

Abstract
Aconitase, the second enzyme of the tricarboxylic acid cycle encoded by ACO1 in the budding yeast Saccharomyces cerevisiae, catalyzes the conversion of citrate to isocitrate. aco1Δ results in mitochondrial DNA (mtDNA) instability. It has been proposed that Aco1 binds to mtDNA and mediates its maintenance. Here we propose an alternative mechanism to account for mtDNA loss in aco1Δ mutant cells. We found that aco1Δ activated the RTG pathway, resulting in increased expression of genes encoding citrate synthase. By deleting RTG1, RTG3, or genes encoding citrate synthase, mtDNA instability was prevented in aco1Δ mutant cells. Increased activity of citrate synthase leads to iron accumulation in the mitochondria. Mutations in MRS3 and MRS4, encoding two mitochondrial iron transporters, also prevented mtDNA loss due to aco1Δ. Mitochondria are the main source of superoxide radicals, which are converted to H2O2 through two superoxide dismutases, Sod1 and Sod2. H2O2 in turn reacts with Fe2+ to generate very active hydroxyl radicals. We found that loss of Sod1, but not Sod2, prevents mtDNA loss in aco1Δ mutant cells. We propose that mtDNA loss in aco1Δ mutant cells is caused by the activation of the RTG pathway and subsequent iron citrate accumulation and toxicity.
1. Introduction
Respiratory metabolism in eukaryotes requires proteins encoded in both the nuclear genome and the mitochondrial genome (mtDNA). Mitochondrial genomes generally encode a small number of proteins, many of which are involved in respiratory metabolism [1, 2]. Maintenance of mtDNA is important for cell growth and survival. Oxidative damage to mtDNA causes respiratory deficiency and human diseases [3–5]. In higher eukaryotes, how the mitochondrial genome is maintained and transmitted is not well understood. However, studies using the budding yeast Saccharomyces cerevisiae have generated an abundance of data on how its mitochondrial genome is maintained [6, 7]. Many nuclear-encoded proteins of diverse functions are required for mtDNA maintenance. When mtDNA is mutated, yeast cells form the so-called “petite” mutants. Yeast can be categorized as those with wild type (rho+ cells), extensively deleted (rho− petites), or with complete loss of (rho0 petites) mtDNA. How mutations in nuclear genes cause defects in mtDNA maintenance is complex and often indirect. For example, mutations in the yeast homolog of frataxin, Yfh1, lead to iron overload in mitochondria, defects in maturation of proteins containing iron-sulfur clusters such as the TCA cycle enzyme aconitase, mtDNA instability and respiratory deficiency, and so forth [8–10]. Yeast studies have suggested that iron citrate toxicity may be responsible for yfh1 mutant phenotypes [11, 12]. Respiratory metabolism generates reactive oxygen species such as superoxide radicals. Superoxide dismutases, Sod1 and Sod2, localized in the cytoplasm and mitochondria, respectively, are responsible for converting superoxide radicals to relatively harmless hydrogen peroxide [13], which can react with ferrous iron (Fe2+) to generate highly reactive hydroxyl radicals through the Fenton reaction. Hydrogen peroxide is detoxified by enzymes such as catalases, converting hydrogen peroxide to oxygen and water [14]. Mutations in yeast catalases and superoxide dismutase lead to oxidative damage and reduced resistance to oxidants [14–16].
Besides resulting in respiratory deficiency, mutations in TCA cycle enzyme encoding genes also lead to variable defects in mtDNA maintenance [17, 18]. The most severe phenotype is caused by mutations in the ACO1 gene encoding aconitase, followed by the IDH1 gene encoding a subunit of mitochondrial isocitrate dehydrogenase [19]. It has been proposed that Aco1 has a novel function in mediating mtDNA maintenance by directly binding mtDNA [20, 21]. Mutations in ACO1 and IDH1 share several growth defect phenotypes, which can be partially rescued by mutations in CIT1, encoding the mitochondrial isoform of citrate synthase [17]. Expression of CIT1, ACO1, IDH1, and IDH2 is under dual control of two transcriptional regulatory complexes, Rtg1/3 and Hap2/3/4/5 [22]. In cells with reduced or defective respiratory functions, expression of these genes is under increased control of Rtg1/3. Rtg1 and Rtg3 are two basic helix-loop-helix transcription factors in the retrograde response pathway that mediates signaling from mitochondria to the nucleus [23]. Activation of Rtg1/3 requires a cytoplasmic protein, Rtg2, which has an N-terminal ATP binding domain in the Hsp70/actin/sugar kinase ATP binding domain superfamily [24]. The retrograde response pathway, also known as the RTG pathway, is activated in response to defects in mitochondrial respiratory function. Cit1, Aco1, and Idh1/2 promote synthesis of α-ketoglutarate, a precursor of glutamate, which is a potent repressor of the RTG pathway [23]. Mutations in ACO1, which lead to both a block in mitochondrial respiratory function and glutamate auxotrophy [25–27], therefore, likely activate the RTG pathway. However, it is not clear whether the RTG pathway contributes to the phenotypes of aco1 mutants.
Mutations in RTG2 and CIT2 have been reported to suppress mtDNA instability due to mutations in YFH1 [12]. In this study, we provide an alternative model to account for mtDNA loss due to an aco1Δ mutation. We found that mutations in either RTG genes, genes encoding citrate synthases, genes encoding mitochondrial iron transporters, or SOD1 suppress aco1Δ-induced mtDNA loss. Therefore, we propose that iron citrate toxicity contributes to aco1Δ mutant phenotypes.
2. Materials and Methods
2.1. Strains, Plasmids, Growth Media, and Growth Conditions
Yeast strains and plasmids used in this study are listed in Tables 1 and 2, respectively. Yeast mutant strains were created by either direct transformation with gene knockout cassettes or through meiotic segregation analysis of heterozygous diploids. Mutations were confirmed by PCR-genotyping, standard genotyping based on selection markers, phenotypic analysis, and/or immunoblotting using antibody against Aco1. The BY4741 rho0 strain was generated by one passage of rho+ cells grown in YPD medium supplemented with 15 μg/mL ethidium bromide. Yeast cells were grown in SD (0.67% yeast nitrogen base plus 2% dextrose), YNBCasD (SD medium plus 1% casamino acids), YNBcasR (0.67% yeast nitrogen base, 1% casamino acids, and 2% raffinose), YPD (1% yeast extract, 2% peptone, 2% dextrose), or YPEthanol (1% yeast extract, 2% peptone, 2% ethanol) medium at 30°C. When necessary, amino acids, adenine, and/or uracil were added to the growth medium at standard concentrations to cover auxotrophic requirements [28].
Table 1.
Strains used in this study.
| Strain | Genotype | Source |
|---|---|---|
| BY4741 | MATa ura3 leu2 his3 met15 | Research genetics |
| BY4731 | MATa ura3 leu2 met15 | Research genetics |
| BY4742 | MATa ura3 leu2 his3 lys2 | Research genetics |
| BY4741 (rho0) | BY4741 rho0 derivative | This study |
| ZLY2630 | BY4741 aco1::kanMX4 | This study |
| ZLY1264 | BY4741 rtg1::kanMX4 | Research genetics |
| ZLY1267 | BY4741 rtg2::kanMX4 | Research genetics |
| ZLY1273 | BY4741 rtg3::kanMX4 | Research genetics |
| ZLY2568 | BY4742 rtg1::LEU2 | This study |
| ZLY143 | BY4731 rtg2::LEU2 | This study |
| ZLY2570 | BY4742 rtg3::URA3 | This study |
| ZLY2648 | BY4741 rtg1::LEU2 aco1::kanMX4 | This study |
| ZLY2631 | BY4741 rtg2::LEU2 aco1::kanMX4 | This study |
| ZLY2652 | BY4741 rtg3::URA3 aco1::kanMX4 | This study |
| ZLY3206 | MATα met? [rho 0 ] | This study |
| CS725-3A | MATa ura3 leu2 his3 met15 cit1/2/3::kanMX4 | [21] |
| ZLY2545 | CS725-3A aco1::HIS3 | [21] |
| RBY353 | BY4741 cit1::kanMX4 | Research genetics |
| RBY355 | BY4741 cit2::kanMX4 | Research genetics |
| RBY356 | BY4741 cit3::kanMX4 | Research genetics |
| ZLY2603 | MATa ura3 leu2 met15 aco1::kanMX4 | This study |
| RBY469 | BY4741 cit1::kanMX4 aco1::kanMX4 | This study |
| RBY277 | BY4741 cit2::kanMX4 aco1::kanMX4 | This study |
| RBY363 | BY4741 cit3::kanMX4 aco1::kanMX4 | This study |
| ZLY854 | BY4741 cit1/2::kanMX4 aco1::kanMX4 | This study |
| BY4741 (mrs3 mrs4) | BY4741 mrs3::kanMX4 mrs4::kanMX4 | [29] |
| ZLY3505 | BY4741 mrs3::kanMX4 mrs4::kanMX4 aco1::HIS3 | This study |
| ZLY4603 | BY4741 sod1::kanMX4 | This study |
| BY4741 (sod2) | BY4741 sod2::kanMX4 | Research genetics |
| TPY1507 | BY4741 sod1::kanMX4 aco1::kanMX4 | This study |
| ZLY3973 | BY4741 sod2::kanMX4 aco1::kanMX4 | This study |
Table 2.
Plasmids used in this study.
| Plasmid | Description | Source |
|---|---|---|
| pRS416 | A yeast centromeric plasmid carrying URA3 selection marker | [30] |
| pUC-rtg1::LEU2 | An rtg1::LEU2 disruption cassette in pUC19 | [31] |
| pUC-rtg2::LEU2 | An rtg2::LEU2 disruption cassette in pUC19 | [31] |
| pUC-rtg3::URA3 | An rtg3::URA3 disruption cassette in pUC19 | [32] |
| pBS-aco1::HIS3 | An aco1::HIS3disruption cassette in pBluescript | This study |
| pCIT2-lacZ | A CIT2-lacZ reporter gene on the plasmid pWCJ (CEN URA3) | This study |
| pRS303-SOD1 | The SOD1 gene was cloned into the integrative plasmid pRS303 | This study |
2.2. Yeast Transformation and β-Galactosidase Activity Assays
Plasmids were transformed into yeast strains using the high-efficiency lithium acetate-PEG method and β-galactosidase assays were carried out as described [28]. For each plasmid and strain combination, assays were conducted in duplicates, and independent experiments were carried out two times. Specific activity of β-galactosidase is expressed as nmols of o-nitrophenol generated from substrate o-nitrophenyl-β-D-galactoside per min per mg protein.
2.3. DAPI Staining of Nuclear and Mitochondrial DNA and FLuorescence Microscopy
DAPI (4′6,-diamidino-2-phenylindole) staining of nuclear and mitochondrial DNA was carried out as described [28]. Briefly, yeast strains were grown in liquid YPD or YNBcasD medium at 30°C overnight to A 600 ~ 0.8. Cells were collected by centrifugation and treated with 1 μg/mL DAPI in 95% ethanol for 30 min and cell pellets were washed with sterile water three times. DAPI-stained DNA molecules in fixed cells were observed by fluorescence microscopy using a Nikon Eclipse E800 microscope equipped with an HBO 100 W/2 mercury arc lamp, a Nikon Plan Fluor 100X objective lens, and epifluorescence with a Nikon UV-2E/C filter set (excitation 340–380 nm and emission 435–485 nm). Digital images were acquired with Photometrics Coolsnap fx CCD camera and Metamorph Imaging Software (Molecular Devices, Sunnyvale, CA) and processed using ImageJ (National Institutes of Health) and Adobe Photoshop (Mountain View, CA).
2.4. Citrate Analysis
Cells were grown in 10 mL YPD medium overnight to ~OD600 1.0. Cultures were chilled in ice-cold water for 22 min and cells were collected by centrifugation at 4°C. Cell pellets were then washed twice in chilled water. Citrate levels were determined using a citrate assay kit (BioVision, CA, USA). Cells were disrupted in 500 μL Assay Buffer in the kit using glass beads method. Cell extract was clarified by centrifugation at 21,000 g at 4°C for 15 min. 20 uL cell extract was analyzed for protein concentration using Bradford assay and the rest of cell extract (~350) was deproteinized in Amicon Ultra 4 column (10 kDa cutoff). Deproteinized extract was analyzed for citrate levels according to the protocol provided by the manufacturer. Citrate levels in different strains were normalized by protein concentration of cellular extract prior to deproteinization. Citrate concentration in the wild-type strain was 7.97 nmols/mg proteins, which was arbitrarily set as 1 unit. Citrate concentration in the wild-type strain determined in this study is similar to ~1.1 nmols/107 cells reported previously [12].
3. Results and Discussion
3.1. aco1Δ Activates the RTG Pathway
Mutations in ACO1 lead to both respiratory deficiency and glutamate starvation, which are expected to activate the RTG pathway. To test this possibility, we determined the effect of an aco1Δ mutation on the expression of a CIT2-lacZ reporter gene, which has been used extensively as a readout of the activity of the RTG pathway [22, 24, 33–35]. Expression of CIT2-lacZ was assessed in wild-type rho+ and aco1Δ mutant cells using β-galactosidase assays. Since aco1Δ cells are rho0 petites, we also determined CIT2-lacZ expression in otherwise wild-type rho0 cells. Cells were grown in rich media with either raffinose or dextrose (D-glucose) as the sole carbon source, which have been used in studies on the RTG pathway and mitochondrial genome maintenance, respectively [12, 20, 21, 36]. In cells grown in raffinose medium, CIT2-lacZ expression was 4-fold higher in rho0 cells compared to rho+ cells, consistent with previous reports that the RTG pathway is activated in rho0 cells [33, 36]. An aco1Δ mutation induced CIT2-lacZ expression slightly higher than in wild-type rho0 cells (Figure 1). In cells grown in dextrose medium, CIT2-lacZ expression in rho0 cells is only marginally higher than in rho+ cells, whereas an aco1Δ mutation almost doubled CIT2-lacZ expression. Activation of the RTG pathway is strain dependent [37]. The lack of induction of the RTG pathway due to loss of mtDNA in dextrose-grown BY4741 background strains used in this study can be partly attributed to a doubling of CIT2-lacZ expression in rho+ cells grown in this medium compared to raffinose medium, which is consistent with activation of the RTG pathway due to compromised mitochondrial respiratory function since dextrose suppresses respiratory metabolism in yeast. Altogether, these data indicate that an aco1Δ mutation leads to activation of the RTG pathway.
Figure 1.
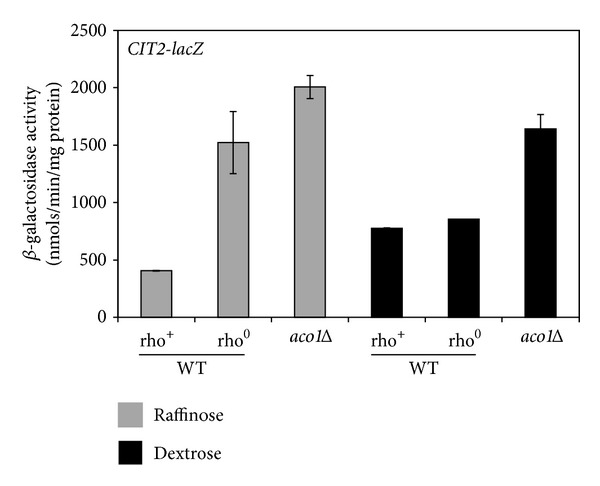
The RTG pathway is activated in aco1Δ mutant cells grown in dextrose medium. Wild-type rho+ (WT, BY4741), its rho0 derivative, and aco1Δ (ZLY2603) mutant strains were transformed with a centromeric plasmid encoding a CIT2-lacZ reporter gene (pCIT2-lacZ) and transformants were grown in YNBcasR (Raffinose) and YNBcasD (Dextrose) medium to mid-logarithmic phase. Cells were collected and β-galactosidase assays were conducted as described in Section 2.
3.2. Mutations in RTG Genes Prevent mtDNA Loss due to aco1Δ
To determine whether activation of the RTG pathway in aco1Δ mutant cells contributes to mtDNA loss, rtg1Δ aco1Δ, rtg2Δ aco1Δ, and rtg3Δ aco1Δ double mutants were constructed and examined for the presence or absence of mtDNA. These double mutant strains were created by crossing respective haploid mutant strains to form heterozygous diploid mutants, which were then sporulated to generate desired haploid segregants. Seven rtg1Δ aco1Δ, seven rtg2Δ aco1Δ, and six rtg3Δ aco1Δ double mutant segregants were obtained. Eight aco1Δ single mutant segregants were also isolated similarly. DAPI, a DNA-specific probe that forms a fluorescent complex [38], was then used to visualize mtDNA using fluorescence microscopy in these mutants along with wild-type rho+ and rho0 strains. In addition to nuclear DNA, DAPI staining revealed punctate cytoplasmic structures of mtDNA in wild-type rho+ cells grown in YPD medium (Figure 2(a)) [38]. In contrast, mtDNA was absent in both wild-type rho0 and aco1Δ mutant cells, consistent with previous reports that Aco1 is required for mtDNA maintenance [17, 18, 21]. Interestingly, mtDNA was maintained in rtg1Δ aco1Δ, rtg2Δ aco1Δ, and rtg3Δ aco1Δ double mutant strains, indicating that Rtg proteins mediate mtDNA instability in aco1Δ mutant cells. The percentage of rho0 cells was quantified from DAPI-stained images and a large majority of rtgΔ aco1Δ double mutant cells were found to contain mtDNA (Figure 2(b)). Among seven rtg2Δ aco1Δ double mutant segregants from heterozygous rtg2Δ/RTG2 aco1Δ/ACO1 diploid mutant cells, all were found to maintain mtDNA. Similarly, all of the seven rtg1Δ aco1 and six rtg3Δ aco1Δ double mutant segregants were also found to be rho+ cells. In contrast, all of the eight aco1Δ single mutant segregants from a heterozygous aco1Δ/ACO1 diploid mutant have lost mtDNA. Together, our data suggest that mtDNA instability in aco1Δ mutant cells may result from activation of the RTG pathway.
Figure 2.

mtDNA is maintained in rtg1Δ aco1, rtg2Δ aco1Δ, and rtg3Δ aco1Δ mutant cells. (a) Cells grown exponentially in YPD medium were stained with DAPI and then examined using fluorescence microscopy. WT (rho+), BY4741; WT (rho0), BY4741 rho0; aco1Δ, ZLY2630; rtg1Δ aco1Δ, ZLY2648; rtg2Δ aco1Δ, ZLY2631; rtg3Δ aco1Δ, ZLY2652. (b) Quantitative analysis of the percentage of rho0 cells in yeast strains based on DAPI-staining images. (c) mtDNA in rtgΔ aco1Δ double mutant cells is functional. Strains as described in (a) as well as rtg1Δ, rtg2Δ, and rtg3Δ single mutants were crossed to a rho0 tester strain (X rho0) of opposite mating type. Diploids were selected and grown on YPD (Dextrose) and YPEthanol (Ethanol) medium. Pictures of cells were taken after 3 days' growth at 30°C.
Damages to mtDNA can lead to extensive deletions (rho−) or point mutations (mit −) [6, 7]. Yeast strains that carry these two types of mutant mitochondrial genomes are respiratory deficient. To determine whether the mtDNA in the rtgΔ aco1Δ double mutant strains is functional, we conducted a complementation assay by crossing wild-type rho+, wild-type rho0, rtg1Δ, rtg2Δ, rtg3Δ, rtg1Δ aco1Δ, rtg2Δ aco1Δ, and rtg3Δ aco1Δ mutant strains to a rho0 tester strain of opposite mating type with wild-type nuclear ACO1 gene and analyzing the respiratory capacity of the resultant diploid strains. rtg1Δ aco1Δ, rtg2Δ aco1Δ, and rtg3Δ aco1Δ mutant strains were unable to utilize carbon sources that require respiratory metabolism such as ethanol because they are defective in the TCA cycle (data not shown), and Figure 2(c) shows that diploids generated from crossing wild-type rho+, rtg1Δ, rtg2Δ, and rtg3Δ strains with the rho0 tester strain were able to grow on ethanol medium. In contrast, diploids from crosses involving wild-type rho0 or the aco1Δ single mutant were unable to grow on ethanol medium, consistent with the absence of mtDNA in these diploids. Remarkably, diploids generated from the rho0 tester strain and rtgΔ aco1Δ double mutants were able to grow on ethanol medium, indicating that mtDNA in rtgΔ aco1Δ cells are functional.
3.3. Mutations in Genes Encoding Citrate Synthase, Primarily CIT1, Prevent mtDNA Loss due to aco1Δ
What is the mechanism of aco1Δ suppression by mutations in RTG genes? The RTG pathway is required for glutamate biosynthesis in cells with reduced respiratory function by regulating expression of CIT1, CIT2, ACO1, IDH1, and IDH2 [23]. It has been shown previously that a cit1Δ mutation partially suppresses mtDNA loss in aco1Δ mutant cells and that mutations in CIT2 and RTG2 rescue respiratory deficiency in yfh1Δ mutant cells [12, 17]. Three genes, CIT1, CIT2, and CIT3, encode citrate synthase in yeast, with CIT1 and CIT3 encoding the two mitochondrial isoforms and CIT2 encoding the peroxisomal isoform [39–41]. Therefore, suppression of mtDNA loss in rtg1Δ aco1Δ, rtg2Δ aco1Δ, and rtg3Δ aco1Δ double mutant cells may be due to reduced expression of genes encoding citrate synthase. To confirm this possibility, we introduced an aco1Δ mutation into a cit1Δ cit2Δ cit3Δ triple mutant in the BY4741 strain background generated by Chen et al. [21]. The presence or absence of mtDNA in the resultant quadruple mutant cells was examined by DAPI staining and fluorescence microscopy. Figure 3(a) shows that the cit1Δ cit2Δ cit3Δ aco1Δ quadruple mutant maintained mtDNA. To determine which citrate synthase-encoding gene(s) is responsible for mtDNA loss in aco1Δ mutant cells, we generated cit1Δ aco1Δ, cit2Δ aco1Δ, and cit3Δ aco1Δ double mutants, as well as an aco1Δ cit1Δ cit2Δ triple mutant by crossing respective haploid mutant strains followed by meiotic segregation analysis. Using DAPI staining and fluorescence microscopy, we found that six out of eight cit1Δ aco1Δ double mutants, zero out of six cit2Δ aco1Δ double mutants, zero out of six cit3Δ aco1Δ double mutants, and six out of six cit1Δ cit2Δ aco1Δ triple mutant strains maintained mtDNA (Figure 3 and data not shown). We also calculated the percentage of rho0 cells in a cit1Δ aco1Δ double, a cit1Δ cit2Δ aco1Δ triple, and a cit1Δ cit2Δ cit3Δ aco1Δ quadruple mutant and found that over 90% cells from these mutants maintained mtDNA (Figure 3(b)). Taken together, these data suggest that citrate synthase is the target of the RTG pathway that mediates mtDNA instability in aco1Δ mutant cells and that Cit1 is primarily responsible for this phenotype in the BY4741 strain background.
Figure 3.
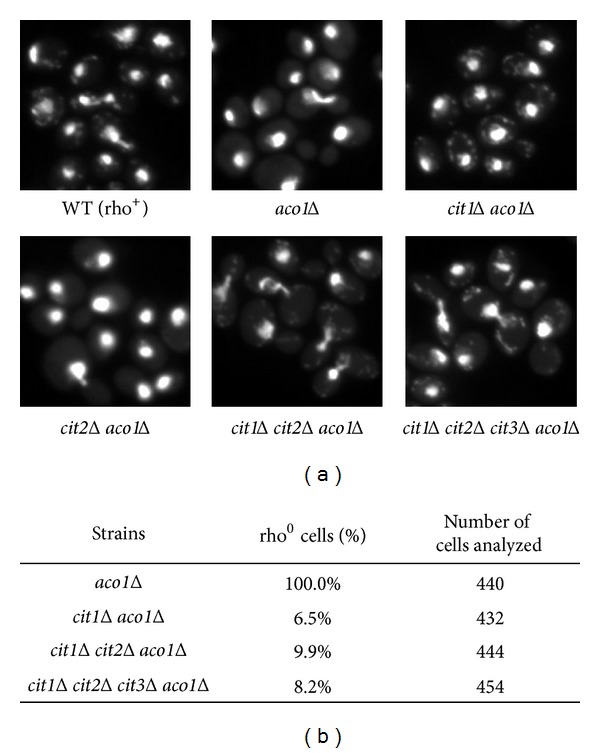
Mutations in genes encoding citrate synthase prevent mtDNA loss due to aco1Δ. (a) Cells grown in YPD medium to mid-logarithmic phase were stained with DAPI and examined using fluorescence microscopy. WT (rho+), BY4741; aco1Δ, ZLY2630; cit1Δ aco1Δ, RBY469; cit2Δ aco1Δ, RBY277; cit1Δ cit2Δ aco1Δ, ZLY854; cit1Δ cit2Δ cit3Δ aco1Δ, ZLY854; rtg2Δ aco1Δ, ZLY2545. (b) Quantitative analysis of the percentage of rho0 cells in yeast strains based on DAPI-staining images.
3.4. Mutations in Genes Encoding Mitochondrial Iron Transporters Mrs3 and Mrs4 Prevent mtDNA Loss due to aco1Δ
It has been proposed that iron citrate toxicity contributes to oxidative damage and mtDNA loss in yfh1Δ mutant cells, which have higher levels of cellular and mitochondrial iron [8, 12]. Mutations in RTG2 and CIT2 reduce petite formation in yfh1Δ mutants by lowering cellular citrate and iron levels. Suppression of mtDNA loss in aco1Δ mutant cells by mutations in RTG genes and genes encoding citrate synthase prompted us to test whether mitochondrial iron overload is responsible for mtDNA instability in aco1Δ mutants. Mitochondrial iron transport is mediated by iron transporters Mrs3 and Mrs4 [11, 29, 42, 43], mutations of which rescue mtDNA loss in yfh1Δ mutants. To this end, we generated an mrs3Δ mrs4Δ aco1Δ triple mutant by introducing an aco1Δ mutation into an mrs3Δ mrs4Δ double mutant. DAPI staining of the triple mutant showed that mtDNA was maintained (Figure 4). Furthermore, quantitative analysis showed that the percentage of rho0 cells in the triple mutant was 0.4%, slightly lower than 2.5% in wild-type rho+ cells. The mrs3Δ mrs4Δ aco1Δ triple mutant was also mated to a rho0 tester strain and the resulting diploids were streaked onto plates with ethanol as the sole carbon source. We found that the diploids could grow on ethanol medium, indicating that mtDNA in the mrs3Δ mrs4Δ aco1Δ triple mutant is functional (data not shown). Together, this data supports the notion that mtDNA loss in aco1Δ mutant cells is due to iron citrate toxicity.
Figure 4.
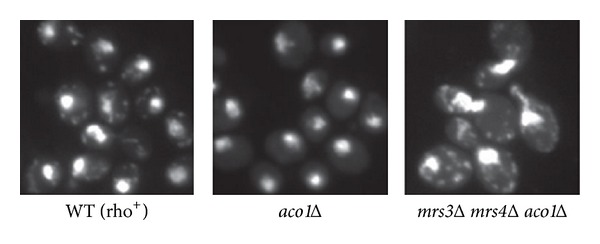
Mutations in MRS3 and MRS4, encoding mitochondrial iron transporters, prevent mtDNA loss due to aco1Δ. Cells grown in YPD medium to mid-logarithmic phase were stained with DAPI and examined using fluorescence microscopy. WT (rho+), BY4741; aco1Δ, ZLY2630; mrs3Δ mrs4Δ aco1Δ, ZLY3505.
It has been reported that the supplementation of exogenous iron (1 mM FeSO4) or raising pH of the growth medium reduces petite frequency in aco1Δ mutant cells grown in YPGalactose medium [17]. Thus, Lin et al. proposed that some of the effects of elevated citrate levels in aco1Δ are due to the ionized form of this metabolite rather than to the formation of a citrate/iron chelate that could produce oxidative damage to mtDNA. These observations seem to contradict our conclusion. However, iron homeostasis is a highly complicated process [44]. It was not established that exogenous iron actually increased iron levels in the mitochondria in Lin et al.'s study. It is also possible that growth conditions (YPD in this study versus YPGalactose in Lin et al.'s study) and/or strain backgrounds (BY4741 versus W303-1B) may affect mtDNA loss associated with aco1Δ mutations.
3.5. Iron Citrate Toxicity Correlates with mtDNA Loss in aco1Δ Mutant Cells
To affirm our hypothesis that iron citrate toxicity contributes to mtDNA loss in aco1Δ mutant cells, we determined citrate levels in wild-type and various mutant strains and found that aco1Δ increased the citrate level by 10.5-fold (Figure 5), which is consistent with an 11.9-fold increase reported by Lin et al. [17]. This increase is not due to that aco1Δ mutants are rho0 cells since the citrate level in wild-type rho0 cells was only 16% higher than that of wild-type rho+ cells. Mutations in RTG1, RTG2, and RTG3 reduced citrate levels in aco1Δ background cells by 59–85%. Similarly, cit1Δ reduced citrate concentration by 62% in aco1Δ background cells, which is comparable to an 89% decrease reported by Lin et al. A double mutation in CIT1 and CIT2 reduced citrate concentration by 98% in aco1Δ background cells, and an additional mutation in CIT3 did not significantly further reduce citrate levels. Clearly, there is strong correlation between the suppression of mtDNA loss phenotype and lower citrate levels in aco1Δ background strains. We also determined the effect of an mrs3Δ mrs4Δ double mutation on citrate levels. In comparison to a wild-type rho+ strain, citrate concentration in an mrs3Δ mrs4Δ double mutant was 53% lower. In contrast, citrate concentration in an mrs3Δ mrs4Δ aco1Δ triple mutant is 24% higher than that of an aco1Δ mutant. Since mrs3Δ mrs4Δ aco1Δ mutant cells maintained mtDNA, we propose that high levels of citrate per se are not sufficient to lead to mtDNA loss and that citrate toxicity requires certain levels of iron in the mitochondria. Together with our genetic data, these biochemical results suggest that iron citrate toxicity accounts for mtDNA loss in aco1Δ mutant cells.
Figure 5.
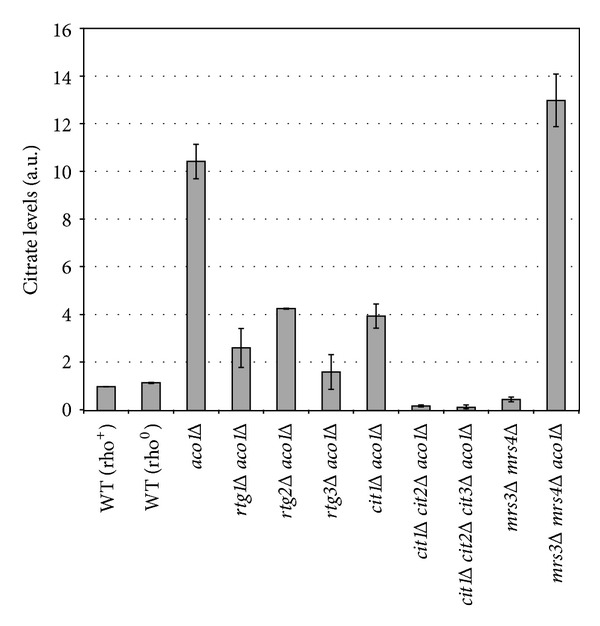
Citrate levels in wild-type (WT) and indicated mutant strains. Citrate concentration was determined as described in Section 2. The results are the average of two independent experiments.
3.6. A Mutation in SOD1, but Not SOD2, Prevents mtDNA Loss due to aco1Δ
Our data suggest that high levels of citrate cause mtDNA instability in aco1Δ mutant cells likely due to iron citrate toxicity. Iron reacts with hydrogen peroxide in the Fenton reaction to produce highly active, potent hydroxyl radicals, which cause oxidative damage to mitochondria. Hydrogen peroxide is partly produced by the superoxide dismutases, Sod1 in the cytoplasm and Sod2 in the mitochondrial matrix [12]. We hypothesized that a reduced production of hydrogen peroxide due to mutations in SOD1 or SOD2 might suppress mtDNA loss in aco1Δ mutant cells by lowering the amount of hydroxyl radicals produced via the Fenton reaction. Therefore, we generated sod1Δ aco1Δ and sod2Δ aco1Δ double mutants by crossing respective haploid mutant strains followed by meiotic segregation analysis. The resultant double mutant strains were analyzed for mtDNA presence by DAPI staining and fluorescence microscopy. 33 out of 34 sod1Δ aco1Δ double mutant strains generated maintained mtDNA while 6 out of 6 sod2Δ aco1Δ double mutants lost mtDNA (Figure 6 and data not shown). These data suggest that hydrogen peroxide generated from reactions catalyzed by Sod1 contributes to mtDNA loss in aco1Δ mutant cells. Mutations in SOD1 also cause oxidative damage due to accumulation of superoxide radicals [15, 16]. However, since an sod1Δ mutation suppressed mtDNA loss in aco1Δ mutant cells, we propose that hydroxyl radicals are more damaging to mtDNA than superoxide radicals. How would mutations in the cytosolic isoform of superoxide dismutase rescue a mitochondrial defect? It has been shown that a small fraction of Sod1 is localized in the intermembrane space of mitochondria, which protects cells from mitochondrial oxidative damage [45–47]. Why does not sod2Δ suppress mtDNA loss associated with aco1Δ? It is possible that loss of Sod2 leads to increased levels of superoxide radicals in the mitochondria, which in the presence of ferrous ions would cause any hydrogen peroxide produced in the mitochondria to be converted to the hydroxyl radicals via the Fenton/Haber Weiss reactions that damage mtDNA.
Figure 6.
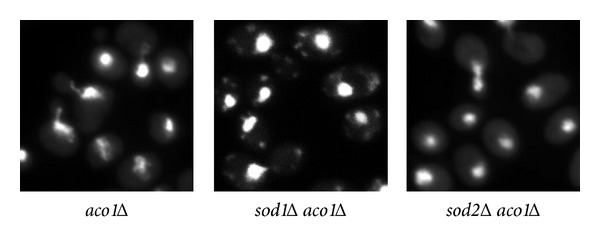
A mutation in SOD1, but not SOD2, suppresses mtDNA loss due to aco1Δ. Cells grown in YPD medium to mid-logarithmic phase were stained with DAPI and examined using fluorescence microscopy. aco1Δ, ZLY2630; sod1Δ aco1Δ, TPY1507; sod2Δ aco1Δ, ZLY3973.
3.7. Loss of mtDNA in aco1Δ Mutant Cells Is Growth Medium Dependent
It has been proposed that mtDNA loss in aco1Δ mutant cells is due to lack of physical protection of mtDNA by Aco1 [20]. One key piece of evidence that supports this hypothesis is the observation that the expression of two catalytically inactive Aco1 mutants, Aco1C382S and Aco1C445S, under the control of the ADH1 promoter from the pRS416 centromeric plasmid, prevented mtDNA loss in aco1Δ mutant cells. To maintain the plasmids, transformants were grown in YNBcasD medium. In light of discovery that iron citrate toxicity contributes to mtDNA loss, one alternative explanation for mtDNA retention in cells expressing ACO1 C382S and ACO1 C445S mutant alleles is due to differences in growth medium, YNBcasD versus YPD. Accordingly, we transformed aco1Δ/ACO1 heterozygous diploid mutant cells with empty pRS416 vector and transformants were sporulated and dissected on YPD or YNBcasD medium. Eleven aco1Δ haploid mutants from a YPD dissection plate were obtained and grown in YPD liquid medium and mtDNA was observed by DAPI staining. We found that all of the eleven aco1Δ segregants lost mitochondrial DNA (Figure 7 and data not shown). In contrast, among nine aco1Δ mutant segregants carrying the empty pRS416 vector from a YNBcasD dissection plate that were grown in YNBcasD liquid medium, seven maintained mtDNA (Figure 7 and data not shown). When these seven aco1Δ mutants containing mtDNA were passed onto YPD plate medium twice and then grown in YPD liquid medium, all lost mtDNA (data not shown). Together, these data suggest that mtDNA loss in aco1Δ mutant cells is growth medium dependent.
Figure 7.

mtDNA loss due to aco1Δ is growth medium dependent. aco1Δ mutant cells (ZLY2630) carrying empty pRS416 vector were grown in YNBcasD or YPD medium to mid-logarithmic phase, stained with DAPI, and examined using fluorescence microscopy.
4. Conclusions
It has been proposed that yeast aconitase (Aco1) physically binds to mtDNA and promotes its maintenance [20, 21]. Our results in this study suggest a different, but not necessarily mutually exclusive, mechanism. We propose that aco1Δ activates the RTG pathway, resulting in increased citrate production through upregulation of genes encoding citrate synthase. Increased levels of citrate lead to iron overload in the mitochondria. Iron then reacts with hydrogen peroxide to generate hydroxyl radicals, which cause oxidative damage to mitochondrial DNA and consequently its instability. Mutations of yeast frataxin (Yfh1) lead to loss of activity of aconitase [48]. Suppression of mtDNA instability due to mutations in both YFH1 and ACO1 by reduced iron citrate levels raises the possibility that mtDNA loss in yfh1 mutant cells may be an indirect consequence of aconitase inactivation. Our data also suggest that the cytosolic superoxide dismutase, Sod1, but not the mitochondrial superoxide dismutase, Sod2, contributes to mtDNA loss in aco1Δ mutant cells.
Conflict of Interests
The authors declare no conflict of interests.
Acknowledgments
The authors would like to thank Dr. Jerry Kaplan at The University of Utah for yeast strains, W. M. Keck Foundation for the Keck Facility, and Robin Rowe for sequencing. This work was supported by grant 1R15GM094772-01A1 (to Z. Liu) from the National Institutes of Health.
References
- 1.Foury F, Roganti T, Lecrenier N, Purnelle B. The complete sequence of the mitochondrial genome of Saccharomyces cerevisiae . FEBS Letters. 1998;440(3):325–331. doi: 10.1016/s0014-5793(98)01467-7. [DOI] [PubMed] [Google Scholar]
- 2.Lang BF, Gray MW, Burger G. Mitochondrial genome evolution and the origin of eukaryotes. Annual Review of Genetics. 1999;33:351–397. doi: 10.1146/annurev.genet.33.1.351. [DOI] [PubMed] [Google Scholar]
- 3.Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nature Reviews Genetics. 2005;6(5):389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greaves LC, Reeve AK, Taylor RW, Turnbull DM. Mitochondrial DNA and disease. Journal of Pathology. 2012;226(2):274–286. doi: 10.1002/path.3028. [DOI] [PubMed] [Google Scholar]
- 5.Siddiqui A, Rivera-Sanchez S, Castro Mdel R, et al. Mitochondrial DNA damage is associated with reduced mitochondrial bioenergetics in Huntington's disease. Free Radical Biology & Medicine. 2012;53(7):1478–1488. doi: 10.1016/j.freeradbiomed.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lipinski KA, Kaniak-Golik A, Golik P. Maintenance and expression of the S. Cerevisiae mitochondrial genome-From genetics to evolution and systems biology. Biochimica et Biophysica Acta. 2010;1797(6-7):1086–1098. doi: 10.1016/j.bbabio.2009.12.019. [DOI] [PubMed] [Google Scholar]
- 7.Contamine V, Picard M. Maintenance and integrity of the mitochondrial genome: a plethora of nuclear genes in the budding yeast. Microbiology and Molecular Biology Reviews. 2000;64(2):281–315. doi: 10.1128/mmbr.64.2.281-315.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Babcock M, De Silva D, Oaks R, et al. Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science. 1997;276(5319):1709–1712. doi: 10.1126/science.276.5319.1709. [DOI] [PubMed] [Google Scholar]
- 9.Koutnikova H, Campuzano V, Foury F, Dollé P, Cazzalini O, Koenig M. Studies of human, mouse and yeast homologues indicate a mitochondrial function for frataxin. Nature Genetics. 1997;16(4):345–351. doi: 10.1038/ng0897-345. [DOI] [PubMed] [Google Scholar]
- 10.Wilson RB, Roof DM. Respiratory deficiency due to loss of mitochondrial dna in yeast lacking the frataxin homologue. Nature Genetics. 1997;16(4):352–357. doi: 10.1038/ng0897-352. [DOI] [PubMed] [Google Scholar]
- 11.Foury F, Roganti T. Deletion of the mitochondrial carrier genes MRS3 and MRS4 suppresses mitochondrial iron accumulation in a yeast frataxin-deficient strain. Journal of Biological Chemistry. 2002;277(27):24475–24483. doi: 10.1074/jbc.M111789200. [DOI] [PubMed] [Google Scholar]
- 12.Chen OS, Hemenway S, Kaplan J. Genetic analysis of iron citrate toxicity in yeast: implications for mammalian iron homeostasis. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(26):16922–16927. doi: 10.1073/pnas.232392299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Culotta VC. Superoxide dismutase, oxidative stress, and cell metabolism. Current Topics in Cellular Regulation C. 2001;36:117–132. doi: 10.1016/s0070-2137(01)80005-4. [DOI] [PubMed] [Google Scholar]
- 14.Grant CM, Perrone G, Dawes IW. Glutathione and catalase provide overlapping defenses for protection against hydrogen peroxide in the yeast Saccharomyces cerevisiae . Biochemical and Biophysical Research Communications. 1998;253(3):893–898. doi: 10.1006/bbrc.1998.9864. [DOI] [PubMed] [Google Scholar]
- 15.Bermingham-McDonogh O, Gralla EB, Valentine JS. The copper, zinc-superoxide dismutase gene of Saccharomyces cerevisiae: cloning, sequencing, and biological activity. Proceedings of the National Academy of Sciences of the United States of America. 1988;85(13):4789–4793. doi: 10.1073/pnas.85.13.4789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu XF, Elashvili I, Gralla EB, Valentine JS, Lapinskas P, Culotta VC. Yeast lacking superoxide dismutase. Isolation of genetic suppressors. Journal of Biological Chemistry. 1992;267(26):18298–18302. [PubMed] [Google Scholar]
- 17.Lin A, Hakala KW, Weintraub ST, McAlister-Henn L. Suppression of metabolic defects of yeast isocitrate dehydrogenase and aconitase mutants by loss of citrate synthase. Archives of Biochemistry and Biophysics. 2008;474(1):205–212. doi: 10.1016/j.abb.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCammon MT, Epstein CB, Przybyla-Zawislak B, McAlister-Henn L, Butow RA. Global transcription analysis of Krebs tricarboxylic acid cycle mutants reveals an alternating pattern of gene expression and effects on hypoxic and oxidative genes. Molecular Biology of the Cell. 2003;14(3):958–972. doi: 10.1091/mbc.E02-07-0422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Merkel M, Kretowicz J. Sensitivity to natamycin in vitro evaluation of Candida spp. and Torulopsis glabrata isolated from vagina. Polski Tygodnik Lekarski. 1985;40(9):257–258. [PubMed] [Google Scholar]
- 20.Chen XJ, Wang X, Butow RA. Yeast aconitase binds and provides metabolically coupled protection to mitochondrial DNA. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(34):13738–13743. doi: 10.1073/pnas.0703078104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen XJ, Wang X, Kaufman BA, Butow RA. Aconitase couples metabolic regulation to mitochondrial DNA maintenance. Science. 2005;307(5710):714–717. doi: 10.1126/science.1106391. [DOI] [PubMed] [Google Scholar]
- 22.Liu Z, Butow RA. A transcriptional switch in the expression of yeast tricarboxylic acid cycle genes in response to a reduction or loss of respiratory function. Molecular and Cellular Biology. 1999;19(10):6720–6728. doi: 10.1128/mcb.19.10.6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Z, Butow RA. Mitochondrial retrograde signaling. Annual Review of Genetics. 2006;40:159–185. doi: 10.1146/annurev.genet.40.110405.090613. [DOI] [PubMed] [Google Scholar]
- 24.Liu Z, Sekito T, Špírek M, Thornton J, Butow RA. Retrograde signaling is regulated by the dynamic interaction between Rtg2p and Mks1p. Molecular Cell. 2003;12(2):401–411. doi: 10.1016/s1097-2765(03)00285-5. [DOI] [PubMed] [Google Scholar]
- 25.Gangloff SP, Marguet D, Lauquin GJ-M. Molecular cloning of the yeast mitochondrial aconitase gene (ACO1) and evidence of a synergistic regulation of expression by glucose plus glutamate. Molecular and Cellular Biology. 1990;10(7):3551–3561. doi: 10.1128/mcb.10.7.3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCammon MT. Mutants of Saccharomyces cerevisiae with defects in acetate metabolism: isolation and characterization of Acn- mutants. Genetics. 1996;144(1):57–69. doi: 10.1093/genetics/144.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vélot C, Haviernik P, Lauquin GJ-M. The Saccharomyces cerevisiae RTG2 gene is a regulator of aconitase expression under catabolite repression conditions. Genetics. 1996;144(3):893–903. doi: 10.1093/genetics/144.3.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Amberg DC, Burke DJ, Strathern JN. Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual. Cold Spring Harbor, NY, USA: Cold Spring Harbor Laboratory; 2005. [Google Scholar]
- 29.Li L, Kaplan J. A mitochondrial-vacuolar signaling pathway in yeast that affects iron and copper metabolism. Journal of Biological Chemistry. 2004;279(32):33653–33661. doi: 10.1074/jbc.M403146200. [DOI] [PubMed] [Google Scholar]
- 30.Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae . Genetics. 1989;122(1):19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rothermel BA, Shyjan AW, Etheredge JL, Butow RA. Transactivation by Rtg1p, a basic helix-loop-helix protein that functions in communication between mitochondria and the nucleus in yeast. Journal of Biological Chemistry. 1995;270(49):29476–29482. doi: 10.1074/jbc.270.49.29476. [DOI] [PubMed] [Google Scholar]
- 32.Jia Y, Rothermel B, Thornton J, Butow RA. A basic helix-loop-helix-leucine zipper transcription complex in yeast functions in a signaling pathway from mitochondria to the nucleus. Molecular and Cellular Biology. 1997;17(3):1110–1117. doi: 10.1128/mcb.17.3.1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chelstowska A, Liu Z, Jia Y, Amberg D, Butow RA. Signalling between mitochondria and the nucleus regulates the expression of a new D-lactate dehydrogenase activity in yeast. Yeast. 1999;15(13):1377–1391. doi: 10.1002/(SICI)1097-0061(19990930)15:13<1377::AID-YEA473>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 34.Liu Z, Sekito T, Epstein CB, Butow RA. RTG-dependent mitochondria to nucleus signaling is negatively regulated by the seven WD-repeat protein Lst8p. EMBO Journal. 2002;20(24):7209–7219. doi: 10.1093/emboj/20.24.7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sekito T, Liu Z, Thornton J, Butow RA. RTG-dependent mitochondria-to-nucleus signaling is regulated by MKS1 and is linked to formation of yeast prion [URE3] Molecular Biology of the Cell. 2002;13(3):795–804. doi: 10.1091/mbc.01-09-0473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liao X, Small WC, Srere PA, Butow RA. Intramitochondrial functions regulate nonmitochondrial citrate synthase (CIT2) expression in Saccharomyces cerevisiae . Molecular and Cellular Biology. 1991;11(1):38–46. doi: 10.1128/mcb.11.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kirchman PA, Kim S, Lai C, Michal Jazwinski S. Interorganelle signaling is a determinant of longevity in Saccharomyces cerevisiae . Genetics. 1999;152(1):179–190. doi: 10.1093/genetics/152.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Williamson DH, Fennell DJ. Visualization of yeast mitochondrial dna with the fluorescent stain ‘DAPI’. Methods in Enzymology C. 1979;56:728–733. doi: 10.1016/0076-6879(79)56065-0. [DOI] [PubMed] [Google Scholar]
- 39.Jia Y-K, Bécam A-M, Herbert CJ. The CIT3 gene of Saccharomyces cerevisiae encodes a second mitochondrial isoform of citrate synthase. Molecular Microbiology. 1997;24(1):53–59. doi: 10.1046/j.1365-2958.1997.3011669.x. [DOI] [PubMed] [Google Scholar]
- 40.Kim KS, Rosenkrantz MS, Guarente L. Saccharomyces cerevisiae contains two functional citrate synthase genes. Molecular and Cellular Biology. 1986;6(6):1936–1942. doi: 10.1128/mcb.6.6.1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lewin AS, Hines V, Small GM. Citrate synthase encoded by the CIT2 gene of Saccharomyces cerevisiae is peroxisomal. Molecular and Cellular Biology. 1990;10(4):1399–1405. doi: 10.1128/mcb.10.4.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mühlenhoff U, Stadler JA, Richhardt N, et al. A specific role of the yeast mitochondrial carriers Mrs3/4p in mitochondrial iron acquisition under iron-limiting conditions. Journal of Biological Chemistry. 2003;278(42):40612–40620. doi: 10.1074/jbc.M307847200. [DOI] [PubMed] [Google Scholar]
- 43.Froschauer EM, Schweyen RJ, Wiesenberger G. The yeast mitochondrial carrier proteins Mrs3p/Mrs4p mediate iron transport across the inner mitochondrial membrane. Biochimica et Biophysica Acta. 2009;1788(5):1044–1050. doi: 10.1016/j.bbamem.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 44.Rutherford JC, Bird AJ. Metal-responsive transcription factors that regulate iron, zinc, and copper homeostasis in eukaryotic cells. Eukaryotic Cell. 2004;3(1):1–13. doi: 10.1128/EC.3.1.1-13.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kawamata H, Manfredi G. Import, maturation, and function of SOD1 and its copper chaperone CCS in the mitochondrial intermembrane space. Antioxidants and Redox Signaling. 2010;13(9):1375–1384. doi: 10.1089/ars.2010.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klöppel C, Michels C, Zimmer J, Herrmann JM, Riemer J. In yeast redistribution of Sod1 to the mitochondrial intermembrane space provides protection against respiration derived oxidative stress. Biochemical and Biophysical Research Communications. 2010;403(1):114–119. doi: 10.1016/j.bbrc.2010.10.129. [DOI] [PubMed] [Google Scholar]
- 47.Sturtz LA, Diekert K, Jensen LT, Lill R, Culotta VC. A fraction of yeast Cu,Zn-superoxide dismutase and its metallochaperone, CCS, localize to the intermembrane space of mitochondria. A physiological role for SOD1 in guarding against mitochondrial oxidative damage. Journal of Biological Chemistry. 2001;276(41):38084–38089. doi: 10.1074/jbc.M105296200. [DOI] [PubMed] [Google Scholar]
- 48.Foury F. Low iron concentration and aconitase deficiency in a yeast frataxin homologue deficient strain. FEBS Letters. 1999;456(2):281–284. doi: 10.1016/s0014-5793(99)00961-8. [DOI] [PubMed] [Google Scholar]