

Abstract
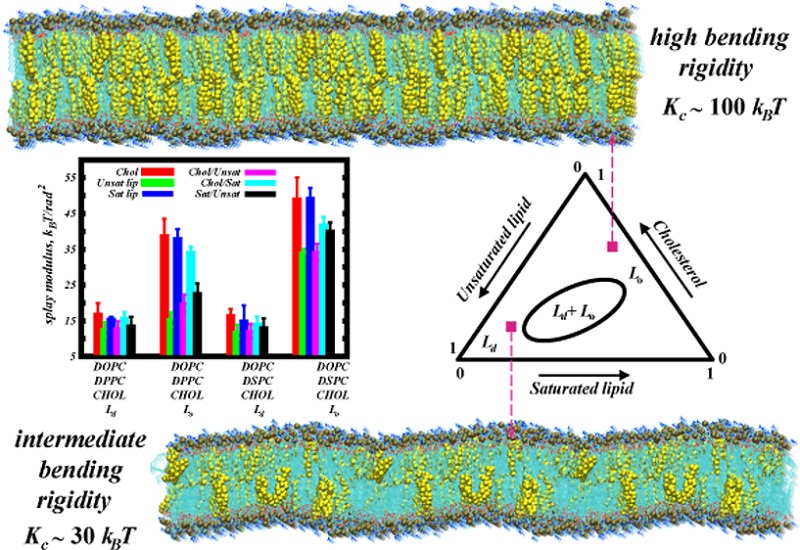
We establish a computational approach to extract the bending modulus, KC, for lipid membranes from relatively small-scale molecular simulations. Fluctuations in the splay of individual pairs of lipids faithfully inform on KC in multicomponent membranes over a large range of rigidities in different thermodynamic phases. Predictions are validated by experiments even where the standard spectral analysis-based methods fail. The local nature of this method potentially allows its extension to calculations of KC in protein-laden membranes.
Elastic properties of lipid membranes have been suggested to play a critical role in the function and organization of membrane proteins.1−4 Yet, a detailed understanding of how the mechanical properties of the lipid bilayer matrix regulate membrane associated proteins is currently missing. A major obstacle in making this link is the lack of robust methodologies able to reliably quantify elastic parameters, such as the bending rigidity, KC, for membranes of different lipid compositions and in different fluidity states (i.e., thermodynamic phases). In this Letter, we establish a general computational approach to determine KC for multicomponent membranes over the entire biologically relevant range of rigidities (in different thermodynamic phases) and illustrate its advantage over currently existing computational methodologies used to extract bending modulii from molecular simulations.
In the past decade, the spectral analysis of bilayer undulations in simulated lipid membranes has emerged as an attractive method for calculating bilayer bending rigidities.5−11 In this approach, thermally excited fluctuations in the bilayer interface shape u(x,y) of a pure lipid membrane are sampled in the course of a molecular dynamics (MD) trajectory, and the energy cost for bilayer deformations is expanded to lower order terms in u(x,y), according to the Helfrich continuum description.12 Analysis of the bending rigidity from the bilayer undulations is then performed in Fourier space in terms of the two-dimensional reciprocal space vector q⃗. The bending modulus is obtained by realizing that, by the equipartition theorem, the spectral amplitude profile ⟨u2(q)⟩ for the small-q modes scales as (kBT/ABOX) × KC × q–4, where ABOX represents the lateral area of the simulation box.5,7
This method has been mostly applied to single-component phospholipid membranes in the disordered (fluid) phase (Ld) and was shown to give KC values that matched well with experiments.5−7,9 However, several limitations of the method have emerged. Specifically, the size of the simulated membrane patch under consideration should be large enough (typically >1000 lipids) to allow for sustained long-wavelength (low q) undulations, and in addition the simulation trajectory subject to the spectral analysis must be sufficiently long to efficiently sample shape fluctuations.5,6 Due to these limitations, a statistical error of 20–25% for the bending modulii values predicted in this way has been reported.6 Importantly, for cholesterol-enriched liquid ordered (Lo) membranes, where the bilayer bending rigidity is expected to be particularly high (>60 kBT), the bending constants obtained with the spectral analysis appear to be significantly underestimated6,10 (as we also discuss in the following).
Recently, taking advantage of the relationship between the bending deformations of a lipid bilayer and the deformation modes originating from lipid splay and tilt, a new class of methods that rely on fluctuations in lipid tilt and splay from molecular simulations has been introduced.10,13−15 Thus, Watson et al.15 suggested modifying the spectral analysis approach to sample in Fourier space the fluctuations in lipid tilt instead of undulations of membrane shape. This allowed for accurate bending constant calculations even when simulations of rather small membrane patches (∼400 lipids) were involved. However, the application of this method has been as well limited to reports on single-component membranes in the relatively soft Ld phase, as the extension of the method to determine KC for multicomponent lipid mixtures has yet to be illustrated on specific examples.
We have been using a molecular-level approach to follow in MD simulations the fluctuations in tilt of all membrane components, and of the splay of all possible pairs of molecules in the membrane.10,14 These probability distributions, in turn, allow one to derive splay and tilt modulii for each molecule or molecular pairs, respectively. In contrast to methods that rely on a spectral analysis of lipid tilt,15 the analysis at the molecular level is inherently local in nature and therefore can easily extend to mixtures of any number of membrane components. Specifically, by calculating the probability of different molecular encounters, the contributions of the different splay terms to the overall rigidity can be determined. This is crucial, because one of the potential advantages for the tilt fluctuation-based methodology is its ability to predict even high membrane rigidities, which are typical for biologically relevant multicomponent lipid mixtures that contain cholesterol in addition to several types of lipids (saturated and unsaturated).16 Prominent examples, as pointed out above, are the cholesterol-rich Lo phases, or membrane microdomains (“rafts”)17 that are expected to have similarly high rigidities.
In this Letter, we show that extensions of our molecular-based method can be used to derive accurate bending rigidities for multicomponent membranes over a wide range of values, reaching even very high values of bending constants (KC ≈ 100 kBT). We show that these predictions fit well the experimentally determined values. To contrast, the traditional spectral analysis technique fails to derive similar predictions for the same (or even larger) membrane patches and simulation times. This sets the molecular-based method in a unique position to faithfully determine bending constants of single or multicomponent membranes over the entire biologically relevant range of rigidities with relative computational ease.
The starting point for our framework is a well equilibrated trajectory from MD simulations of a lipid membrane containing Nlip different molecular (lipid) species. In principle, the system could also contain membrane-associated proteins, but for simplicity in this Letter we focus on pure membrane systems (the possible extensions to protein-decorated membranes are discussed below). The molecular representation of the system can vary from all-atom lipid bilayers immersed in explicit solvent, to a coarser description, where atoms on lipid and solvent components are grouped into coarse-grained beads. To demonstrate that the methodology performs equally well under different molecular representations, we present results from comparative MD studies using two distinct descriptions applied to bilayers having the same lipid compositions (see Table 1): first, all-atom (AA) represented lipid membranes in an explicit water environment (simulated with the NAMD 2.9 package18 and with the CHARMM36 lipid force field19) and, second, membranes represented by the Martini coarse-grained (CG) force field.20 To illustrate the versatility of our approach, we studied several complex ternary lipid mixtures composed of a saturated component (either DPPC (dipalmitoylphosphatidylcholine), or DSPC (distearoylphosphatidylcholine) lipid), an unsaturated component (DOPC, dioleoylphosphatidylcholine, lipid), and cholesterol (Chol). Simulating various concentration and temperature conditions allows studies of these mixtures in both the fluid Ld and liquid-ordered Lo phases21,22 (Table 1).
Table 1. Lipid Systems Studied by MD Simulationsa.
| DOPC/DPPC/CHOL | DOPC/DPPC/CHOL | DOPC/DSPC/CHOL | DOPC/DSPC/CHOL | |
|---|---|---|---|---|
| Ld | Lo | Ld | Lo | |
| 0.66:0.19:0.15 | 0.12:0.58:0.3 | 0.74:0.09:0.17 | 0.12:0.56:0.32 | |
| T = 15 °C | T = 15 °C | T = 22 °C | T = 22 °C | |
| ALL-ATOM (AA) | NT = 400 | NT = 400 | NT = 400 | NT = 400 |
| t = 60 ns | t = 60 ns | t = 60 ns | t = 60 ns | |
| Martini Small (MS) | NT = 512 | NT = 512 | NT = 512 | NT = 512 |
| t = 16 μs | t = 16 μs | t = 16 μs | t = 18 μs | |
| Martini Large (ML) | NT = 2048 | NT = 2048 | NT = 2048 | NT = 2048 |
| t = 6 μs | t = 6 μs | t = 6 μs | t = 6 μs |
T, simulation temperature; NT, number of lipids in the simulation; t, trajectory durations (neglecting initial equilibration phases) used for the analysis.
Because it relies only on spatially localized molecular encounters, one advantage of our approach is that it allows one to use as input relatively small simulated lipid patches without compromising the evaluated value of the bending modulus. The results presented below are for bilayers containing 400–500 lipids in total, but we found the bending modulus values calculated from converged 128-lipid size patches to be within 1% of those obtained with 400–500 lipid-size membranes (see Table S1 in the Supporting Information). In fact, the only requirement for reliable determination of the bending rigidities with our method is to start with a trajectory in which the model membrane patch satisfies its experimentally validated structural properties, and for which the tilt and splay distributions are statistically well sampled.
To extract the elastic constant, KC, from the MD trajectory, we take advantage of the fact that, as previously noted,14 the bending deformation mode in a lipid bilayer is closely related to the splay modulus, χ12, associated with the free energy cost for splaying one lipid molecule with respect to another. Therefore, we proposed that by calculating χ12 for all possible pairs of lipids and by appropriately weighting the corresponding splay contributions, it should be possible to obtain the macroscopic bending modulus parameter. To average these χ12’s, we follow the analogous weighting of Kozlov and Helfrich23 for the bending rigidity of mixed membranes with several components, where each has its own KC when it is pure. Then, weights for each splay component are taken with proportion to the number of molecular encounters found in the simulations. This is somewhat similar to the weighting previously proposed for calculating the spontaneous curvature of binary surfactant mixtures.24 Here, we generalize our formulation to lipid mixtures with an arbitrary number of components, and suggest the following heuristic approximation for calculating the monolayer bending modulus km = KC/2 from MD trajectories based on the molecular pairwise splay contributions:
| 1 |
In the above, χ12ij denotes the splay modulus for the ijth pair type and φij is the number of near-neighboring ij encounter pairs, obtained directly from MD trajectories; finally, φtotal = ∑⟨i,j⟩φij represents the total number of encounters in the simulation for all possible pairwise contributions ⟨i,j⟩ for which the splay is calculated (for example, there are 0.5Nlip(Nlip + 1) = 6 such ij types of contributions for ternary mixtures). We note that the expression in eq 1 is analogous to the Reuss averaging used in effective medium theories for estimating the stiffness of composite biomaterials under uniform stress,25 which is most applicable to fluid mixtures. We also point out that for the special case of a single-component lipid bilayer, the expression in eq 1 simply represents the bending rigidity of the respective pure lipid membrane, as previously reported in ref (14).
To obtain χ12, we first define local lipid director vectors t⃗ in the following manner26 (see Figures S2 and S3 in the Supporting Information): for PC lipids in the all-atom representation, t⃗ is the vector that connects the midpoint between the phosphate and backbone C2 atoms to the geometric center-of-mass of the three terminal carbons on the two lipid chains; in CG Martini simulations, t⃗ for lipids is defined as the vector connecting the geometric center-of-mass of PO4, GL1, and GL2 beads with the geometric center-of-mass of the terminal carbon beads on the two lipid tails. For cholesterol, in the all-atom representation the director vector joins C3 and C17 atoms, and in the CG description, t⃗ connects R1 and R5 ring beads. We note that taking somewhat different definitions of t⃗ vectors (as described for example in ref (26)) kept the bending modulus values within the uncertainties reported in Table 2 (see Figure S4 in the Supporting Information).
Table 2. Bilayer Bending Modulus (KC, in kBT Units) for Different Compositions Obtained from the MD Simulations and OS Experiments.
| DOPC/DPPC/CHOL | DOPC/DPPC/CHOL | DOPC/DSPC/CHOL | DOPC/DSPC/CHOL | |
|---|---|---|---|---|
| Ld | Lo | Ld | Lo | |
| ALL-ATOM (AA)a | 34 ± 3 | 97 ± 4 | 30 ± 2 | 105 ± 5 |
| Martini Small (MS)a | 26 ± 3 | 64 ± 4 | 24 ± 3 | 89 ± 4 |
| Martini Large (ML)b | 23 ± 7 | 39 ± 8 | 27 ± 7 | 44 ± 7 |
| OS experimentsc | 44(+40/–18) | ≥100 | 34(+19/–10) | ≥100 |
The bending constant values were determined from the approach based on fluctuations in the lipid splay.
The bending constant values were determined from the spectral analysis method (see also Figure S8 in the Supporting Information).
For the estimation of the error bars from the OS measurements, see Methods and Figure S1 in the Supporting Information.
In the next step, we evaluate the probability distributions P(α) of the angle α between different t⃗ vectors, i.e., between all possible lipid/lipid and lipid/cholesterol pairs. Since, according to the Helfrich continuum theory,27−29 in the region of small θ lipid tilt angles (θ being defined as the angle between the vector t⃗ and the bilayer normal), the free energy of lipid splay is proportional to (∇·θ)2, the P(α) probability distributions contain only those pairs for which at least one of the participant molecules is tilted by no more than θ = 10° angle with respect to the bilayer normal. Then, it is possible to obtain χ12 from P(α) by performing a quadratic fit in the interval of small α angles to the function PMF(α) = −kBT ln[P(α)/sin α], which describes the two-molecule potential of mean force for splay. Then, χ12’s correspond to the coefficients yielding the best fit.14 Figure 1 illustrates this procedure for the example of the Chol/saturated lipid pair in DOPC/DPPC/Chol and DOPC/DSPC/Chol mixtures simulated in Ld and Lo phases with CG force fields (see Table 1 for system definitions).
Figure 1.
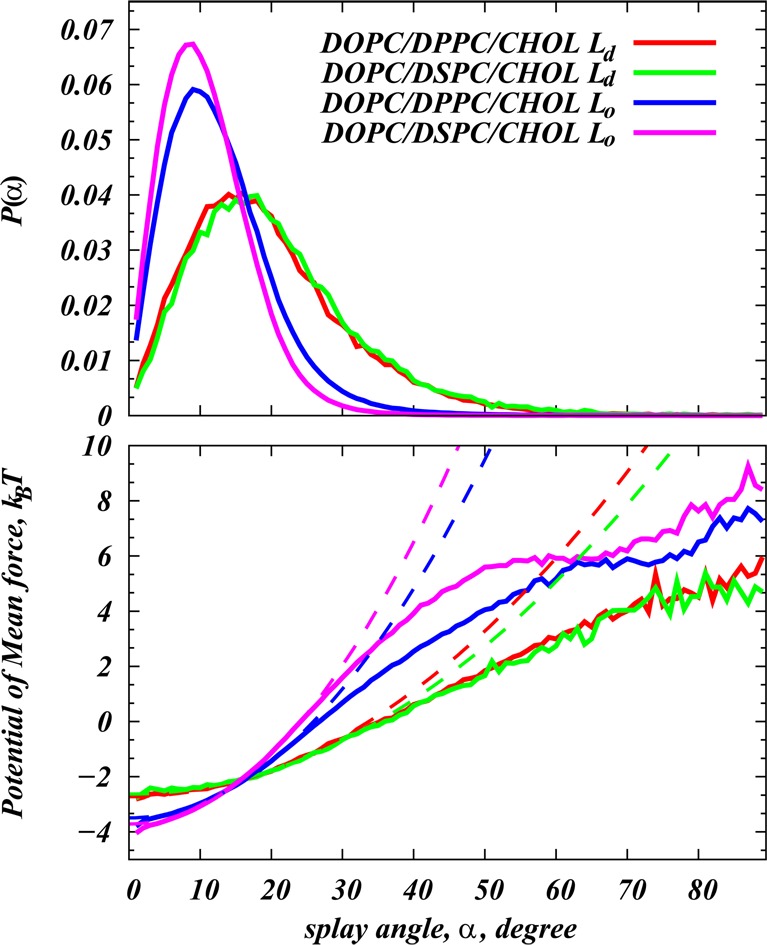
(Top) Normalized probability densities P(α) of finding a pair of cholesterol and saturated lipid (DPPC or DSPC in the respective systems) at an angle α with respect to each other. The data shown are obtained from Martini simulations of DOPC/DPPC/Chol and DOPC/DSPC/Chol mixtures in the Ld and Lo phases (MS simulation set in Table 1). To limit the analysis to near neighbors, for these calculations, only molecules (pairs of Chol) within 10 Å of each other were included, and consideration was given only to those pairs for which at least one of the molecules was tilted by no more than θ = 10° angle with respect to bilayer normal (see text). (Bottom) Potential of mean force profiles (solid curves) calculated from the P(α) distributions (see text). Dashed lines represent the best quadratic fits, from which the corresponding splay moduli χ12 were calculated (see Figure 2). Fits for the Lo and Ld systems were performed in α ∈ [5;20] and α ∈ [10;30] angular intervals, respectively, to limit the fit to a low angle regime and yet include the best sampled regions in the P(α) distribution profiles. For each fitting procedure, the quality of the fit was assessed by the reduced chi-squared parameter, which typically was found to be low (10–3 to 10–4), signifying the good quality of the quadratic fit.
Figure 2 shows calculated splay modulii for all six molecular pairs in the Martini CG simulations of the studied mixtures (MS simulation set in Tables 1 and 2). Table 2 compares the resulting KC values obtained from the MS and AA simulations as well as those obtained from the osmotic stress (OS) experiments on the same mixtures and under the same composition–temperature conditions (see Supporting Methods for details of the OS experiments). Overall, KC values determined from the simulations by using the molecular splay-based methodology (AA and MS rows in Table 2) are in quantitative agreement with the experimentally determined bending rigidities (OS row in Table 2) both in Ld and in Lo phases. The OS data indicate that the bending rigidities of DOPC/DPPC/Chol and DOPC/DSPC/Chol mixtures vary several fold between the Ld and Lo phases and in the liquid ordered phase reach values as high as ∼100 kBT, consistent with earlier experimental measurements on various binary lipid mixtures in the liquid ordered phase.30,31 Strikingly, the experimental measurements reveal that the addition of relatively small amounts of saturated lipids (9% DSPC or 19% DPPC) to DOPC/Chol mixtures in the fluid phase significantly stiffens these bilayers, as the KC values for the ternary mixtures in the Ld phase are appreciably higher than the bending rigidities (∼18 kBT) reported for fluid DOPC/Chol mixtures in the 0–30% Chol concentration range.30 We note that uncertainties in the experimental KC data are large due to the reduction of bending fluctuations in the presence of cholesterol. Therefore, for Lo phases that are enriched in cholesterol and the saturated lipid, we were only able to determine a lower limit of KC ≥ 100 kBT.
Figure 2.
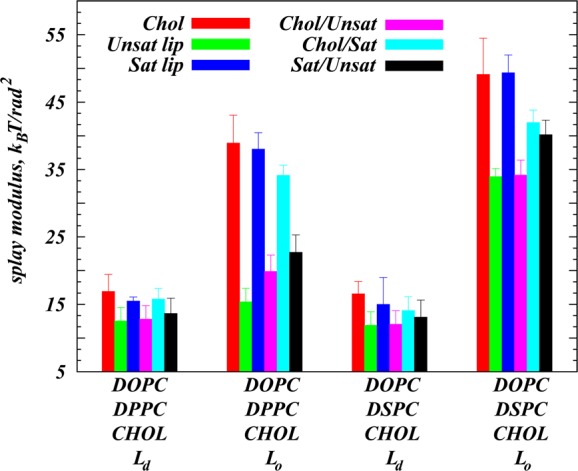
Splay modulii χ12 for all the possible molecular pairings derived from Martini simulations of DOPC/DPPC/Chol and DOPC/DSPC/Chol mixtures in the Ld and Lo phases (MS simulation set in Table 1). The data for different pairs are shown in the following colors: Chol/Chol in red, DOPC/DOPC in green, pairs of saturated lipids (either DPPC or DSPC in the respective systems) in blue, Chol/DOPC in purple, pairs of Chol and saturated lipid in cyan, and the DOPC with saturated lipid pair in black. Error bars represent the standard deviations obtained from fits (see Figure 1) performed in different angular intervals and on different trajectory segments (see Supporting Material for more details).
As revealed from Table 2, our computational methodology not only faithfully captures the experimentally determined trends, but also predicts KC in remarkable agreement with the experimental values over the entire range of bending rigidities. Only for the DOPC/DPPC/Chol mixture in the Lo phase simulated with the Martini force field did our method yield lower values (64 ± 4 kBT) than the experimentally measured bending rigidity. The KC value determined for the same system in the AA representation (97 ± 6 kBT) was within the uncertainties of the OS data. This discrepancy, as well as the general tendency for the Martini-based simulations to yield somewhat smaller values for KC compared to the AA data (yet still within experimental uncertainties) likely results from the different representation of DPPC and longer-tailed DOPC and DSPC lipids under AA and Martini force fields. (Note that in the all-atom description, 16-carbon palmitoyl tails are shorter by two methylene groups compared to 18-carbon stearoyl or oleoyl chains, but in Martini the difference between 16- and 18-carbon chains is magnified by the presence of an additional CG bead in the 18-carbon tails that usually represents not two but three to four heavy atoms.)
Importantly, in both AA and Martini simulations our method reliably captures the experimentally measured dramatic (at least 3-fold) increase in KC between the Ld and Lo phases. To test whether these trends in the bending rigidity are detectable using the standard spectral analysis approach (described above), we performed the undulation-based analysis for trajectories of large (2048 lipid) membrane patches of the same lipid composition simulated with the Martini CG force-fields (ML simulations in Tables 1 and 2) and extracted the corresponding elastic constants (see Supporting Methods). As shown in Table 2 (ML row), this methodology predicted KC values in quantitative agreement with the OS data in the Ld phase but failed to do so for the Lo phase mixtures. Using the spectral undulation analysis, the increase in the predicted bending rigidity compared to the fluid phase was less pronounced, and at most 1.6-fold higher. In addition, in agreement with previously reported studies (see, for example, ref (6)), the bending rigidity values calculated with this analysis could only be determined with relatively large uncertainties, especially in the Lo phase.
The underestimation of the bilayer bending rigidity with the spectral analysis method in the liquid ordered phases has been reported before6,10 and can be explained by the fact that this technique generally relies on extensive sampling of membrane undulations. But such deformation modes are highly suppressed in the Lo phase membranes due to cholesterol’s stiffening effects (as also seen experimentally) and therefore hardly assessed in the simulations, even for significantly large bilayer patches subjected to very long simulation times, as is the case here. On the other hand, the improved methodology we have established takes advantage of the localized splay deformation modes between pairs of molecules. In fact, the lipid splay is the most significant microscopic mode of deformations that drives global membrane response to bending.15,26,27 Therefore, inherently local in nature, the presented analysis based on the lipid splay should be universally applicable to membranes of arbitrary lipid composition irrespective of the thermodynamic phase (i.e., bending rigidity regime).
Another unique aspect of our methodology is that it offers a quantitative view of how splaying of different pairs of molecules in multicomponent lipid mixtures contribute to the overall bending rigidity. As an illustration, in Figure 2 we show the splay modulii, χ12, for all the molecular pairings in the simulated mixtures (the data were collected from the MS simulation set). The plot reveals that, in contrast to the Ld phase where various pairs contribute to the bending rigidity equally (no detectable difference in χ12 among different pairs), in the Lo phase several molecular pairings clearly show a higher impact on overall rigidity. Indeed, pairs that involve cholesterol and saturated lipid (Chol/Chol, Chol/Sat Lip, and Sat Lip/Sat Lip) are characterized by relatively large χ12’s compared to the others, indicating a higher free energy cost for splaying these pairs. Interestingly, Unsat Lip/Unsat Lip and Chol/Unsat Lip pairs are the “softest” for splay deformations, as their respective χ12’s were the lowest. These trends are in line with the cholesterol aligning field concept introduced recently (reviewed in ref (14)), which suggests that upon increasing Chol concentration, sterol molecules transition from a “laying down” (in fluid membranes) to “standing up” configuration (in relatively ordered bilayers).13 This nematic-like arrangement of sterols under Lo conditions produces a strong aligning field quantifiable by the large tilt modulus of the membrane components (see Figure S5 in the Supporting Information). As we showed recently for binary PC/Chol mixtures14 and illustrate here for the ternary systems, this sterol orientational field has a strong effect on the alignment of saturated lipids (high tilt modulii in Figure S5 for saturated lipids in Lo mixtures). Taken together with the splay modulus data, our results suggest that the strong orientational coupling between cholesterol and saturated lipids in the liquid ordered phase determines the high KC in Lo membranes.
The unique feature of our computational framework that relies on local splaying interactions has yet another additional advantage. This locality allows the method to be naturally extended, for example, to lipid membranes decorated by a concentration of partial or transmembrane (TM) insertions, such as peptides or even multihelical TM proteins. For these and similar systems, the standard spectral analysis technique is hard to apply, since the long-range membrane undulations, required as input for the method, will be confounded by the presence of a concentration of proteins. To contrast, by defining the lipid directors on possibly curved surfaces near inserted proteins,29,32,33 our technique could potentially help to overcome the specific challenge of calculating local (or “effective”) bending moduli for protein-laden membranes from local splay information and aid in providing answers related to the ways different insertions alter the splay properties of lipid membranes.
In conclusion, in this Letter we have established a versatile computational approach to quantify the bending modulus for lipid membranes from relatively small-scale molecular simulations. The reliance of the method on local splay interactions between different lipid pairs allows one to accurately evaluate KC in multicomponent lipid membranes in different thermodynamic phases, even where the standard spectral analysis methods break down. Fully validated by experiments, the approach affords several unique possibilities, including the quantitative determination of how splaying of different molecular pairings in multicomponent lipid mixtures contribute to the overall bending rigidity, and the expected relative computational ease of expandability to protein-decorated membranes.
Acknowledgments
We thank Harel Weinstein for insightful discussions. G.K. is supported by HRH Prince Alwaleed Bin Talal Bin Abdulaziz Alsaud Institute of Computational Biomedicine at Weill Medical College of Cornell University. Computational resources of the David A. Cofrin Center for Biomedical Information in the HRH Prince Alwaleed Bin Talal Bin Abdulaziz Alsaud Institute for Computational Biomedicine are also gratefully acknowledged. D.H. acknowledges support from the Stephanie Gross intramural fund. The Fritz Haber Center is supported by the Minerva Foundation, Munich, Germany. Experimental work was supported by the Austrian Science Funds FWF, grant no. P24459-B20 to G.P. We thank Heinz Amenitsch and Michael Rappolt for experimental support at the SAXS beamline at Elettra.
Supporting Information Available
Supporting methods, one table, and eight figures are available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Zimmerberg J.; Kozlov M. M. Nat. Rev. Mol. Cell Biol. 2006, 7, 9. [DOI] [PubMed] [Google Scholar]
- Soubias O.; Teague W. E. Jr.; Hines K. G.; Mitchell D. C.; Gawrisch K. Biophys. J. 2010, 99, 817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen O. S.; Koeppe R. E. II. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 107. [DOI] [PubMed] [Google Scholar]
- Marsh D. Biophys. J. 2007, 93, 3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl E.; Edholm O. Biophys. J. 2000, 79, 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofsass C.; Lindahl E.; Edholm O. Biophys. J. 2003, 84, 2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt E. G.; Braun A. R.; Sachs J. N.; Nagle J. F.; Edholm O. Biophys. J. 2011, 100, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen B. N.; Schlesinger P. H.; Baker N. A. J. Am. Chem. Soc. 2009, 131, 4854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu S. W.; Vasudevan S.; Jakobsson E.; Mashl R. J.; Scott H. L. Biophys. J. 2003, 85, 3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khelashvili G. R., M.; Chiu S.-W.; Pabst G.; Harries D. Soft Matter 2011, 7, 10299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz R.; Gompper G.; Lipowsky R. Phys. Rev. Lett. 1999, 82, 221. [Google Scholar]
- Helfrich W. Z. Naturforsch., C 1973, 28, 693. [DOI] [PubMed] [Google Scholar]
- Khelashvili G.; Pabst G.; Harries D. J. Phys. Chem. B 2010, 114, 7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khelashvili G.; Harries D. J. Phys. Chem. B 2013, 117, 2411. [DOI] [PubMed] [Google Scholar]
- Watson M. C.; Brandt E. G.; Welch P. M.; Brown F. L. Phys. Rev. Lett. 2012, 109, 028102. [DOI] [PubMed] [Google Scholar]
- Marsh D. Biochim. Biophys. Acta 2009, 1788, 2114. [DOI] [PubMed] [Google Scholar]
- Lingwood D.; Simons K. Science 2010, 327, 46. [DOI] [PubMed] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kale L.; Schulten K. J. Comput. Chem. 2005, 26, 1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauda J. B.; Venable R. M.; Freites J. A.; O’Connor J. W.; Tobias D. J.; Mondragon-Ramirez C.; Vorobyov I.; MacKerell A. D.; Pastor R. W. J. Phys. Chem. B 2010, 114, 7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrink S. J.; Risselada H. J.; Yefimov S.; Tieleman D. P.; de Vries A. H. J. Phys. Chem. B 2007, 111, 7812. [DOI] [PubMed] [Google Scholar]
- Uppamoochikkal P.; Tristram-Nagle S.; Nagle J. F. Langmuir 2010, 26, 17363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heberle F. A.; Wu J.; Goh S. L.; Petruzielo R. S.; Feigenson G. W. Biophys. J. 2010, 99, 3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov M. M.; Helfrich W. Langmuir 1992, 8, 2792. [Google Scholar]
- Safran S. A.; Pincus P. A.; Andelman D.; MacKintosh F. C. Phys. Rev. A 1991, 43, 1071. [DOI] [PubMed] [Google Scholar]
- Lakes R.Composite Biomaterials. In The Biomedical Engineering Handbook, 2nd ed.; Bronzino J. D., Ed.; CRC Press: Boca Raton, FL, 2000. [Google Scholar]
- May E. R.; Narang A.; Kopelevich D. I. Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys. 2007, 76, 021913. [DOI] [PubMed] [Google Scholar]
- Kozlovsky Y.; Kozlov M. M. Biophys. J. 2002, 82, 882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May S.; Kozlovsky Y.; Ben-Shaul A.; Kozlov M. M. Eur. Phys. J. E: Soft Matter Biol. Phys. 2004, 14, 299. [DOI] [PubMed] [Google Scholar]
- Fosnaric M.; Iglic A.; May S. Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys. 2006, 74, 051503. [DOI] [PubMed] [Google Scholar]
- Pan J.; Tristram-Nagle S.; Nagle J. F. Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys. 2009, 80, 021931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracia R. S.; Bezlyepkina N.; Knorr R. L.; Lipowsky R.; Dimova R. Soft Matter 2010, 6, 1472. [Google Scholar]
- Zemel A.; Ben-Shaul A.; May S. J. Phys. Chem. B 2008, 112, 6988. [DOI] [PubMed] [Google Scholar]
- Campelo F.; McMahon H. T.; Kozlov M. M. Biophys. J. 2008, 95, 2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.