rf columns on a CombiFlash Rf system (Teledyne ISCO Inc.). Diastereomeric ratios were determined by integration of crude NMR spectra. Optical rotations were measured on a Jasco P-2000 polarimeter using a 100 mm path-length cell at 589 nm. 1H and 13C NMR spectra were recorded on a Varian Mercury 300 (at 300 MHz and 75 MHz respectively), a Varian 400 (at 400 MHz and 100 MHz respectively) or a Varian Inova 500 (at 500 MHz and 125 MHz respectively), and are reported relative to internal chloroform (1H, δ = 7.26, 13C, δ = 77.0). Data for 1H NMR spectra are reported as follows: chemical shift (δ ppm) (multiplicity, coupling constant (Hz), integration). Multiplicity and qualifier abbreviations are as follows: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad, app = apparent. IR spectra were recorded on a Perkin Elmer Paragon 1000 spectrometer and are reported in frequency of absorption (cm−1). Preparatory HPLC was performed with either an Agilent 1100 or 1200 Series HPLC utilizing an Agilent Zorbax RX-SIL 5mm column (9.4 × 250 mm). Analytical SFC was performed with a Mettler SFC supercritical CO2 analytical chromatography system with Chiralcel AD-H, OJ-H columns (4.6 mm × 25 cm). Melting points were determined using a Büchi B-545 capillary melting point apparatus and the values reported are uncorrected. HRMS were acquired using either an Agilent 6200 Series TOF with an Agilent G1978A Multimode source in electrospray ionization (ESI), atmospheric pressure chemical ionization (APCI) or mixed (MM) ionization mode, or obtained from the Caltech Mass Spectral Facility.
4.2 Preparation of indole substrates
4.2.1 1,3,4-trimethyl-1H-indole
Procedure for Vilsmeier-Haack reaction followed by LiAlH4 reduction was adapted from Petit et al.16 In a flame-dried flask under nitrogen, POCl3 (0.42 mL, 4.6 mmol) was added at 0 °C to 4-methyl-1H-indole (0.5 g, 3.8 mmol) in DMF (7.6 mL). The reaction was stirred at room temperature overnight. 2N NaOH(aq) was then added, the solution was stirred for 2 h, then poured into EtOAc. The organic layer was washed with brine, dried (Na2SO4), filtered, and concentrated. The crude aldehyde was carried forward without further purification.
In a flame-dried flask under nitrogen, a solution of the Vilsmeier-Haack product (0.39 g, 2.5 mmol) in THF (5 mL) was added dropwise to a suspension of LiAlH4 (0.19 g, 5 mmol) in THF (1.6 mL). The reaction was heated to reflux for 4 h, then cooled to room temperature and stirred overnight. The reaction was diluted with Et2O and cooled to 0 °C. Water (0.19 mL) was added slowly, then 15% NaOH(aq) (0.19 mL), then water (0.6 mL) were added. The mixture was warmed to room temperature and stirred for 15 minutes. Some MgSO4 was added, the mixture was stirred for 15 minutes, filtered and concentrated. The crude indole was carried forward without further purification.
In a flame-dried flask, the indole (0.3 g, 2.1 mmol) was dissolved in THF (13 mL). Sodium hydride (60% w/w, 124 mg, 3.1 mmol) was added in one portion, then methyl iodide (0.26 mL, 4.1 mmol) was added dropwise. The reaction was stirred at room temperature until consumption of starting material was observed by TLC. The reaction was diluted with ethyl acetate and the excess NaH was quenched with water. The organic layer was separated, and the aqueous layer was extracted 3× with ethyl acetate. The combined organic layers were washed with brine, dried (MgSO4), filtered and concentrated under reduced pressure. The crude residue was purified by flash chromatography. 44% yield over three steps.
1H NMR (500 MHz, CDCl3) δ 7.12-7.05 (m, 2H), 6.81 (ddd, J = 6.6, 1.4, 0.8 Hz, 1H), 6.76 (d, J = 0.9 Hz, 1H), 3.69 (s, 3H), 2.72 (s, 3H), 2.51 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 137.5, 131.4, 126.9, 126.7, 121.5, 120.0, 110.9, 107.0, 32.5, 20.0, 12.8; IR (NaCl/thin film): 2918, 1608, 1573, 1551, 1497, 1453, 1417, 1313, 1250, 1205, 1157, 1057, 767, 739 cm−1.
4.2.2 1,3,7-trimethyl-1H-indole
Prepared according to the procedure for 1,3,4-trimethyl-1H-indole. Spectral data matches that reported in the literature.17
4.2.3 1,3-dimethyl-5-reverse prenyl-1H-indole
In a glovebox, Pd2(dba)3 (51 mg, 6 mmol) and SPhos (91 mg, 22 mmol) in THF (3 mL) were stirred at room temperature for 1h until a dark yellow homogeneous solution was formed. The solution was then transferred to a Schlenk tube, and 5-iodo-1,3-dimethyl-1H-indole (300 mg, 1.11 mmol), trifluorosilane18 (256 mg in 2.1 mL THF, 1.66 mmol), TBAF (1 M solution in THF, 1.66 mL) and THF (7 mL) were added. The reaction mixture was heated to 60°C for 36 hours. Additional portions of TBAF (1M solution in THF, 0.55 mL) were added at 12-hour intervals. 5% EtOAc in hexanes was then added to the reaction, and the mixture was filtered through a silica gel plug. The solution was concentrated, and the crude orange oil was purified by flash chromatography (5% to 9% EtOAc in hexanes) to give the reverse prenylated indole as a light yellow oil (177 mg, 0.84 mmol, 76% yield). 1H NMR (500 MHz, CDCl3) 7.51 (t, J = 0.9 Hz, 1H), 7.24 (dd, J = 8.7, 1.83 Hz, 1H), 7.21 (d, J = 8.5 Hz, 1H), 6.80 (d, J = 1.0 Hz, 1H), 6.13 (dd, J = 17.5, 10.6 Hz, 1H), 5.09 (dd, J = 17.6, 1.5 Hz, 1H), 5.04 (dd, J = 10.5, 1.5 Hz, 1H), 3.71 (s, 3H), 2.32 (d, J = 1.0 Hz, 3H), 1.49 (s, 6H); 13C NMR (126 MHz, CDCl3) 149.1, 138.8, 135.4, 128.3, 126.7, 120.6, 115.5, 110.1, 109.9, 108.5, 41.1, 32.5, 28.8, 9.5; IR (NaCl/thin film): 3080, 2964, 2920, 1634, 1489, 1455, 1425, 1387, 1376, 1365, 1292, 1256, 1201, 1152, 1053, 1004, 909, 874, 788; HRMS (MM) calc'd for C15H19N [M+H]+ 214.1590, found 214.1592.
4.3 General procedure for the formal (3 + 2) cycloaddition of indoles and acrylates
To a flame-dried flask was added indole (0.20 mmol, 1.00 equiv), acrylate (0.20 mmol, 1.00 equiv), and (R)-3,3'-dichloro-BINOL (0.04 mmol, 0.20 equiv). The flask was charged with CH2Cl2 (1.5 mL), followed by addition of SnCl4 (0.24 mmol, 1.20 equiv unless specifically indicated, 1 M in CH2Cl2), then stirred at room temperature. The reaction was quenched by diluting with 1 mL MeCN and 1 mL 1 M HCl, followed by addition of 5 mL H2O. The aqueous layer was extracted with diethyl ether (3 × 15 mL) and the combined organic layers were washed with 3 N NaOH(aq) (10 mL). The combined organic layers were dried (MgSO4), filtered and concentrated. The crude residue was purified by flash chromatography.
4.3.1 Pyrroloindoline 21a
The dr was determined to be 14:1 by 1H NMR analysis of the crude reaction mixture. The crude residue was purified by flash chromatography (5→10% ethyl acetate/hexanes) to yield 49.4 mg (73% yield) of 21a. The enantiomeric excess was determined to be 91% by chiral SFC analysis (AD-H, 2.5 mL/min, 7% IPA in CO2, λ = 254 nm): tR(major) = 2.9 min tR(minor) = 2.4 min. Spectral data matches that reported in the literature.7
4.3.2 Pyrroloindoline 21b
The dr was determined to be 7:1 by 1H NMR analysis of the crude reaction mixture. The crude residue was purified by flash chromatography (0→10% ethyl acetate/hexanes) to yield 49.1 mg (69% yield) of 21b. The enantiomeric excess was determined to be 92% by chiral SFC analysis (AD-H, 2.5 mL/min, 7% IPA in CO2, λ = 254 nm): tR(major) = 3.1 min tR(minor) = 2.3 min. The major diastereomer was separated by flash chromatography (0→10% ethyl acetate/hexanes). 1H NMR (500 MHz, CDCl3; compound exists as a 3.7:1 mixture of rotamers, the major rotamer is denoted by *, minor rotamer denoted by§) δ 7.08 (t, J = 7.8 Hz, 1H*, 1H§), 6.65-6.57 (m, 1H§), 6.55 (d, J = 7.6 Hz, 1H*), 6.45-6.40 (m, 1H§), 6.38 (d, J = 7.8 Hz, 1H*), 5.49 (s, 1H*), 5.29 (s, 1H§), 4.74 (d, J = 9.3 Hz, 1H*), 4.45 (m, 1H§), 3.83 (s, 3H*), 3.78 (s, 3H§), 3.12 (s, 3H*), 2.85 (s, 3H§), 2.68 (dd, J = 13.3, 9.7 Hz, 1H*), 2.64-2.59 (m, 1H§), 2.53 (dd, J = 13.3, 1.6 Hz, 1H*), 2.33 (s, 3H§), 2.31 (s, 3H*), 2.20-2.10 (m, 1H§), 1.60 (s, 3H§), 1.47 (s, 3H*); 13C NMR (125 MHz, CDCl3; compound exists as a 3.7:1 mixture of rotamers, the major rotamer is denoted by *, minor rotamer denoted by §) δ 172.7*, 170.5§, 159.0§ (q, JC-F = 37.2 Hz), 150.6*, 149.7§, 133.1§, 132.7*, 131.6*, 130.9§, 128.8*§, 122.6§, 121.5*, 116.1* (q, JC-F = 288.3 Hz), 107.3§, 106.2*, 94.0*, 92.0§, 61.3§, 60.0*, 53.1*, 52.6§, 50.0*, 42.7*, 39.4§, 37.4*, 34.6§, 24.1*, 23.3§, 18.5*§; IR (NaCl/thin film): 3047, 2957, 2930, 2880, 2825, 1752, 1701, 1596, 1477, 1434, 1385, 1356, 1338, 1293, 1263, 1254, 1216, 1204, 1155, 1097, 1064, 1020, 989, 854, 772, 744, 727 cm−1;[a]D25 = −158.8 (c = 1.01, CHCl3). HRMS (APCI) calc'd for C17H19F3N2O3 [M+H]+ 357.1421, found 357.1426.
4.3.3 Pyrroloindoline 21c
The dr was determined to be 13:1 by 1H NMR analysis of the crude reaction mixture. The crude residue was purified by flash chromatography (0→10% ethyl acetate/hexanes) to yield 51.3 mg (72% yield) of 21c. The enantiomeric excess was determined to be 89% by chiral SFC analysis (AD-H, 2.5 mL/min, 5% IPA in CO2, λ = 254 nm): tR(major) = 4.4 min tR(minor) = 2.7 min. The major diastereomer was separated by flash chromatography (0→10% ethyl acetate/hexanes). 1H NMR (500 MHz, CDCl3; compound exists as a 1.9:1 mixture of rotamers, the major rotamer is denoted by *, minor rotamer denoted by §) δ 6.98 (d, J = 7.4 Hz, 1H*, 1H§), 6.87 (s, 1H§), 6.84 (s, 1H*), 6.50 (d, J = 7.8 Hz, 1H§), 6.43 (d, J = 8.0 Hz, 1H*), 5.57 (s, 1H*), 5.27 (s, 1H§), 4.73 (d, J = 9.3 Hz, 1H*), 4.41 (t, J = 7.6 Hz, 1H§), 3.82 (s, 3H*), 3.76 (s, 3H§), 3.05 (s, 3H*), 2.86 (s, 3H§), 2.59 (dd, J = 13.3, 9.7 Hz, 1H*), 2.55-2.48 (m, 1H§), 2.35 (dd, J = 13.5, 2.2 Hz, 1H*), 2.28 (br s, 3H§), 2.20-2.10 (m, 1H§), 1.49 (s, 3H§), 1.38 (s, 3H*); 13C NMR (125 MHz, CDCl3; compound exists as a 1.9:1 mixture of rotamers, the major rotamer is denoted by *, minor rotamer denoted by §) δ 172.6*, 170.7§, 159.1§ (q, JC-F = 37.0 Hz), 147.3*, 147.2§, 134.4*§, 129.7§, 129.2§, 129.1*, 128.2*, 122.3*§, 116.1* (q, JC-F = 288.2 Hz), 109.9§, 108.2*, 93.8*, 92.1§, 61.2§, 60.3*, 53.2§, 53.0*, 52.5§, 49.2*, 44.0*, 40.3§, 37.4*, 35.5§, 23.5*, 23.2§, 20.8*§; IR (NaCl/thin film): 2958, 2924, 2873, 2822, 1750, 1699, 1618, 1500, 1435, 1384, 1356, 1339, 1288, 1257, 1201, 1152, 1117, 1094, 1057, 1035, 986, 874, 844, 807, 761, 728 cm−1; [a]D25 = −128.4 (c = 1.08, CHCl3). HRMS (APCI) calc'd for C17H19F3N2O3 [M+H]+ 357.1421, found 357.1407.
4.3.4 Pyrroloindoline 21d
The dr was determined to be 14:1 by 1H NMR analysis of the crude reaction mixture. The crude residue was purified by flash chromatography (0→10% ethyl acetate/hexanes) to yield 56.3 mg (79% yield) of 21d. The enantiomeric excess was determined to be 89% by chiral SFC analysis (AD-H, 2.5 mL/min, 5% IPA in CO2, λ = 254 nm): tR(major) = 3.4 min tR(minor) = 2.8 min. The major diastereomer was separated by flash chromatography (0→10% ethyl acetate/hexanes). 1H NMR (500 MHz, CDCl3; compound exists as a 2.4:1 mixture of rotamers, the major rotamer is denoted by *, minor rotamer denoted by §) δ 6.99-6.93 (m, 1H§), 6.91 (d, J = 7.4 Hz, 1H*), 6.65 (d, J = 6.7 Hz, 1H§), 6.58 (d, J = 7.3 Hz, 1H*), 6.41 (s, 1H§), 6.34 (s, 1H*), 5.60 (s, 1H*), 5.31 (s, 1H§), 4.72 (d, J = 9.0 Hz, 1H*), 4.48-4.39 (m, 1H§), 3.82 (s, 3H*), 3.77 (s, 3H§), 3.06 (s, 3H*), 2.86 (s, 3H§), 2.58 (dd, J = 13.2, 9.2 Hz, 1H*), 2.52-2.45 (m, 1H§), 2.39-2.33 (m, 1H*), 2.32 (br s, 3H*, 3H§), 2.10-2.00 (m, 1H§), 1.49 (s, 3H§), 1.38 (s, 3H*); 13C NMR (125 MHz, CDCl3; compound exists as a 2.4:1 mixture of rotamers, the major rotamer is denoted by *, minor rotamer denoted by §) δ 172.6*, 170.6§, 159.1§ (q, JC-F = 37.9 Hz), 149.6*, 149.4§, 139.0§, 138.9*, 131.5*, 131.4§, 121.2*§, 120.6§, 119.3*, 116.1* (q, JC-F = 288.4 Hz), 110.4§, 108.9*, 93.5*, 91.9§, 61.3§, 60.3*, 53.0*, 52.9§, 52.5§, 49.0*, 44.0*, 40.5§, 36.7*, 34.6§, 23.6*, 23.0§, 21.7*; IR (NaCl/thin film): 2958, 2929, 2875, 2813, 1750, 1697, 1617, 1594, 1499, 1435, 1382, 1356, 1341, 1294, 1257, 1203, 1190, 1148, 1111, 1094, 1059, 1034, 1006, 985, 877, 852, 803, 763, 729 cm−1; [a]D25 = −115.4 (c = 1.54, CHCl3). HRMS (MM) calc'd for C17H19F3N2O3 [M+H]+ 357.1421, found 357.1434.
4.3.5 Pyrroloindoline 21e
The dr was determined to be 15:1 by 1H NMR analysis of the crude reaction mixture. The crude residue was purified by flash chromatography (5→25% ethyl acetate/hexanes) to yield 48.1 mg (68% yield) of 21e (major diastereomer only). The enantiomeric excess was determined to be 93% by chiral SFC analysis (AD-H, 2.5 mL/min, 7% IPA in CO2, λ = 254 nm): tR(major) = 3.6 min tR(minor) = 2.5 min. 1H NMR (500 MHz, CDCl3; compound exists as a 6.1:1 mixture of rotamers, the major rotamer is denoted by *, minor rotamer denoted by §) δ 7.05-7.00 (m, 1H*), 7.00-6.93 (m, 2H*, 1H§), 6.91 (d, J = 7.2 Hz, 1H§), 6.82 (t, J = 7.4 Hz, 1H§), 5.27 (s, 1H§), 5.14 (d, J = 1.6 Hz, 1H*), 4.59 (dd, J = 9.1, 2.3 Hz, 1H§), 4.06 (dd, J = 11.2, 6.6 Hz, 1H*), 3.80 (s, 3H§), 3.72 (s, 3H*), 3.26 (s, 3H§), 2.99 (s, 3H*), 2.67 (dd, J = 12.6, 6.6 Hz, 1H*), 2.57 (dd, J = 13.4, 9.2 Hz, 1H§), 2.30 (s, 3H§), 2.22 (s, 3H*), 2.13-2.03 (m, 1H*, 1H§), 1.47 (s, 3H*), 1.42 (s, 3H§); 13C NMR (125 MHz, CDCl3; compound exists as a 6.1:1 mixture of rotamers, the major rotamer is denoted by *, minor rotamer denoted by §) δ 172.6§, 171.1*, 158.7*, 149.9*, 148.8§, 136.6*§, 131.4§, 131.0*, 126.6*, 124.01*, 122.6§, 121.2§, 119.6*, 119.4§, 116.0* (q, JC-F = 285.8 Hz), 95.6§, 92.5*, 60.3*, 59.2§, 55.1*, 52.9§, 52.4*, 49.7§, 44.0§, 41.9§, 41.0*, 38.6*, 26.3*, 26.0§, 18.9§, 17.5*; IR (NaCl/thin film): 2963, 1753, 1684, 1437, 1359, 1269, 1162, 1120, 1103, 1086, 1067, 977 cm−1; [a]D25 = −27.0 (c = 0.91, CH2Cl2). HRMS (MM) calc'd for C17H19F3N2O3 [M+H]+ 357.1421, found 357.1434.
4.3.6 Pyrroloindoline 21f
The dr was determined to be 10:1 by 1H NMR analysis of the crude reaction mixture. The crude residue was purified by flash chromatography (5→25% ethyl acetate/hexanes) to yield 41.4 mg (58% yield) of 21f. The enantiomeric excess was determined to be 92% by chiral SFC analysis (AD-H, 2.5 mL/min, 5% IPA in CO2, λ = 254 nm): tR(major) = 3.7 min tR(minor) = 2.3 min. The major diastereomer was separated by flash chromatography (0→10% ethyl acetate/hexanes). 1H NMR (500 MHz, CDCl3; compound exists as a 2.0:1 mixture of rotamers, the major rotamer is denoted by *, minor rotamer denoted by §) δ 6.92-6.82 (m, 1H*, 1H§), 6.81-6.76 (m, 1H§), 6.74 (dd, J = 8.0, 2.7 Hz, 1H*), 6.50 (dd, J = 8.6, 4.1 Hz, 1H§), 6.40 (dd, J = 8.6, 4.1 Hz, 1H*), 5.60 (s, 1H*), 5.31 (s, 1H§), 4.74 (d, J = 9.3 Hz, 1H*), 4.43 (t, J = 7.8 Hz, 1H§), 3.82 (s, 3H*), 3.77 (s, 3H§), 3.04 (s, 3H*), 2.85 (s, 3H§), 2.58 (dd, J = 13.5, 9.6 Hz, 1H*), 2.53-2.45 (m, 1H§), 2.40-2.32 (m, 1H*), 2.06 (dd, J = 13.3, 6.6 Hz, 1H§), 1.49 (s, 3H§), 1.38 (s, 3H*); 13C NMR (125 MHz, CDCl3; compound exists as a 2.0:1 mixture of rotamers, the major rotamer is denoted by *, minor rotamer denoted by §) δ 172.4*, 170.5§, 159.0§ (q, JC-F = 35.8 Hz), 157.9*, 156.7§, 156.0*, 145.6*, 145.4§, 135.8§ (d, JC-F = 8.7 Hz), 135.6* (d, JC-F = 7.5 Hz), 116.0* (q, JC-F = 289.2 Hz), 115.1§, 114.8* (d, JC-F = 23.2 Hz), 110.5§ (d, JC-F = 7.3 Hz), 110.2§ (d, JC-F = 24.3 Hz), 109.4§ (d, JC-F = 24.1 Hz), 109.3* (d, JC-F = 24.4 Hz), 108.6* (d, JC-F = 8.1 Hz), 105.9§ (d, JC-F = 8.0 Hz), 93.8*, 92.1§, 61.1§, 60.2*, 53.2§, 53.1*, 52.6§, 49.2§, 43.8*, 40.1§, 37.6*, 35.5§, 23.4*, 22.9§; IR (NaCl/thin film): 2959, 2880, 2825, 1750, 1699, 1611, 1495, 1436, 1386, 1356, 1339, 1270, 1229, 1202, 1178, 1152, 1118, 1091, 1052, 1034, 986, 872, 845, 808, 756, 728 cm−1; [a]D25 = −108.5 (c = 1.08, CHCl3). HRMS (APCI) calc'd for C16H16F4N2O3 [M+H]+ 361.1170, found 361.1187.
4.3.7 Pyrroloindoline 21g
The dr was determined to be 10:1 by 1H NMR analysis of the crude reaction mixture. The crude residue was purified by flash chromatography (0→10% ethyl acetate/hexanes) to yield 44.8 mg (61% yield) of 21g. The enantiomeric excess was determined to be 90% by chiral SFC analysis (AD-H, 2.5 mL/min, 5% IPA in CO2, λ = 254 nm): tR(major) = 3.8 min tR(minor) = 2.5 min. The major diastereomer was separated by flash chromatography (0→10% ethyl acetate/hexanes). 1H NMR (500 MHz, CDCl3; compound exists as a 1.8:1 mixture of rotamers, the major rotamer is denoted by *, minor rotamer denoted by §) δ 6.75-6.70 (m, 1H*, 1H§), 6.67-6.65 (m, 1H§), 6.64 (d, J = 2.4 Hz, 1H*), 6.55 (d, J = 8.5 Hz, 1H§), 6.45 (d, J = 8.5 Hz, 1H*), 5.55 (s, 1H*), 5.24 (s, 1H§), 4.74 (d, J = 9.4 Hz, 1H*), 4.38 (t, J = 8.0 Hz, 1H§), 3.81 (s, 3H*), 3.77 (s, 3H§), 3.76 (s, 3H*, 3H§), 3.04 (s, 3H*), 2.86 (s, 3H§), 2.59 (dd, J = 13.5, 9.6 Hz, 1H*), 2.52 (dd, J = 13.0, 8.7 Hz, 1H§), 2.35 (dd, J = 13.5, 2.5 Hz, 1H*), 2.09-2.00 (m, 1H§), 1.48 (s, 3H§), 1.38 (s, 3H*); 13C NMR (125 MHz, CDCl3; compound exists as a 1.8:1 mixture of rotamers, the major rotamer is denoted by *, minor rotamer denoted by §) δ 172.5*, 170.7§, 159.0§ (q, JC-F = 37.1, 36.6 Hz), 154.6§, 153.6*, 143.7*, 143.6§, 135.9§, 135.7*, 116.1* (q, JC-F = 288.3 Hz), 113.3§, 113.0*, 111.4§, 109.02*, 108.98*, 108.8§, 94.1*, 92.3§, 61.0§, 60.3*, 56.0*, 55.9§, 53.6§, 53.1*, 52.6§, 49.3*, 43.9*, 39.9§, 38.1*, 36.8§, 23.5*§; IR (NaCl/thin film): 2958, 2833, 1750, 1691, 1598, 1497, 1434, 1384, 1356, 1341, 1281, 1259, 1231, 1203, 1154, 1093, 1062, 1031, 986, 870, 844, 808, 756, 728 cm−1; [a]D25 = −103.2 (c = 0.82, CHCl3). HRMS (APCI) calc'd for C17H19F3N2O4 [M+H]+ 373.1370, found 373.1383.
4.3.8 Pyrroloindoline 21h
The dr was determined to be 13:1 by 1H NMR analysis of the crude reaction mixture. The crude residue was purified by flash chromatography (5→20% ethyl acetate/hexanes) to yield 74.1 mg (86% yield) of 21h. The enantiomeric excess was determined to be 87% by chiral SFC analysis (AD-H, 2.5 mL/min, 8% IPA in CO2, λ = 254 nm): tR(major) = 5.3 min tR(minor) = 8.1 min. The major diastereomer was separated by flash chromatography (5% ethyl acetate/hexanes). 1H NMR (500 MHz, CDCl3; compound exists as a 2.6:1 mixture of rotamers, the major rotamer is denoted by *, minor rotamer denoted by §) δ 7.32-7.15 (m, 4H*, 4H§), 7.08 (br t, J = 7.7 Hz, 3H*, 3H§), 6.95-6.75 (m, 1H*, 1H§), 6.65-6.50 (m, 1H*, 1H§), 5.72 (br s, 1H*), 5.46 (br s, 1H§), 4.65 (br d, J = 6.3 Hz, 1H*), 4.33 (br s, 1H§), 3.78 (br s, 3H*, 3H§), 3.13 (br s, 3H*), 2.90 (br s, 3H§), 2.76-1.87 (m, 6H*, 6H§); 13C NMR (125 MHz, CDCl3; compound exists as a 2.6:1 mixture of rotamers, the major rotamer is denoted by *, minor rotamer denoted by §) δ 172.6*, 170.6§, 159.0* (q, JC-F = 37.5 Hz), 150.3*§, 141.1*§, 131.9*, 131.7§, 129.0*§, 128.5*§, 128.2*§, 126.1*§, 122.0*, 121.2* (q, JC-F = 278.2 Hz), 119.0*, 117.2§, 114.9§, 109.8§, 108.3*, 90.5*, 89.2§; IR (NaCl/thin film): 3026, 2952, 1751, 1701, 1607, 1491, 1437, 1355, 1204, 1151, 985, 749 cm−1; [a]D25 = −128.3 (c = 1.22, CH2Cl2). HRMS (MM) calc'd for C23H23F3N2O3 [M+H]+ 433.1734, found 433.1750.
4.3.9 Pyrroloindoline 21i
The dr was determined to be 5:1 by 1H NMR analysis of the crude reaction mixture. The crude residue was purified by flash chromatography (5→15% ethyl acetate/hexanes) to yield 61.1 mg of white needles 21i (84% yield). The enantiomeric excess was determined to be 92% by chiral SFC analysis (AD-H, 2.5 mL/min, 5% IPA in CO2, λ = 254 nm): tR(major) = 7.8 min tR(minor) = 3.5 min. The major and minor diastereomers were separated by reverse phase preparatory HPLC (50→95% acetonitrile/water, 0.05% trifluoroacetic acid). 1H NMR (500 MHz, CDCl3; compound exists as a 4.9:1 mixture of rotamers, the major rotamer is denoted by *, minor rotamer denoted by §) δ 7.14 (td, J = 7.7, 1.3 Hz, 1H*, 1H§), 7.03 (br d, J = 7.3 Hz, 1H*, 1H§), 6.90-6.80 (br s, 1H§), 6.76 (t, J = 7.4 Hz, 1H*), 6.67-6.58 (br s, 1H§), 6.54 (d, J = 7.9 Hz, 1H*), 5.81 (dtd, J = 16.5, 11.6, 11.1, 6.2 Hz, 1H*, 1H§), 5.74 (s, 1H*), 5.51 (br s, 1H§), 5.27 (br d, J = 17.1 Hz, 1H*, 1H§), 5.14 (br d, J = 10.2 Hz, 1H*, 1H§), 4.72 (d, J = 9.1 Hz, 1H*), 4.34 (br s, 1H§), 4.25 (dd, J = 16.5, 3.6 Hz, 1H*), 4.04 (dd, J = 16.5, 6.2 Hz, 1H*), 4.00-3.94 (m, 1H§), 3.82 (s, 3H*), 3.76 (s, 3H§), 2.62 (dd, J = 13.3, 9.7 Hz, 1H*), 2.58-2.49 (m, 1H§), 2.40 (dd, J = 13.5, 2.6 Hz, 1H*), 2.11 (br s, 1H§), 1.48 (s, 3H§), 1.39 (s, 3H*); 13C NMR (125 MHz, CDCl3; compound exists as a 4.9:1 mixture of rotamers, the major rotamer is denoted by *, minor rotamer denoted by §) δ 172.6*, 170.6§, 158.9§ (q, JC-F = 36.9 Hz), 148.4*, 134.7*, 133.8*, 133.3§, 128.7*, 121.7§, 121.5*, 120.3§, 118.8*, 117.8§, 116.8*, 116.0* (q, JC-F = 288.4 Hz), 110.8§, 108.5*, 91.3*, 89.6§, 60.9§, 59.9*, 53.0*, 52.5§, 51.8*, 50.4§, 49.4*, 44.3*, 40.8§, 23.7*, 23.4§; IR (NaCl/thin film): 3053, 2958, 2877, 1751, 1700, 1691, 1685, 1642, 1608, 1487, 1437, 1384, 1356, 1340, 1309, 1257, 1205, 1151, 1106, 1093, 1027, 991, 925, 841, 817, 792, 744 cm−1; [a]D25 = −146.2 (c = 1.60, CHCl3). HRMS (APCI) calc'd for C18H19F3N2O3 [M+H]+ 369.1421, found 369.1416.
4.3.10 Pyrroloindoline ent-endo-21i
1H NMR (500 MHz, CDCl3; compound exists as a 15.0:1 mixture of rotamers, the major rotamer is denoted by *, minor rotamer denoted by §) δ 7.11-7.07 (m, 1H§), 7.06 (td, J = 7.6, 1.3 Hz, 1H*), 7.06-7.03 (m, 1H§), 6.98 (dd, J = 7.3, 0.7 Hz, 1H*), 6.73 (td, J = 7.4, 1.0 Hz, 1H §), 6.66 (td, J = 7.4, 1.0 Hz, 1H*), 6.46-6.44 (m, 1H§), 6.44 (d, J = 7.9 Hz, 1H*), 5.87 (dddd, J = 17.1, 10.3, 5.9, 5.1 Hz, 1H*), 5.82-5.75 (m, 1H§), 5.57 (s, 1H*), 5.53 (d, J = 1.4 Hz, 1H§), 5.30 (dq, J = 17.2, 1.7 Hz, 1H*), 5.22-5.19 (m, 1H§), 5.16 (dq, J = 10.2, 1.5 Hz, 1H*), 5.10 (dd, J = 9.5, 4.5 Hz, 1H§), 4.75 (dt, J = 8.4, 1.3 Hz, 1H*), 4.24-4.13 (m, 2H*), 3.85 (ddd, J = 49.3, 17.2, 5.1 Hz, 2H§), 3.53 (s, 3H§), 3.17 (s, 3H*), 2.85 (d, J = 13.0 Hz, 1H*), 2.50 (dd, J = 13.2, 4.5 Hz, 1H§), 2.39 (dd, J = 13.0, 8.4 Hz, 1H*), 2.25 (dd, J = 13.3, 9.6 Hz, 1H§), 1.44 (s, 3H§), 1.42 (s, 3H*); 13C NMR (125 MHz, CDCl3; compound exists as a 15.0:1 mixture of rotamers, the major rotamer is denoted by *, minor rotamer denoted by §) δ 170.0*, 156.8§ (q, JC-F = 36.8 Hz), 149.3*, 147.8§, 134.2*, 133.2§, 132.6§, 132.0*, 128.9*, 128.6§, 122.5*, 121.7§, 118.7§, 118.0*, 117.1§, 116.3*, 116.1* (q, JC-F = 288.6 Hz), 108.0§, 106.8*, 88.7§, 88.1*, 60.3§, 60.1* (q, JC-F = 3.1 Hz), 52.6§, 52.5§, 52.4*, 50.6*, 49.0*, 46.6§, 42.5*, 41.4§, 25.7*, 22.9§; IR (NaCl/thin film): 3055, 2954, 2869, 1760, 1742, 1699, 1607, 1490, 1447, 1436, 1338, 1317, 1274, 1254, 1208, 1183, 1144, 1105, 1093, 1032, 999, 942, 922, 887, 842, 859, 742 cm−1; [a]D25 = +188.1 (c = 0.275, CHCl3). HRMS (APCI) calc'd for C18H19F3N2O3 [M+H]+ 369.1421, found 369.1429.
4.3.11 Pyrroloindoline 21j
The dr was determined to be 18:1 by 1H NMR analysis of the crude reaction mixture. The crude residue was purified by flash chromatography (0→10% ethyl acetate/hexanes) to yield 68.6 mg (84% yield) of 21j. The enantiomeric excess was determined to be 92% by chiral SFC analysis (OD, 2.5 mL/min, 3% IPA in CO2, λ = 254 nm): tR(major) = 6.5 min tR(minor) = 5.6 min. The major diastereomer was separated by flash chromatography (0→10% ethyl acetate/hexanes). 1H NMR (500 MHz, CDCl3; compound exists as a 2.1:1 mixture of rotamers the major rotamer is denoted by *, minor rotamer denoted by §) δ 7.16 (d, J = 8.1 Hz, 1H*, 1H§), 7.02 (s, 1H§), 6.99 (s, 1H*), 6.52 (d, J = 7.7 Hz, 1H§), 6.45 (d, J = 8.2 Hz, 1H*), 6.00 (dd, J = 17.3, 10.5 Hz, 1H*, 1H§), 5.60 (s, 1H*), 5.31 (s, 1H§), 5.07 – 4.96 (m, 2H*, 2H§), 4.73 (d, J = 9.3 Hz, 1H*), 4.47 – 4.41 (m, 1H§), 3.82 (s, 1H*), 3.77 (s, 1H§), 3.06 (s, 1H*), 2.86 (s, 1H§), 2.60 (dd, J = 13.1, 9.9 Hz, 1H*), 2.52 (t, J = 10.7 Hz, 1H§), 2.38 (d, J = 12.5 Hz, 1H*), 2.14 – 1.98 (m, 1H§), 1.57 – 1.31 (m, 9H*, 9H§); 13C NMR (126 MHz, CDCl3) ; IR (NaCl/thin film): 3081, 2965, 2874, 2822, 1753, 1698, 1618, 1496, 1434, 1359, 1283, 1257, 1204, 1156, 1117, 1054, 995, 912, 844, 813; [a]D25 = −115 (c = 0.450, CHCl3). HRMS (ESI) calc'd for C21H25F3N2O3 [M+H]+ 3 411.1890, found 411.1901.
4.4 General procedure for the formal (3 + 2) cycloaddition/in situ reduction
To a flame-dried flask was added indole (0.20 mmol, 1.00 equiv), acrylate (0.20 mmol, 1.00 equiv), and (R)-3,3'-dichloro-BINOL (0.04 mmol, 0.20 equiv). The flask was charged with CH2Cl2 (1.5 mL), followed by addition of SnCl4 (0.24 mmol, 1.20 equiv unless specifically indicated, 1 M in CH2Cl2). NaBH4 (0.30 mmol, 1.50 equiv) was then added, and the reaction was stirred at room temperature for 24 h (unless specifically indicated). The reaction was quenched by diluting with 1 mL MeCN and 1 mL 1 M HCl, followed by addition of 5 mL H2O. The aqueous layer was extracted with ethyl acetate (3 × 15 mL) and the combined organic layers were washed with saturated NaHCO3(aq) (10 mL). The aqueous layer was extracted with ethyl acetate (10 mL). The combined organic layers were dried (Na2SO4), filtered and concentrated. The crude residue was purified by flash chromatography.
4.4.1 Indoline 28a
Prepared from 1,3-dimethyl-1H-indole 19 and methyl 2-trifluoroacetamidoacrylate using the general procedure. The dr was determined to be 15:1 by 1H NMR analysis of the crude reaction mixture. The crude residue was purified by flash chromatography (5→25% ethyl acetate/hexanes) to yield 64.1 mg (93% yield) of 28a, a pale yellow oil. The enantiomeric excess of the major diastereomer was determined to be 92% by chiral SFC analysis (AD-H, 2.5 mL/min, 5% IPA in CO2, λ = 254 nm): tR(major) = 9.9 min tR(minor) = 5.9 min. The major diastereomer was separated by flash chromatography (5% ethyl acetate/hexanes). 1H NMR (500 MHz, CDCl3) δ 7.23 (br d, J = 5.9 Hz, 1H), 7.14 (td, J = 7.7, 1.2 Hz, 1H), 6.99 (dd, J = 7.3, 0.8 Hz, 1H), 6.76 (td, J = 7.4, 0.8 Hz, 1H), 6.54 (d, J = 7.9 Hz, 1H), 4.27 (br td, J = 7.7, 4.7 Hz, 1H), 3.65 (s, 3H), 3.31 (d, J = 9.1 Hz, 1H), 2.98 (d, J = 9.1 Hz, 1H), 2.74 (s, 3H), 2.21 (dd, J = 14.7, 4.7 Hz, 1H), 2.15 (dd, J = 14.7, 8.2 Hz, 1H), 1.40 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 171.1, 156.6 (q, JC-F = 37.5 Hz), 152.1, 153.3, 128.5, 122.4, 119.0, 115.6 (q, JC-F = 287.9 Hz), 108.4, 68.3, 52.7, 51.2, 42.8, 42.1, 35.8, 26.1; IR (NaCl/thin film): 3319, 2956, 2858, 2811, 1751, 1718, 1607, 1559, 1491, 1452, 1209, 1179, 744 cm−1; [a]D25 = +79.6 (c = 1.32, CH2Cl2). HRMS (MM) calc'd for [M+H]+ C16H19F3N2O3 345.1421, found 345.1423.
4.4.2 Indoline 28b
Prepared from 1,3,4-trimethyl-1H-indole and methyl 2-trifluoroacetamidoacrylate using the general procedure. The dr was determined to be 11:1 by 1H NMR analysis of the crude reaction mixture. The crude residue was purified by flash chromatography (5→25% ethyl acetate/hexanes) to yield 56.8 mg (79% yield, yellow oil) of 28b as a single diastereomer. The enantiomeric excess of the major diastereomer was determined to be 93% by chiral SFC analysis (AD-H, 2.5 mL/min, 6% IPA in CO2, λ = 254 nm): tR(major) = 3.9 min tR(minor) = 3.5 min. 1H NMR (500 MHz, CDCl3) δ 7.64 (br d, J = 4.9 Hz, 1H), 7.06 (t, J = 7.7 Hz, 1H), 6.54 (d, J = 7.6 Hz, 1H), 6.42 (d, J = 7.9 Hz), 4.10 (ddd, J = 9.0, 6.5, 4.2 Hz, 1H), 3.65 (s, 3H), 3.32 (d, J = 9.3 Hz, 1H), 2.95 (d, J = 9.2 Hz, 1H), 2.71 (s, 3H), 2.45 (dd, J = 15.0, 4.0 Hz, 1H), 2.30 (s, 3H), 2.13 (dd, J = 14.8, 8.9 Hz, 1H), 1.50 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 171.0, 156.7 (q, JC-F = 37.5 Hz), 152.8, 134.5, 131.5, 128.8, 122.4, 115.6 (q, JC-F = 287.9 Hz), 106.7, 68.6, 52.7, 51.7, 43.7, 40.9, 35.9, 26.6, 18.7; IR (NaCl/thin film): 3315, 2956, 2812, 1750, 1710, 1593, 1559, 1484, 1457, 1209, 1179, 774 cm−1; [a]D25 = +66.0 (c = 1.04, CH2Cl2). HRMS (MM) calc'd for C17H21F3N2O3 [M+H]+ 359.1577, found 359.1591.
4.4.3 Indoline 28c
Prepared from 1,3,5-trimethyl-1H-indole and methyl 2-trifluoroacetamidoacrylate using the general procedure. The dr was determined to be 17:1 by 1H NMR analysis of the crude reaction mixture. The crude residue was purified by flash chromatography (10→25% ethyl acetate/hexanes) to yield 52.6 mg (73% yield) of 28c, a yellow oil. The enantiomeric excess of the major diastereomer was determined to be 89% by chiral SFC analysis (AD-H, 2.5 mL/min, 6% IPA in CO2, λ = 254 nm): tR(major) = 4.8 min tR(minor) = 3.4 min. The major diastereomer was separated by flash chromatography (7% ethyl acetate/hexanes). 1H NMR (500 MHz, CDCl3) δ 7.43 (br d, J = 4.8 Hz, 1H), 6.95 (d, J = 7.9 Hz, 1H), 6.81 (s, 1H), 6.47 (d, J = 7.9 Hz, 1H), 4.16 (q, J = 6.5 Hz, 1H), 3.67 (s, 3H), 3.27 (d, J = 9.1 Hz, 1H), 2.91 (d, J = 9.1 Hz, 1H), 2.70 (s, 3H), 2.25 (s, 3H), 2.17 (d, J = 6.5 Hz, 2H), 1.39 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 171.0, 156.7 (q, JC-F = 37.5 Hz), 150.0, 135.5, 128.9, 128.8, 123.3, 115.6 (q, JC-F = 287.9 Hz), 108.7, 69.0, 52.7, 51.5, 42.7, 42.1, 36.4, 26.07, 20.7; IR (NaCl/thin film): 3326, 2955, 2922, 2863, 2806, 1752, 1719, 1555, 1499, 1452, 1209, 1163, 806 cm−1; [a]D25 = +42.3 (c = 0.87, CH2Cl2). HRMS (MM) calc'd for C17H21F3N2O3 [M+H]+ 359.1577, found 359.1565.
4.4.4 Indoline 28d
Prepared from 1,3,6-trimethyl-1H-indole and methyl 2-trifluoroacetamidoacrylate using the general procedure. The dr was determined to be 17:1 by 1H NMR analysis of the crude reaction mixture. The crude residue was purified by flash chromatography (5→30% ethyl acetate/hexanes) to yield 65.0 mg (91% yield) of 28d, a yellow oil. The enantiomeric excess of the major diastereomer was determined to be 92% by chiral SFC analysis (AD-H, 2.5 mL/min, 5% IPA in CO2, λ = 254 nm): tR(major) = 11.1 min tR(minor) = 5.0 min. The major diastereomer was separated by flash chromatography (5% ethyl acetate/hexanes). 1H NMR (500 MHz, CDCl3) δ 7.29 (d, J = 5.7 Hz, 1H), 6.88 (d, J = 7.5 Hz, 1H), 6.58 (d, J = 7.5 Hz, 1H), 6.37 (s, 1H), 4.22 (td, J = 7.5, 4.9 Hz), 3.66 (s, 3H), 3.29 (d, J = 9.1 Hz, 1H), 2.96 (d, J = 9.1 Hz, 1H), 2.72 (s, 3H), 2.30 (s, 3H), 2.20-2.11 (m, 2H), 1.38 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 171.1, 156.6 (q, JC-F = 37.5 Hz), 152.3, 138.6, 132.5, 122.2, 119.8, 115.6 (q, JC-F = 287.8 Hz), 109.3, 68.6, 52.7, 51.4, 42.5, 42.2, 35.8, 26.2, 21.6; IR (NaCl/thin film): 3321, 2956, 2923, 2870, 2804, 1750, 1716, 1615, 1557, 1497, 1455, 1208, 1179, 802 cm−1; [a]D25 = +76.0 (c = 1.56, CH2Cl2). HRMS (MM) calc'd for C17H21F3N2O3 [M+H]+ 359.1577, found 359.1577.
4.4.5 Indoline 28e
Prepared from 1,3,7-trimethyl-1H-indole and methyl 2-trifluoroacetamidoacrylate using the general procedure. The dr was determined to be 17:1 by 1H NMR analysis of the crude reaction mixture. The crude residue was purified by flash chromatography (5→30% ethyl acetate/hexanes) to yield 58 mg (81% yield) of 28e, a pale yellow oil. The enantiomeric excess of the major diastereomer was determined to be 94% by chiral SFC analysis (AD-H, 2.5 mL/min, 6% IPA in CO2, λ = 254 nm): tR(major) = 4.3 min tR(minor) = 3.3 min. The major diastereomer was separated by flash chromatography (5% ethyl acetate/hexanes). 1H NMR (500 MHz, CDCl3) δ 7.44 (d, J = 4.7 Hz, 1H), 6.90 (d, J = 7.5 Hz, 1H), 6.86 (d, J = 7.4 Hz, 1H), 6.73 (t, J = 7.4 Hz, 1H), 4.14-4.08 (m, 1H), 3.66 (s, 3H), 3.29 (d, J = 9.6 Hz, 1H), 2.97 (d, J = 9.6 Hz, 1H), 2.93 (s, 3H), 2.37 (s, 3H), 2.17-2.06 (m, 2H), 1.40 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 171.1, 156.7 (q, JC-F = 37.5 Hz), 149.9, 136.1, 131.8, 121.2, 120.4, 120.3, 115.6 (q, JC-F = 287.8 Hz), 69.7, 52.7, 51.3, 42.8, 42.3, 39.5, 26.6, 19.5; IR (NaCl/thin film): 3322, 2959, 2924, 1750, 1713, 1557, 1480, 1456, 1412, 1208, 1180, 1071, 750 cm−1; [a]D25 = +84.9 (c = 1.20, CH2Cl2). HRMS (APCI) calc'd for C17H21F3N2O3 [M+H]+ 359.1577, found 359.1595.
4.4.6 Indoline 28f
Prepared from 5-fluoro-1,3-dimethyl-1H-indole and methyl 2-trifluoroacetamidoacrylate using the general procedure. The dr was determined to be 13:1 by 1H NMR analysis of the crude reaction mixture. The crude residue was purified by flash chromatography (10→30% ethyl acetate/hexanes) to yield 57.0 mg (79% yield) of 28f, a pale yellow oil. The enantiomeric excess of the major diastereomer was determined to be 90% by chiral SFC analysis (AD-H, 2.5 mL/min, 6% IPA in CO2, λ = 254 nm): tR(major) = 3.9 min tR(minor) = 2.9 min. The major diastereomer was separated by preparatory TLC (40% CH2Cl2/hexanes then 50% CH2Cl2/hexanes). 1H NMR (300 MHz, CDCl3) δ 7.31 (br d, JC-H = 5.8 Hz, 1H), 6.83 (td, JC-H = 8.8, 2.6 Hz, 1H), 6.72 (dd, JC-H = 8.2, 2.6 Hz, 1H), 6.44 (dd, JC-H = 8.5, 4.1 Hz, 1H), 4.25 (td, JC-H = 7.7, 4.8 Hz, 1H), 3.68 (s, 3H), 3.31 (d, JC-H = 9.2 Hz, 1H), 2.97 (d, JC-H = 9.2 Hz, 1H), 2.70 (s, 3H), 2.21 (dd, JC-H = 14.7, 4.8 Hz, 1H), 2.12 (dd, JC-H = 14.7, 8.1 Hz, 1H), 1.38 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 171.0, 157.2 (d, JC-F = 237.2 Hz), 156.6 (q, JC-F = 37.8 Hz), 148.4, 137.1 (d, JC-F = 7.1 Hz), 116.7, 114.5 (d, JC-F = 23.3 Hz), 110.1 (d, JC-F = 24.0 Hz), 108.8 (d, JC-F = 8.1 Hz), 68.6, 52.8, 51.1, 42.8, 42.0, 36.4, 26.1; IR (NaCl/thin film): 3319, 2958, 2866, 2811, 1745, 1711, 1552, 1494, 1468, 1267, 1210, 1179, 808 cm−1; [a]D25 = +63.7 (c = 0.62, CH2Cl2). HRMS (ESI) calc'd for C16H18F4N2O3 [M+H]+ 363.1326, found 363.1334.
4.4.7 Indoline 28g
Prepared from 5-methoxy-1,3-dimethyl-1H-indole and methyl 2-trifluoroacetamidoacrylate using the general procedure. The reaction was allowed to run for 18.5 h. The dr was determined to be 14:1 by 1H NMR analysis of the crude reaction mixture. The crude residue was purified by flash chromatography (10→30% ethyl acetate/hexanes) to yield 68.5 mg (91% yield) of 28g, a yellow oil. The enantiomeric excess of the major diastereomer was determined to be 88% by chiral SFC analysis (AD-H, 2.5 mL/min, 7% IPA in CO2, λ = 254 nm): tR(major) = 7.1 min tR(minor) = 3.6 min. 1H NMR (500 MHz, CDCl3) δ 7.81 (br d, JC-H = 5.0 Hz, 1H), 6.70 (dd, JC-H = 8.4, 1.9 Hz, 1H), 6.62 (d, JC-H = 1.8 Hz, 1H), 6.50 (d, JC-H = 8.3 Hz, 1H), 4.01 (dd, JC-H = 13.1, 6.3 Hz, 1H), 3.74 (s, 3H), 3.67 (s, 3H), 3.28 (d, JC-H = 8.6 Hz, 1H), 2.89 (d, JC-H = 9.1 Hz, 1H), 2.68 (s, 3H), 2.23-2.08 (m, 2H), 1.30 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 171.0, 156.8 (q, JC-F = 37.5 Hz), 154.1, 146.1, 137.0, 115.6 (q, JC-F = 287.8 Hz), 113.1, 110.8, 109.6, 69.0, 55.8, 52.7, 51.5, 43.0, 42.0, 36.9, 26.2; IR (NaCl/thin film): 3319, 2955, 2804, 1751, 1718, 1555, 1496, 1468, 1214, 1179, 1031 cm−1; [a]D25 = +26.3 (c = 1.24, CH2Cl2). HRMS (APCI) calc'd for C17H21F3N2O4 [M+H]+ 375.1526, found 375.1542.
4.4.8 Indoline 28h
Prepared from 1-allyl-3-methyl-1H-indole and methyl 2-trifluoroacetamidoacrylate using the general procedure, except the formal (3 + 2) cycloaddition was allowed to run for 24 h before adding NaBH4. After adding NaBH4, the reaction was allowed to run for another 24 h. The dr was determined to be 5:1 by 1H NMR analysis of the crude reaction mixture. The crude residue was purified by flash chromatography (5→20% ethyl acetate/hexanes) to yield 60.0 mg (81% yield) of 28h, a yellow oil. The enantiomeric excess of the major diastereomer was determined to be 90% by chiral SFC analysis (AD-H, 2.5 mL/min, 5% IPA in CO2, λ = 254 nm): tR(major) = 7.2 min tR(minor) = 6.2 min. Isolated as a 5:1 mixture of diastereomers; the major diastereomer is denoted by *, minor diastereomer denoted by §. 1H NMR (500 MHz, CDCl3) δ 8.26 (br d, J = 8.5 Hz, 1H§), 7.16-7.05 (m, 2H*, 1H§), 6.98 (ddd, J = 7.4, 1.2, 0.5 Hz, 1H*, 1H§), 6.78 (td, J = 7.4, 1.0 Hz, 1H§), 6.72 (td, J = 7.4, 1.0 Hz, 1H*), 6.64 (d, J = 7.9 Hz, 1H§), 6.56 (d, J = 7.9 Hz, 1H*), 5.93-5.83 (m, 1H*, 1H§), 5.32-5.20 (m, 1H*, 1H§), 4.81-4.75 (m, 1H§), 4.40 (td, J = 7.6, 5.0, 1H*), 3.79 (ddt, J = 15.0, 5.9, 1.4 Hz, 1H*, 1H§), 3.73-3.70 (m, 1H§), 3.65-3.59 (m, 1H*), 3.63 (s, 3H*), 3.42 (s, 3H§), 3.31 (d, J = 9.3 Hz, 1H*), 3.28 (d, J = 9.6 Hz, 1H§), 3.07 (d, J = 9.6 Hz, 1H§), 3.05 (d, J = 9.3 Hz, 1H*), 2.35 (ddd, J = 14.8, 5.9, 0.6 Hz, 1H§), 2.23 (dd, J = 14.6, 4.9 Hz, 1H*), 2.18 (dd, J = 14.9, 4.6 Hz, 1H§), 2.13 (dd, J = 14.7, 7.8 Hz, 1H*), 1.41 (s, 3H§), 1.38 (s, 3H*); 13C NMR (100 MHz, CDCl3) δ 171.2*, 170.1§, 156.6* (q, JC-F = 37.6 Hz), 150.8*, 135.4*, 133.3*, 132.4§, 128.6§, 128.4*, 123.3§, 122.5*, 119.4§, 118.9§, 118.7*, 118.1*, 115.5* (q, JC-F = 287.9 Hz), 109.9§, 108.4*, 65.2§, 65.1*, 52.7*, 52.5§, 52.1§, 51.8*, 50.9*, 50.5§, 42.7§, 42.6*, 42.5§, 42.1*, 27.8§, 26.1*; IR (NaCl/thin film): 3316, 2957, 2923, 1750, 1718, 1605, 1554, 1487, 1460, 1437, 1209, 1165, 744 cm−1; [a]D25 = +33.412 (c = 1.62, CH2Cl2). HRMS (MM) calc'd for C18H21F3N2O3 [M+H]+ 371.1577, found 371.1582.
4.4.9 Indoline 28i
Prepared from 3-butyl-1-methyl-1H-indole and methyl 2-trifluoroacetamidoacrylate using the general procedure. The dr was determined to be >20:1 by 1H NMR analysis of the crude reaction mixture. The crude residue was purified by flash chromatography (5→20% ethyl acetate/hexanes) to yield 63.4 mg (82% yield) of 28i, a pale yellow oil. The enantiomeric excess of the major diastereomer was determined to be 92% by chiral SFC analysis (AD-H, 2.5 mL/min, 5% IPA in CO2, λ = 254 nm): tR(major) = 7.2 min tR(minor) = 5.3 min. The major diastereomer was separated by flash chromatography (5% ethyl acetate/hexanes). 1H NMR (500 MHz, CDCl3) δ 7.30 (br d, JC-H = 5.5 Hz, 1H), 7.13 (td, JC-H = 7.7, 1.2 Hz, 1H), 6.96 (dd, JC-H = 7.3, 0.7 Hz, 1H), 6.75 (t, JC-H = 7.3 Hz, 1H), 6.53 (d, JC-H = 7.8 Hz, 1H), 4.22-4.15 (m, 1H), 3.64 (s, 3H), 3.24 (d, JC-H = 9.3 Hz, 1H), 3.08 (d, JC-H = 9.3 Hz, 1H), 2.74 (s, 3H), 2.23 (dd, JC-H = 14.7, 8.4 Hz, 1H), 2.17 (dd, JC-H = 14.7, 4.7 Hz, 1H), 1.86-1.77 (m, 1H), 1.69-1.57 (m, 1H), 1.40-1.24 (m, 3H), 1.19-1.08 (m, 1H), 0.89 (t, JC-H = 7.2 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 171.2, 156.6 (q, JC-F = 37.5 Hz), 152.3, 134.3, 128.6, 123.0, 119.0, 115.5 (q, JC-F = 287.8 Hz), 108.4, 66.4, 52.7, 51.2, 46.2, 40.6, 39.3, 35.9, 26.5, 23.2, 14.0; IR (NaCl/thin film): 3319, 2956, 2932, 2860, 2809, 1751, 1718, 1606, 1559, 1491, 1465, 1207, 1178, 743 cm−1; [a]D25 = +62.0 (c = 1.15, CH2Cl2). HRMS (ESI) calc'd for C19H25F3N2O3 [M+H]+ 387.1890, found 387.1902.
4.4.10 Indoline 28j
Prepared from 1-methyl-3-phenethyl-1H-indole and methyl 2-trifluoroacetamidoacrylate using the general procedure, except the formal (3 + 2) cycloaddition was allowed to run for 24 h before adding NaBH4. After adding NaBH4, the reaction was allowed to run for another 24 h. The dr was determined to be 12:1 by 1H NMR analysis of the crude reaction mixture. The crude residue was purified by flash chromatography (5→20% ethyl acetate/hexanes) to yield 77.6 mg (89% yield) of 28j. The enantiomeric excess of the major diastereomer was determined to be 89% by chiral SFC analysis (AD-H, 2.5 mL/min, 7% IPA in CO2, λ = 254 nm): tR(major) = 9.5 min tR(minor) = 8.1 min. The major diastereomer was separated by flash chromatography (10% ethyl acetate/hexanes). 1H NMR (400 MHz, CDCl3) δ 7.30-7.22 (m, 3H), 7.21-7.11 (m, 4H), 6.99 (dd, JC-H = 7.4, 1.0 Hz, 1H), 6.75 (td, JC-H = 7.4, 0.7 Hz, 1H), 6.55 (d, JC-H = 7.9 Hz, 1H), 4.27 (dt, JC-H = 12.6, 6.4 Hz, 1H), 3.63 (s, 3H), 3.30 (d, JC-H = 9.3 Hz, 1H), 3.17 (d, JC-H = 9.3 Hz, 1H), 2.76 (s, 3H), 2.68 (td, JC-H = 13.0, 5.0 Hz, 1H), 2.47 (td, JC-H = 12.9, 4.7 Hz, 1H), 2.34-2.22 (m, 2H), 2.13 (ddd, JC-H = 13.7, 12.3, 5.1 Hz, 1H), 1.97 (ddd, JC-H = 13.8, 12.6, 4.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 171.1, 156.6 (q, JC-F = 37.5 Hz), 152.4, 141.7, 133.5, 128.7, 128.5, 128.2, 126.0, 122.9, 119.0, 115.5 (q, JC-F = 287.8 Hz), 108.4, 66.0, 52.7, 51.1, 46.3, 41.5, 40.7, 35.8, 30.8; IR (NaCl/thin film): 3317, 2951, 2858, 2812, 1749, 1716, 1606, 1555, 1494, 1453, 1208, 1178, 745 cm−1; [a]D26 = +23.1 (c = 0.87, CH2Cl2). HRMS (MM) calc'd for C23H25F3N2O3 [M+H]+ 435.1890, found 435.1882.
4.4.11 Indoline 28k
Prepared from 1-methyl-3-(2-((triisopropylsilyl)oxy)ethyl)-1H-indole and methyl 2-trifluoroacetamidoacrylate using the general procedure, except the formal (3 + 2) cycloaddition was allowed to run for 20.5 h before adding NaBH4. After adding NaBH4, the reaction was allowed to run for another 24 h. The dr was determined to be >20:1 by 1H NMR analysis of the crude reaction mixture. The crude residue was purified by flash chromatography (5→30% ethyl acetate/hexanes) to yield 68.3 mg (64% yield) of 28k, a yellow oil. The enantiomeric excess of the major diastereomer was determined to be 85% by chiral SFC analysis (AD-H, 2.5 mL/min, 5% IPA in CO2, λ = 254 nm): tR(major) = 8.7 min tR(minor) = 7.5 min. 1H NMR (500 MHz, CDCl3) δ 7.51 (br s, 1H), 7.15 (t, JC-H = 7.6 Hz, 1H), 7.00 (d, JC-H = 7.2 Hz, 1H), 6.75 (t, JC-H = 7.1 Hz, 1H), 6.54 (d, JC-H = 7.3 Hz, 1H), 4.22-4.15 (m, 1H), 3.81 (dd, JC-H = 6.96, 5.35 Hz, 2H), 3.64 (s, 3H), 3.34-3.26 (m, 1H), 3.26-3.18 (m, 1H), 2.74 (s, 3H), 2.46-2.36 (m, 1H), 2.24 (dd, JC-H = 14.7, 3.7 Hz, 1H), 2.11-1.94 (m, 2H), 1.08-1.03 (m, 21H); 13C NMR (125 MHz, CDCl3) δ 171.1, 156.7 (q, JC-F = 37.6 Hz), 152.1, 134.3, 128.7, 123.0, 118.9, 115.6 (q, JC-F = 287.8 Hz), 108.5, 67.6, 60.0, 52.6, 51.2, 45.3, 41.2, 39.5, 18.0, 18.0, 11.9; IR (NaCl/thin film): 3323, 2943, 2866, 1719, 1606, 1552, 1491, 1463, 1207, 1175, 1104, 882, 742 cm−1; [a]D25 = +27.4 (c = 0.85, CH2Cl2). HRMS (APCI) calc'd for C26H41F3N2O4Si [M+H]+ 531.2860, found 531.2883.
4.5 General procedure for in situ monitoring of the formal (3 + 2) cycloaddition by 1H NMR
In the glovebox, a 1 M solution of 1,3-dimethylindole, a 1 M solution of benzyl trifluoroacetamidoacrylate (with 0.3 equiv 1,4-diethylbenzene as an internal standard), a 0.72 M solution of SnCl4, and a 0.0675 M solution of (R)-BINOL in CD2Cl2 were made. To an oven-dried NMR tube equipped with a Teflon-lined cap were added 90 μL of the indole solution, 90 μL of the acrylate + internal standard solution, 267 μL of the (R)-BINOL solution, and 186 μL of CD2Cl2. A 1H NMR spectrum was taken (1 scan) to determine the initial ratio of substrates, (R)-BINOL, and internal standard. Immediately before beginning the collection of kinetics data, SnCl4 was added via a microsyringe through the Teflon cap of the NMR tube. The tube was inverted once, then quickly inserted into the instrument.
The concentration of acrylate over the course of the reaction was determined by integrating its resonance at 6.3 ppm, then normalizing by the internal standard's resonance at 2.74 ppm.
Scheme 6.

Re-exposure of pyrroloindoline exo-8a to SnCl4 and NaBH4
Table 1.
Catalyst screen.

| entry | BINOL derivative | yield (%)a | d.r.b | ee (%)c |
|---|---|---|---|---|
| 1 | H (7) | 88 | 10:1 | 90 |
| 2 | 6,6'-(OMe)2 (22) | 63 | 6:1 | 82 |
| 3 | 6,6'-Br2 (23) | 65 | 8:1 | 86 |
| 4 | 3,3'-Me2 (24) | 63 | 8:1 | 90 |
| 5 | 3,3'-Br2 (17) | 63 | 16:1 | 90 |
| 6 | 3,3'-Cl2 (25) | 73 | 14:1 | 91 |
Isolated yield of exo:endo mixture.
Determined by 1H NMR analysis of crude reaction mixture.
Determined by HPLC using chiral stationary phase.
Acknowledgements
We thank Dr. David vanderVelde for assistance with NMR structure determination, as well as Prof. Brian Stoltz, Dr. Scott Virgil, and the Caltech Center for Catalysis and Chemical Synthesis for access to analytical equipment. We also thank Sigma-Aldrich for a kind donation of chemicals. NMR spectra were obtained on a spectrometer funded by the NIH (RR027690). Fellowship support was provided by Natural Sciences and Engineering Research Council (NSERC) of Canada (J. N., PGS D scholarship). S. E. R. is a fellow of the Alfred P. Sloan Foundation and a Camille Dreyfus Teacher-Scholar. Financial support from the California Institute of Technology, the NIH (NIGMS RGM097582A), and the donors of the ACS Petroleum Research Fund is gratefully acknowledged.
Abbreviations used
- BINOL
1,1'-Bi(2-naphthol)
- IPA
isopropanol
- dba
dibenzylidineacetone
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
5. References and Notes
- 1.Selected indoline natural products: kapakahines A-D, Yeung BKS, Nakao Y, Kinnel RB, Carney JR, Yoshida WY, Scheuer PJ, Kelly-Borges M. J. Org. Chem. 1996;61:7168. doi: 10.1021/jo960725e.. Creasey WA. Monoterpenoid Indole Alkaloids. In: Saxton JE, Taylor EC, editors. The Chemistry of Heterocyclic Compounds. Part 4. Vol. 25. Wiley; New York: 1994. pp. 715–754. strychnine: Supplement to.. Siddiqui S, Siddiqui RH. J. Ind. Chem. Soc. 1931;8:667. ajmaline:. Subramaniam G, Hiraku O, Hayashi M, Koyano T, Komiyama K, Kam T-S. J. Nat. Prod. 2007;70:1783. doi: 10.1021/np0703747. aspidophylline A:
- 2.(a) Tuntiwachwuttikul P, Taechowisan T, Wanbanjob A, Thadaniti S, Taylor WC. Tetrahedron. 2008;64:7583. [Google Scholar]; (b) Hauser D, Weber HP, Sigg HP. Helv. Chim. Acta. 1970;53:1061. doi: 10.1002/hlca.19700530521. [DOI] [PubMed] [Google Scholar]; (c) Ding G, Jiang LH, Guo LD, Chen XL, Zhang H, Che YS. J. Nat. Prod. 2008;71:1861. doi: 10.1021/np800357g. [DOI] [PubMed] [Google Scholar]; (d) Zheng C-J, Kim C-J, Bae KS, Kim Y-H, Kim W-G. J. Nat. Prod. 2006;69:1816. doi: 10.1021/np060348t. [DOI] [PubMed] [Google Scholar]; (e) Mansour M, Lemenoli L, Levy J, Lemen J. Phytochem. 1974;13:2861. [Google Scholar]; (f) Massiot G, Thepenier P, Jacquier MJ, Lemenolivier L, Delaude C. Heterocycles. 1989;29:1435. [Google Scholar]; (g) Goodson JA, Henry TA. J. Chem. Soc., Transactions. 1925;127:1640. [Google Scholar]; (h) Polonovski M. Bull. Soc. Chim. Fr. 1916;19:46. [Google Scholar]
- 3.Selected indoline syntheses: Boger DL, Coleman RS. J. Org. Chem. 1984;49:2240.. Uchiyama M, Kameda M, Mishima O, Yokoyama N, Koike M, Kondo Y, Sakamoto T. J. Am. Chem. Soc. 1998;120:4934.. Nicolaou KC, Roecker AJ, Pfefferkorn JA, Cao G-Q. J. Am. Chem. Soc. 2000;122:2966.. Bailey WF, Mealy MJ. J. Am. Chem. Soc. 2000;122:6787.. Viswanathan R, Prabhakaran EN, Plotkin MA, Johnston JN. J. Am. Chem. Soc. 2003;125:163. doi: 10.1021/ja0284308.. Yamada K, Kurokawa T, Tokuyama H, Fukuyama T. J. Am. Chem. Soc. 2003;125:6630. doi: 10.1021/ja035303i.. Danheiser RL, Dunetz JR. J. Am. Chem. Soc. 2005;127:5776. doi: 10.1021/ja051180l.. Correa A, Tellitu I, Domínguez E, SanMartin R. J. Org. Chem. 2006;71:8316. doi: 10.1021/jo061486q.. Gilmore CD, Allan KM, Stoltz BM. J. Am. Chem. Soc. 2008;130:1558. doi: 10.1021/ja0780582.. Boal BW, Schammel AW, Garg NK. Org. Lett. 2009;11:3458. doi: 10.1021/ol901383j.
- 4.(a) Taniguchi M, Hino T. Tetrahedron. 1981;37:1487. [Google Scholar]; (b) Bruncko M, Crich D, Samy R. J. Org. Chem. 1994;59:5543. [Google Scholar]; (c) Marsden SP, Depew KM, Danishefsky SJ. J. Am. Chem. Soc. 1994;116:11143. [Google Scholar]
- 5.(a) Bui T, Syed S, Barbas CF. J. Am. Chem. Soc. 2009;131:8758. doi: 10.1021/ja903520c. [DOI] [PubMed] [Google Scholar]; (b) Austin JF, Kim SG, Sinz CJ, Xiao WJ, MacMillan DWC. Proc. Natl. Acad. Sci. U.S.A. 2004;101:5482. doi: 10.1073/pnas.0308177101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Ashimori A, Matsuura T, Overman LE, Poon DJ. J. Org. Chem. 1993;58:6949. [Google Scholar]; (b) Huang A, Kodanko JJ, Overman LE. J. Am. Chem. Soc. 2004;126:14043. doi: 10.1021/ja046690e. [DOI] [PubMed] [Google Scholar]; (c) Trost BM, Zhang Y. J. Am. Chem. Soc. 2006;128:4590. doi: 10.1021/ja060560j. [DOI] [PubMed] [Google Scholar]; (d) Ma S, Han X, Krishnan S, Virgil SC, Stoltz BM. Angew. Chem., Int. Ed. 2009;48:8037. doi: 10.1002/anie.200902943. [DOI] [PubMed] [Google Scholar]; (e) Trost BM, Quancard J. J. Am. Chem. Soc. 2006;128:6314. doi: 10.1021/ja0608139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Repka LM, Ni J, Reisman SE. J. Am. Chem. Soc. 2010;132:14418. doi: 10.1021/ja107328g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.An alternative scenario would involve a moderately enantioselective, irreversible conjugate addition, followed by a highly selective, catalyst-controlled irreversible protonation
- 9.(a) Ishihara K, Kaneeda M, Yamamoto H. J. Am. Chem. Soc. 1994;116:11179. [Google Scholar]; (b) Ishihara K, Nakamura S, Kaneeda M, Yamamoto H. J. Am. Chem. Soc. 1996;118:12854. [Google Scholar]; (c) Ishihara K, Kurihara H, Yamamoto H. J. Am. Chem. Soc. 1996;118:3049. [Google Scholar]; (d) Ishihara K, Nakashima D, Hiraiwa Y, Yamamoto H. J. Am. Chem. Soc. 2003;125:24. doi: 10.1021/ja021000x. [DOI] [PubMed] [Google Scholar]; (e) Rauniyar V, Zhai HM, Hall DG. J. Am. Chem. Soc. 2008;130:8481. doi: 10.1021/ja8016076. [DOI] [PubMed] [Google Scholar]
- 10.Kieffer ME, Repka LM, Reisman SE. J. Am. Chem. Soc. 2012;134:5131. doi: 10.1021/ja209390d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mills JE, Maryanoff CA, McComsey DF, Stanzione RC, Scott L. J. Org. Chem. 1987;52:1857–1859. [Google Scholar]
- 12.(a) Kuwano R, Sato K, Kurokawa T, Karube D, Ito Y. J. Am. Chem. Soc. 2000;122:7614, 7615. [Google Scholar]; (b) Baeza A, Pfaltz A. Chem. Eur. J. 2010;16:2036–2039. doi: 10.1002/chem.200903105. [DOI] [PubMed] [Google Scholar]; (c) Rueping M, Brinkmann C, Antonchick AP, Atodiresei I. Org. Lett. 2010;12:4604–4607. doi: 10.1021/ol1019234. [DOI] [PubMed] [Google Scholar]; (d) Wang D-S, Chen Q-A, Li W, Yu C-B, Zhou Y-G, Zhang X. J. Am. Chem. Soc. 2010;132:8909. doi: 10.1021/ja103668q. [DOI] [PubMed] [Google Scholar]; (e) Xiao Y-C, Wang C, Yao Y, Sun J, Chen Y-C. Angew. Chem. Int. Ed. 2011;50:10661. doi: 10.1002/anie.201105341. [DOI] [PubMed] [Google Scholar]
- 13.Zhu Q, Lu Y. Angew. Chem. Int. Ed. 2010;49:7753. doi: 10.1002/anie.201003837. [DOI] [PubMed] [Google Scholar]
- 14.Nakanishi M, Katayev D, Besnard C, Kündig EP. Angew. Chem. Int. Ed. 2011;50:7438. doi: 10.1002/anie.201102639. [DOI] [PubMed] [Google Scholar]
- 15.Arp FO, Fu GC. J. Am. Chem. Soc. 2006;128:14264. doi: 10.1021/ja0657859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petit S, Duroc Y, Larue V, Giglione, Léon C, Soulama C, Denis A, Dardel F, Meinnel T, alArtaud I. ChemMedChem. 2009;4:261. doi: 10.1002/cmdc.200800251. [DOI] [PubMed] [Google Scholar]
- 17.Bajwa GS, Brown RK. Can. J. Chem. 1969;47:785. [Google Scholar]
- 18.Kira M, Hino T, Sakurai H. Tetrahedron Lett. 1989;30:1099. [Google Scholar]
- 19.Rodriguez JG, Lafuente A, Garcia-Almaraz P. J. Heterocycl. Chem. 2000;37:1281. [Google Scholar]