

Abstract
Lipid modifications aid in regulating (and misregulating) protein function and localization. However, efficient methods to screen for a lipid's ability to modify proteins are not readily available. We present a strategy to identify protein-reactive lipids and apply it to a neurodevelopmental disorder, Smith-Lemli-Opitz syndrome (SLOS). Alkynyl surrogates were synthesized for polyunsaturated fatty acids, phospholipids, cholesterol, 7-dehydrocholesterol (7-DHC), and a 7-DHC-derived oxysterol. To probe for protein-reactive lipids, we used click chemistry to biotinylate the alkynyl tag and detected the lipid-adducted proteins with streptavidin Western blotting. In Neuro2a cells, the trend in amount of protein adduction followed known rates of lipid peroxidation (7-DHC >> arachidonic acid > linoleic acid >> cholesterol), with alkynyl-7-DHC producing the most adduction among alkynyl lipids. 7-DHC reductase-deficient cells, which cannot properly metabolize 7-DHC, exhibited significantly more alkynyl-7-DHC-protein adduction than control cells. Model studies demonstrated that a 7-DHC peroxidation product covalently modifies proteins. We hypothesize that 7-DHC generates electrophiles that can modify the proteome, contributing to SLOS's complex pathology. These probes and methods would allow for analysis of lipid-modified proteomes in SLOS and other disorders exhibiting 7-DHC accumulation. More broadly, the alkynyl lipid library would facilitate exploration of lipid peroxidation's role in specific biological processes in numerous diseases.
Keywords: cholesterol, 7-dehydrocholesterol, oxysterol, lipid peroxidation, lipid electrophiles
Palmitoylation, N-myristoylation, cholesterylation, and glycosylphosphatidylinositol-anchor addition are a few of several known modes of protein lipidation (1, 2). These modifications affect a protein's stability, cellular localization, and biological activities (3, 4). In addition to directly modifying proteins, lipids are susceptible to reaction with molecular oxygen, yielding oxidation products that also have diverse biological effects. We recently provided evidence that 7-dehydrocholesterol (7-DHC), the immediate biosynthetic precursor of cholesterol (5), is one of Nature's most oxidizable lipids (6). Elevated levels of 7-DHC are a hallmark of Smith-Lemli-Opitz syndrome (SLOS), a disorder that arises from mutations in the gene encoding 7-DHC reductase (Dhcr7), the last enzyme in the cholesterol biosynthesis pathway (7–9). This defect in cholesterol biosynthesis causes a broad array of phenotypical, physiological, and neurological abnormalities (7, 9, 10). It is not clear whether the abnormalities are due to 7-DHC buildup, cholesterol deficiency, or some other cause. A plausible hypothesis is that 7-DHC or its peroxidation products are biologically active and contribute to the pathology of SLOS (11–14).
Our analytical studies provided evidence that 7-DHC undergoes free-radical chain oxidation to yield 5α, 6α-epoxide (DHCEp) as well as a variety of other oxysterol products, some of which are derived by either nonenzymatic or enzymatic reactions within cells (15–18). For example, DHCEp can metabolize to 3β,5α-dihydroxycholest-7-en-6-one (DHCEO), a biomarker of oxidative stress in SLOS models, as shown in Fig. 1. DHCEp is very reactive, with the epoxide moiety undergoing hydrolysis in aqueous environments and ring-opening in the presence of alcohols.
Fig. 1.

Conversion of 7-DHC to DHCEO in SLOS cells and tissues. Lipid peroxidation and enzymatic oxidation yield the oxysterol DHCEO, which is a biomarker of oxidative stress in SLOS.
While peroxidation of cholesterol and 7-DHC generates oxysterols, peroxidation of polyunsaturated fatty acids, such as arachidonic acid (20:4, ω-6) and linoleic acid (18:2, ω-6), results in different assorted by-products, including the electrophilic 4-hydroxynonenal (HNE) (19). HNE has been shown to react with nucleophilic protein residues to afford stable covalent adducts (20–22). Protein modification with HNE may be involved in the development of various diseases, including Parkinson's disease, Alzheimer's disease, and atherosclerosis, in addition to altered cell signaling (23–26). HNE-protein adduction has been studied extensively, with protein targets of modification and sites of adduction within modified proteins identified (21, 27).
An effective strategy that we have developed to study protein modification involves incorporating an alkyne functional group into a lipid by organic synthesis, thus generating a surrogate for the naturally occurring compound (21, 28, 29). We report here on the preparation of a library of synthetic alkynyl fatty acids, phospholipids, and sterols and the incubation of these lipid surrogates in Neuro2a cells. Subsequent Huisgen-Sharpless cycloaddition (click reaction) (30, 31) of cell lysates with an azido-biotin reagent (21) enabled us to visualize adducted proteins by streptavidin Western blotting, thereby showing the overall level of proteome modification by the alkynyl lipid. A comparison of protein adduction by alkynyl 7-DHC (a-7-DHC) in control and SLOS model cells reveals increased protein adduction in the SLOS model. Model peptide and protein adduction studies suggest one potential 7-DHC oxidation product likely to be responsible for covalent protein modification in the SLOS model cell culture.
MATERIALS AND METHODS
Synthetic procedures
1H and 13C NMR spectra were collected on a 300 or 400 MHz NMR. High-resolution mass spectrometry (HRMS) analyses were carried out at the University of Notre Dame. Purification by column chromatography was carried out on silica gel, and TLC plates were visualized with phosphomolybdic acid. The synthesis of a-HNE (21), a-PA, a-DPPC, and a-PLPC have been previously published (28). The synthesis of a-LA and a-AA were previously reported (28), but an improved synthetic route is described here. Full synthetic procedures for the remaining alkynyl lipids in Fig. 2A are included in the supplementary data.
Fig. 2.

Library of alkynyl lipid probes. A: Synthesized probes include fatty acids, phospholipids, and sterols that were tested for adduction to the Neuro2a proteome. B: Overview of synthetic route to a-Chol and a-DHC.
Cell cultures
Neuroblastoma cell line Neuro2a was purchased from American Type Culture Collection (Rockville, MD). Neuro2a cells were maintained in DMEM (Life Technologies) and supplemented with L-glutamine, 10% FBS (Thermo Scientific HyClone), and penicillin/streptomycin at 37°C and 5% CO2. According to the vendor, the FBS contained cholesterol at concentrations of 32 mg/100 ml. This translates into a final cholesterol concentration of 32 μg/ml in our culture medium. To evaluate the role of exogenous cholesterol on gene expression, cells were also cultured with medium containing 10% cholesterol-deficient serum (Thermo Scientific HyClone Lipid Reduced FBS). This FBS medium did not have detectable cholesterol levels. The Neuro2a cells were subcultured once a week, and the culture medium was changed every two days. Neuro2a cells were cultured for two days before transfections. Cells were transfected with Dhcr7 pGIPZ plasmids using a Nucleofector instrument and Nucleofector kit V (Amaxa GmbH, Cologne, Germany) optimized for use with Neuro2a cells. Briefly, 2 × 106 cells were resuspended in 100 μl of transfection buffer, shRNA was added, and cells were electroporated using program T-24. The cells were grown for 24 h following transfection, and the stable cell line was established using puromycin as selection marker. The expression of Dhcr7 was monitored by quantitative RT-PCR, and the amount of 7-DHC, with MS.
Cell viability assay
Neuro2a cells were plated 6,000/well in 96-well plates and allowed to adhere on the plate for 24 h. The medium was removed, and the cells were treated with lipids of interest at different concentrations dissolved in 2% FBS DMEM. After 24 h of incubation, the medium was removed, and the cells were washed once with cold PBS. Cell viability was then evaluated using the CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega), a colorimetric method for determining the number of viable cells in proliferation or cytotoxicity (see supplementary data for further details). A premixed solution of 2% FBS DMEM containing CellTiter 96® AQueous One Solution Reagent (5/1 v/v of medium to assay reagent) was added to culture wells (120 μl/well). After 1 h of incubation at 37°C, the absorbance at 490 nm was recorded using a 96-well plate reader. The absorbance at 490 nm is directly proportional to the number of living cells in culture.
Alkynyl lipid treatment in Neuro2a cells
Neuro2a cells were grown in DMEM supplemented with L-glutamine, 10% FBS, and penicillin/streptomycin at 37°C and 5% CO2. Cells were plated 2 × 106 in 10 cm plates, then allowed to settle and grow for 24 h. The cells were then incubated in the presence of alkynyl probes (20 μM) in 2% FBS DMEM for 24 h. The harvested cells were centrifuged at 800 g for 5 min, then washed with cold PBS, pH 7.4. Cell pellets were stored at −40°C until further processing (either lipid extraction and HPLC-MS analysis or protein biotinylation).
Extraction, separation, and HPLC-MS analysis of free sterols and sterol esters
To the cell pellets was added Folch solution [5 ml chloroform/methanol (2/1) containing 0.001M BHT and PPh3], followed by the addition of NaCl aqueous solution (0.9%, 1 ml). d7-7-DHC, d7-7-DHC-LA, d7-Chol, and d7-Chol-LA were added as internal standards. The resulting mixture was vortexed for 1 min and centrifuged for 5 min. The lower organic phase was recovered, dried under a stream of nitrogen, redissolved in methylene chloride, and subjected to separation with NH2-SPE [500 mg cartridge; the column was conditioned with 4 ml of hexane, and the neutral lipids containing oxysterols were eluted with 4 ml of chloroform/2-propanol (2/1, v/v)]. The eluted fractions were then dried under nitrogen and reconstituted for HPLC-MS analysis. All processed samples were analyzed by a Thermofinnigan TSQ Quantum Ultra equipped with a Finnigan Surveyor Autosampler Plus. Reverse-phase HPLC was performed on a Phenomenex Luna C18 column (150 × 2 mm, 3 µM), preequilibrated in MeCN (solvent A)/CH2Cl2 (solvent B) (87.5/12.5). The samples were injected and eluted (0.2 ml/min) with an increasing concentration of solvent B [time (min)/per percentage of solvent B: 4/12.5, 7/47.5, 28/50.0, 32/12.5, 35/12.5] (32). The MS was operated in the positive-ion mode using atmospheric pressure chemical ionization (APCI) in the selected reaction monitoring (SRM) mode. MS parameters were optimized for d7-7-DHC and d7-7-DHC-LA and were as follows: auxiliary gas pressure was set at 5 psi, sheath gas pressure was 30 psi, utilizing nitrogen for both. Discharge current was set at 20 eV, and the vaporizer temperature was set at 230°C. Collision-induced dissociation was optimized at 16 eV under 1.0 mTorr of argon. Data acquisition and analysis were performed using Xcalibur software, version 2.0 (San Jose, CA). Assignment of the oleate, palmitate, and arachidonate sterol esters was previously established using independently synthesized standards (32).
Biotinylation of alkynyl lipid-adducted proteins in Neuro2a cells
Cell pellets were lysed on ice for 20 min in 1 ml of cold lysis buffer supplemented with 150 mM NaCl, protease inhibitor mixture, and phosphatase inhibitor mixture (1.0 mM sodium fluoride, 1.0 mM sodium molybdate, 1.0 mM sodium orthovanadate, 10.0 mM β-glycerophosphate) for each plate. The lysate was cleared by centrifugation at 10,000 g for 10 min to remove cellular debris. The total protein concentration was determined using standard BCA assay (Pierce). To detect the alkynyl lipid-modified proteins in Neuro2a, cell lysates from alkynyl probe-treated cells were reduced with sodium borohydride, 5 mM final concentration, for 1 h at room temperature. Sodium borohydride reduction was employed to stabilize any protein-lipid electrophile adducts that may form reversibly, such as Michael and Schiff base adducts. Then the sodium borohydride was quenched by acidifying to pH 6. Subsequently, all click reagents were added to the reduced cell lysates, azido-biotin (21) (0.2 mM), tris(3-hydroxypropyltriazolylmethyl)amine (THPTA) (33) (0.2 mM), CuSO4 (1 mM), and sodium ascorbate (1 mM), and the reaction was vortexed and allowed to react at room temperature for 2 h. The reaction mixture was precipitated using cold acetone (acetone/water, 6/1, v/v) to remove all excess chemicals. The cell pellets were reconstituted in 100 μl of LDS sample buffer including DTT (50 mM).
Immunoblot analysis of protein adducts with alkyne probes
The reconstituted proteins were resolved using 10% NuPAGE Novex BisTris® gel (Invitrogen, Carlsbad, CA). Precision Plus ProteinTM KaleidoscopeTM standards (10–250 kDa, Bio-Rad) were run on the same gel for reference. The proteins were electrophoretically transferred to a polyvinylidene difluoride membrane (Invitrogen) and probed with streptavidin conjugated with Alexa Fluor 680®. Biotinylated proteins were visualized using Odyssey Infrared Imaging System™ and Odyssey software as described by the manufacturer (Licor, Lincoln, NE). Integrated intensities were obtained with the Odyssey software and can be found in the supplementary data.
Modification of AcTpepK (Ac-AVAGKAGAR) by 7-DHCEp and a-DHCEp
A solution of AcTpepK (1 mM) and 7-DHCEp or a-7-DHCEp (5 mM) in pH 7.4 phosphate buffer:CH3CN (100 µl, 1:1 by volume) was incubated at 37°C for 1 h. The reaction mixture was reduced with 2 M NaBH4 (5 µl), then neutralized with 10% HCl (10 µl) for HPLC/MS analysis. Reverse-phase HPLC was performed on a Supelco Discovery C18 column (150 × 2.1 mm, 5 µM) using a mobile phase consisting of A: 0.05% TFA in H2O and B: 0.05% TFA in CH3CN. The unreacted peptide and adducts were eluted with a gradient of 5% to 35% B over 20 min, then to 100% B over 5 min and held for 20 min, and back to 5% B over 5 min. The MS was operated in the positive-ion mode using electrospray ionization with conditions optimized for the AcTpepK.
Modification of cytochrome c by 7-DHC/7-DHCEp
Cytochrome c (cyt c; from equine heart, 40 µM) was incubated in the presence of 7-DHC (1.25 mM) or 7-DHCEp (400 µM) in 10 mM ammonium bicarbonate buffer (pH 7.4) at room temperature overnight with stirring. For the 7-DHC experiment, 10% MeCN was added to aid in lipid solubilization, and the reaction was performed under O2 (1 atm). After stirring overnight, samples were stored at −40°C until MALDI-TOF MS analysis.
MALDI-TOF MS analysis of sterol-modified cyt c
Modified cyt c sample (0.5 μl) and a saturated solution of sinapinic acid (3,5-dimethoxy-4-hydroxycinnamic acid) in CH3CN/H2O/TFA (50/50/0.1, v/v/v) (1 μl) were spotted onto a MALDI target plate, immediately mixed by pipetting up and down twice, and allowed to dry before analysis. MALDI-TOF MS analyses were performed with a PerSeptive Biosystems Voyager-DE STR MALDI-TOF equipped with a pulsed N2 laser. Protein spectra were collected in positive ion linear mode with an accelerating voltage of 20 kV. Each spectrum was the accumulation of 1,000 laser shots, with a laser intensity of 2,200–2,300 that was optimized for each spectrum to provide the best signal-to-noise ratio. The MALDI-TOF MS spectra were processed using the Data Explorer software.
a-7-DHC treatment in control versus Dhcr7-deficient Neuro2a cells
Neuro2a cells were maintained in DMEM and supplemented with L-glutamine, 10% FBS (Thermo Scientific HyClone), and penicillin/streptomycin at 37°C and 5% CO2. Cells were plated 7.5 × 105 in 6 cm plates, then allowed to settle and grow for 24 h. Both control and Dhcr7-deficient Neuro2a cells were treated with a-7-DHC at concentrations ranging 0–5 μM in DMEM with N-2 supplement. After incubation at 37°C for 24 h, cells were harvested and lysed, and the total protein concentration was determined using standard BCA assay (Pierce). The lysate was reduced with NaBH4 (5 mM) to stabilize any adducts that may have formed and neutralized. Subsequently, all click reagents were added to the reduced cell lysates, azido-biotin (21) (0.2 mM), THPTA (33) (0.2 mM), CuSO4 (1 mM), and sodium ascorbate (1 mM), and the reaction was vortexed and allowed to react at room temperature for 2 h.
RESULTS
Synthesis of alkynyl lipid probes
Analogs of several lipids and lipid electrophiles have been synthesized with an alkyne group introduced as a functional tag (Fig. 2A). For each lipid, the alkyne is incorporated in a terminal chain position, facilitating a subsequent click reaction (21). For the alkynyl fatty acids and phospholipids, this triple bond substitution results in a net loss of four hydrogens from the naturally occurring lipid. In the alkynyl sterols and oxysterols prepared, the triple bond replaces the C24 isopropyl group.
While full synthetic procedures for all alkynyl lipids are included in the supplementary data [and synthetic approaches to some of the alkynyl analogs have been published previously (21, 28)], we highlight efficient syntheses of the novel alkynyl cholesterol (a-Chol), a-7-DHC (Fig. 2B), and a-DHCEp here. a-Chol was prepared starting from commercially available 3β-hydroxy-5-cholenic acid; a key synthetic step was the deoxygenation of the propargyl alcohol intermediate to the corresponding alkyne. Traditional reductive methods resulted in undesired side reactions: elimination of water to yield an enyne and isomerization of the alkyne to an allene. One-pot cobalt protection of the alkyne, acid-catalyzed borane reduction, and final deprotection of the alkyne afforded a-Chol in good yield (34). a-Chol was smoothly converted to a-7-DHC via oxidation to alkynyl 7-ketocholesterol and a subsequent Shapiro reaction to form the diene. mCPBA oxidation of a-7-DHC provided a-DHCEp, the alkynyl analog of DHCEp.
Alkynyl compounds show cellular metabolism comparable to their naturally occurring analogs
Since the naturally occurring lipid and its alkynyl analog are structurally very similar, we expected to observe minimal perturbation of the lipid's cellular metabolism. The suitability of a-Chol and a-7-DHC as surrogates for cholesterol and 7-DHC, respectively, was tested by comparing their cellular toxicity, incorporation, and esterification. Neuro2a neuroblastoma cells were treated with cholesterol, a-Chol, 7-DHC, and a-7-DHC, and cellular viability was determined after 24 h (supplementary Fig. I). a-Chol and cholesterol were nontoxic at all concentrations tested (≤40 μM). Although more toxic than cholesterol and a-Chol, a-7-DHC exhibited similar toxicity to its natural analog, 7-DHC.
To test the incorporation and metabolism of the compounds of interest, Neuro2a cells were treated with a-Chol or a-7-DHC using a concentration that does not affect cell viability (2.5 μM), and cell extracts were analyzed after 24 h by reverse-phase HPLC-MS with SRM. Upon treatment with a-Chol, cells incorporate and esterify the alkynyl analog (supplementary Fig. II). Similarly, upon incubation with a-7-DHC, cells successfully incorporate the alkynyl lipid (Fig. 3A). Using methods previously established in our group, we were able to separate and analyze endogenous cholesterol, a-Chol, a-7-DHC, and their corresponding esters in one chromatographic run (32). Under these analytical conditions, cholesterol and its esters fragment to give a common ion having m/z 369, generated by loss of water or the fatty ester tail, respectively. Accordingly, a-Chol and its esters fragment to afford a carbocation with m/z 351, and a-7-DHC and its esters undergo fragmentation to yield a carbocation with m/z 349. The fatty acid constituent profiles of a-Chol esters and the endogenous cholesterol esters (displayed in Fig. 3E, F) are comparable, indicating that the alkynyl probe is metabolized similarly to natural cholesterol. Most of the a-7-DHC is converted to a-Chol (Fig. 3B) and its corresponding alkynyl cholesteryl esters (Fig. 3E), exhibiting metabolism typical of the naturally occurring 7-DHC. There are no a-7-DHC esters formed at this concentration of treatment (Fig. 3D). Both the cell viability and incorporation results confirm that the alkynyl derivatives are suitable surrogates for the naturally occurring sterols.
Fig. 3.

Incorporation and metabolism of a-7-DHC in Neuro2a cells. Neuro2a cells were incubated in the presence of 2.5 μM a-7-DHC for 24 h in 2% FBS DMEM. Free sterols and sterol esters were analyzed by HPLC-MS operated in SRM mode. A–C: Chromatograms showing free sterols. D–F: Chromatograms showing sterol esters (expansions of A–C, respectively). A and D: Molecules that fragment to give an ion with m/z 349, a-7-DHC, and its esters. B and E: Molecules that fragment to give an ion with m/z 351, a-Chol, and its esters OA, oleate; PA, palmitate; SA, stearate. C and F: Molecules that fragment to give m/z 369, cholesterol, and its esters.
Biotinylation allows for detection of Neuro2a cellular proteome modification with alkynyl lipid probes
The unique characteristic of the lipid analogs used in this study is the presence of a terminal alkyne moiety, which enables the click reaction with an azido-biotin reagent (Fig. 4) (21, 27, 35). With evidence that a-Chol and a-7-DHC incorporate into cells and with previous work demonstrating that alkynyl fatty acid and alkynyl phospholipid probes can be incorporated and metabolized as well (28, 29), Neuro2a cells were treated with each of our alkynyl fatty acids, phospholipids, and sterols shown in Fig. 2A. An overview of the procedure is outlined in Fig. 4, with details described in Materials and Methods. To visualize the biotinylated adducts (alkynyl lipid-modified proteins with biotin tag), we used Western blotting, employing a fluorescent streptavidin conjugate (Fig. 5). Integrated intensities from the Western blot analyses of Neuro2a protein adduction with the alkynyl lipid probes indicated the degree of alkynyl lipid modification of the Neuro2a proteome (supplementary Fig. III).
Fig. 4.

General experimental workflow, including the azido-biotin reagent used for click biotinylation of alkynyl lipid-modified proteins.
Fig. 5.
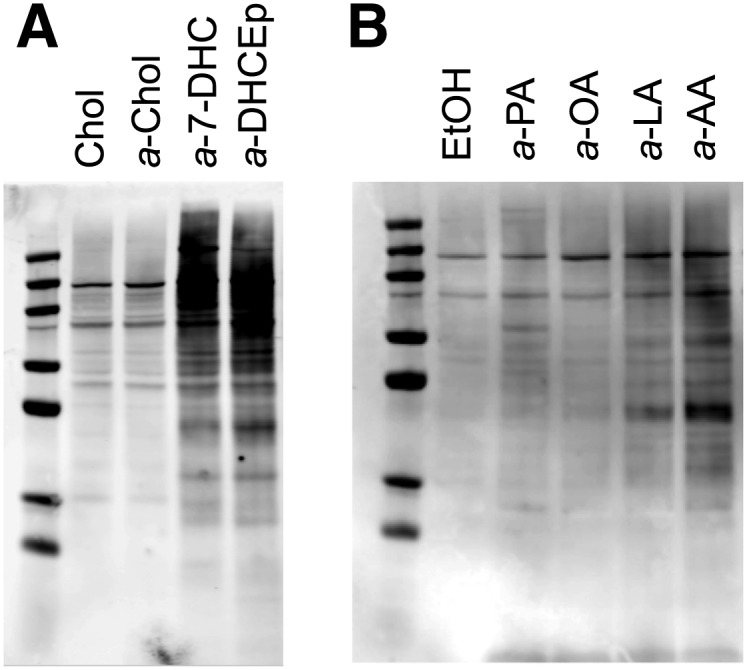
Neuro2a protein adduction with alkynyl lipids. Neuro2a cells were incubated in the presence of alkynyl lipid (20 μM) for 24 h in 2% FBS DMEM. Proteins adducted with alkynyl lipid were ligated with biotin via click reaction. The degree of adduction is determined by Western blotting using the streptavidin-AlexaFluor 680 conjugate. The fluorophore was detected using the Odyssey Infrared Imaging System (Licor). A: Western blot comparison of Neuro2a proteome modification by alkynyl sterols. Chol was used as a control treatment. B: Western blot comparison of Neuro2a proteome modification by alkynyl fatty acids. Ethanol was used as a control treatment.
a-7-DHC exhibited the greatest amount of Neuro2a proteome adduction among all alkynyl lipids (Fig. 5A). Because a-7-DHC led to a significant amount of protein adduction, an alkynyl analog of DHCEp (a-DHCEp), a major product of 7-DHC peroxidation, was prepared and tested for protein adduction. a-DHCEp also showed a considerable amount of protein modification, demonstrating that 7-DHC oxidation products can act as reactive electrophiles. a-Chol afforded negligible protein adduction.
As shown in Fig. 5B, within the alkynyl fatty acid class, the amount of protein adduction followed: 19a-arachidonic acid (a-AA) > 17a-linoleic acid (a-LA) > 17a-oleic acid (a-OA) ≈ 15a-palmitic acid (a-PA). The alkynyl phospholipids yielded little adduction relative to the ethanol control (data not shown). The oxidizability of each lipid correlates with the amount of Neuro2a proteome modification by each lipid: a higher rate of lipid peroxidation generally results in a greater amount of lipid-protein adduction (6).
Direct evidence for model peptide and protein adduction
To confirm that alkynyl lipids covalently bind proteins, we used in vitro studies, incubating lipids with a peptide and small protein; the two lipid species giving the greatest amount of Neuro2a proteome modification (a-7-DHC and a-DHCEp) were utilized in these model studies. When DHCEp or a-DHCEp is reacted with a model peptide containing one nucleophilic site, we observed compounds by HPLC-MS that have masses corresponding to the respective peptide-oxysterol adduct (supplementary Fig. IV).
Cyt c, a 12 kDa protein containing 19 lysine residues and 3 histidine residues, was chosen as an illustrative protein for DHCEp (400 Da) adduction studies. Cyt c (40 μM) was incubated with DHCEp (400 μM) at room temperature for 18 h and subsequently analyzed by MALDI-MS (Fig. 6A). Focusing on the region displaying doubly charged cyt c species, a protein-DHCEp adduct shifted +200 m/z units from the unmodified protein was observed. A peak representing cyt c-oxysterol adduction plus loss of the cyt c heme group during MS analysis was also seen, providing further support for modification.
Fig. 6.

Reaction of cyt c with 7-DHCEp and 7-DHC. MALDI-MS analysis of (A) protein adduct with 7-DHCEp and (B) protein adduct with 7-DHC.
A similar adduction experiment employing the highly oxidizable 7-DHC, rather than one of its preformed oxysterols (DHCEp), was performed. Cyt c (40 μM) was incubated with 7-DHC (384 Da) (1.25 mM) under an oxygen atmosphere (1 atm) at room temperature for 18 h and subsequently analyzed by MALDI-MS (Fig. 6B). 7-DHC showed an adduction profile similar to that of DHCEp. A peak in the range of doubly charged cyt c species with a m/z shift of +200 appeared after incubation with 7-DHC. This result is consistent with 7-DHC undergoing lipid peroxidation to form an electrophilic oxysterol (384 Da + 16 Da), such as DHCEp, which subsequently modifies cyt c. A peak representing cyt c-7-DHC oxysterol adduction and subsequent loss of the heme group during MS analysis was also observed. These model experiments verified that we are able to monitor the modification of a single protein or peptide by an oxysterol.
Protein adduction increases in the cellular SLOS model
In SLOS, mutations in the gene encoding Dhcr7 result in an inactive enzyme and an accumulation of 7-DHC (36). Here, we tested whether increased 7-DHC and the presence of 7-DHC-derived oxysterols would lead to increased lipidation of a cellular proteome. Dhcr7-deficient Neuro2a cells were generated as described previously and utilized as a model for SLOS study (37). Using the a-7-DHC probe, the extent of protein modification in control Neuro2a cells and Dhcr7-deficient Neuro2a cells was compared. Biotinylated proteins were visualized using streptavidin Western blotting (Fig. 7). At treatment concentrations ≥ 2.5 μM of a-7-DHC, Dhcr7-deficient cells clearly showed an increased amount of protein adduction relative to control Neuro2a cells. Control Neuro2a cells treated with a-7-DHC at 0–5 μM showed little protein adduction, because at these concentrations, a-7-DHC is effectively being converted to a-Chol. These results suggest that increased lipid peroxidation affects the cellular proteome. Future proteomics studies will identify the a-7-DHC-modified proteins in SLOS models.
Fig. 7.

a-7-DHC protein adduction in control Neuro2a versus Dhcr7-deficient Neuro2a cells. Neuro2a cells were incubated in the presence of a-7-DHC (0–5 μM) or a-Chol (20 μM) for 24 h. Alkynyl lipid-adducted proteins were ligated with biotin via click reaction. The degree of adduction is determined by Western blotting using the streptavidin-AlexaFluor 680 conjugate. The fluorophore was detected using the Odyssey Infrared Imaging System (Licor).
DISCUSSION
Because of the potent biological activities of a variety of lipids and their corresponding oxidation products, we have designed several alkynyl lipid and alkynyl lipid electrophile probes to survey their reactivity with proteins. Employing an alkyne functional group as the tag generally results in minimal perturbation of the lipid's biological activity, as the naturally occurring lipid and the alkynyl analog are structurally very similar. Upon incubating cells in the presence of an alkynyl lipid, an azido-biotin reagent (21) is clicked to any alkynyl lipid-modified proteins, and subsequent Western blotting with streptavidin fluorophore detection indicates the relative level of protein adduction.
Cell incorporation experiments validate a-7-DHC and a-Chol as appropriate biological surrogates for their respective nonalkynyl analogs. When comparing the relative amounts of protein adduction by all alkynyl lipids, the results mirror the rates of peroxidation: 7-DHC >> arachidonic acid > linoleic acid >> cholesterol (Fig. 5) (6). 7-DHC is known to be 10-fold more oxidizable than arachidonic acid (6). Correspondingly, significantly more protein adduction is observed in cells treated with a-7-DHC than in cells treated with a-AA and the other fatty acids. For fatty acids and phospholipids, the relative amount of protein adduction is dependent on the number of bis-allylic methylene centers in the lipid [arachidonic acid (3 centers) > linoleic acid (1) > oleic acid (0) ≈ palmitic acid (0)]. While the amount of protein adduction seems to be influenced by the oxidizability of the lipid probe, there are additional mechanisms of modification that should be considered when analyzing our results. S-acylation with palmitic acid and other longer chain fatty acids, both saturated and unsaturated, cholesterylation, and cysteine-alkyne reaction2 are a few known modes of protein adduction that may contribute to the overall modification of the Neuro2a proteome (1, 38, 39). Nonetheless, the substantial amounts of protein adduction observed for a-AA, a-7-DHC, and a-DHCEp (the two lipids in our study most susceptible to peroxidation and an electrophilic a-7-DHC oxidation product) imply that lipid oxidizability is a significant factor in assessing a lipid's propensity to modify proteins.
Modification of proteins with cholesterol-derived oxysterols has been associated with a number of diseases, including multiple sclerosis, antibody light chain misfolding disorders, and neurodegenerative disorders (40–43). Free-radical oxidation of 7-DHC provides assorted oxysterols, which have been shown to have important biological effects, such as cytotoxicity (15). In the context of SLOS, a mixture of 7-DHC-derived oxysterols induced changes in gene expression in control Neuro2a cells that are similar to those identified in Dhcr7-deficient Neuro2a cells (44). DHCEp is a major product of 7-DHC peroxidation and can metabolize to another oxysterol, DHCEO, a biomarker for 7-DHC oxidation that can affect neural development in a SLOS model mouse (16, 45). Because some of the 7-DHC-derived oxysterols are electrophilic, such as DHCEp, it seems reasonable to consider covalent modification of proteins as a contributing factor in the pathogenesis of SLOS.
Two key experiments directly correlate 7-DHC peroxidation and oxysterol formation to protein adduction. In the first experiment, incubation of 7-DHC or 7-DHCEp in the presence of a single protein, cyt c, provides a similar adduction profile. Because the adduction behavior is comparable, this suggests that 7-DHC autoxidizes to form oxysterols, such as DHCEp, which subsequently modify cyt c (Fig. 6). In the second experiment, 7-DHC shows greater amounts of protein adduction in Dhcr7-deficient Neuro2a than in control Neuro2a cells (Fig. 7). In deficient cells, the enzyme that converts 7-DHC to cholesterol is inactivated, resulting in a buildup of oxidizable 7-DHC. Treatment with a higher concentration of a-7-DHC (20 μM) in control Neuro2a cells led to protein adduction (Fig. 5A), suggesting that Dhcr7 is overwhelmed under these conditions and that a-7-DHC is accumulating. These experiments directly correlate accumulation of 7-DHC to increased protein modification, supporting the hypothesis that these covalent modifications may be involved in the mechanism of SLOS. We will report in due course on a-7-DHC-adducted proteins in Neuro2a cells using our photocleavable azido-biotin pulldown protocol (21). Any differential adduction of specific proteins between Dhcr7-deficient and control Neuro2a cells may shed light on the pathogenesis of SLOS. Protein adduction by 7-DHC oxysterols may also have bearing on the progression of other diseases that show elevated levels of 7-DHC, such as X-linked dominant chondrodysplasia punctata (CDPX2), cerebrotendinous xanthomatosis (CTX), and breast cancer (8, 46, 47).
In conclusion, the general strategy of using alkynyl probes in conjunction with click biotinylation and streptavidin Western blot visualization is an efficient way to assay the relative propensity of lipids to modify proteins. The rapid readout of results and ease of the azido-biotin tagging process make this an appealing approach for studying protein adduction in any system of interest. In our study of an alkynyl lipid library, a-7-DHC showed the greatest amount of Neuro2a protein modification, a finding that may have implications in the pathogenesis of SLOS. Current work is underway to identify and isolate any a-7-DHC-adducted proteins in control and Dhcr7-deficient Neuro2a cells and determine the sites of modification.
Supplementary Material
Footnotes
Abbreviations:
- 7-DHC
- 7-dehydrocholesterol
- a-7-DHC
- alkynyl 7-dehydrocholesterol
- a-AA
- 19a-arachidonic acid
- a-Chol
- alkynyl cholesterol
- a-DHCEp
- alkynyl DHC epoxide
- a-DPPC
- 1-palmitoyl-2-15a-palmitoyl-sn-glycero-phosphocholine
- a-HNE
- 8a-4-hydroxynonenal
- a-LA
- 17a-linoleic acid
- a-OA
- 17a-oleic acid
- a-PA
- 15a-palmitic acid
- a-PLPC
- 1-palmitoyl-2-17a-linoleoyl-sn-glycero-phosphocholine
- a-POPC
- 1-15a-palmitoyl-2-oleoyl-sn-glycero-phosphocholine
- APCI
- atmospheric pressure chemical ionization
- Chol
- cholesterol
- cyt c
- cytochrome c
- DHCEO
- 3β,5α-dihydroxycholest-7-en-6-one
- DHCEp
- 7-dehydrocholesterol 5α, 6α-epoxide
- Dhcr7
- 7-dehydrocholesterol reductase
- HNE
- 4-hydroxynonenal
- SLOS
- Smith-Lemli Opitz syndrome
- SRM
- selected reaction monitoring
This work was supported by National Institutes of Health Grant R01-HD-064727 (to N.A.P.), Vanderbilt Kennedy Center for Research on Human Development (Z.K.), and the São Paulo Research Foundation (FAPESP) (to T.C.G.-M.).
The online version of this article (available at http://www.jlr.org) contains supplementary data in the form of four figures and seven schemes.
REFERENCES
- 1.Nadolski M. J., Linder M. E. 2007. Protein lipidation. FEBS J. 274: 5202–5210 [DOI] [PubMed] [Google Scholar]
- 2.Hang H. C., Linder M. E. 2011. Exploring protein lipidation with chemical biology. Chem. Rev. 111: 6341–6358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Resh M. D. 2012. Targeting protein lipidation in disease. Trends Mol. Med. 18: 206–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Triola G., Waldmann H., Hedberg C. 2012. chemical biology of lipidated proteins. ACS Chem. Biol. 7: 87–99 [DOI] [PubMed] [Google Scholar]
- 5.Nes W. D. 2011. Biosynthesis of cholesterol and other sterols. Chem. Rev. 111: 6423–6451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu L., Davis T. A., Porter N. A. 2009. Rate constants for peroxidation of polyunsaturated fatty acids and sterols in solution and in liposomes. J. Am. Chem. Soc. 131: 13037–13044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tint G. S., Seller M., Hughes-Benzie R., Batta A. K., Shefer S., Genest D., Irons M., Elias E., Salen G. 1995. Markedly increased tissue concentrations of 7-dehydrocholesterol combined with low levels of cholesterol are characteristic of the Smith-Lemli-Opitz syndrome. J. Lipid Res. 36: 89–95 [PubMed] [Google Scholar]
- 8.Porter F. D., Herman G. E. 2011. Malformation syndromes caused by disorders of cholesterol synthesis. J. Lipid Res. 52: 6–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kelley R. I., Hennekam R. C. M. 2000. The Smith-Lemli-Opitz syndrome. J. Med. Genet. 37: 321–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee R. W. Y., Tierney E. 2011. Hypothesis: the role of sterols in autism spectrum disorder. Autism Res. Treat. 2011: 653570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sikora D. M., Pettit-Kekel K., Penfield J., Merkens L. S., Steiner R. D. 2006. The near universal presence of autism spectrum disorders in children with Smith-Lemli-Opitz syndrome. Am. J. Med. Genet. A. 140: 1511–1518 [DOI] [PubMed] [Google Scholar]
- 12.Gaoua W., Chevy F., Roux C., Wolf C. 1999. Oxidized derivatives of 7-dehydrocholesterol induce growth retardation in cultured rat embryos: a model for antenatal growth retardation in the Smith-Lemli-Opitz syndrome. J. Lipid Res. 40: 456–463 [PubMed] [Google Scholar]
- 13.Richards M. J., Nagel B. A., Fliesler S. J. 2006. Lipid hydroperoxide formation in the retina: correlation with retinal degeneration and light damage in a rat model of Smith-Lemli-Opitz syndrome. Exp. Eye Res. 82: 538–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Valencia A., Rajadurai A., Carle A. B., Kochevar I. E. 2006. 7-Dehydrocholesterol enhances ultraviolet A-induced oxidative stress in keratinocytes: roles of NADPH oxidase, mitochondria, and lipid rafts. Free Radic. Biol. Med. 41: 1704–1718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu L., Korade Z., Porter N. A. 2010. Oxysterols from free radical chain oxidation of 7-dehydrocholesterol: product and mechanistic studies. J. Am. Chem. Soc. 132: 2222–2232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu L., Korade Z., Rosado D. A., Liu W., Lamberson C. R., Porter N. A. 2011. An oxysterol biomarker for 7-dehydrocholesterol oxidation in cell/mouse models for Smith-Lemli-Opitz syndrome. J. Lipid Res. 52: 1222–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu L., Liu W., Sheflin L. G., Fliesler S. J., Porter N. A. 2011. Novel oxysterols observed in tissues and fluids of AY9944-treated rats: a model for Smith-Lemli-Opitz syndrome. J. Lipid Res. 52: 1810–1820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu L., Korade Z., Rosado D. A., Mirnics K., Porter N. A. 2013. Metabolism of oxysterols derived from nonenzymatic oxidation of 7-dehydrocholesterol in cells. J. Lipid Res. 54: 1135–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schneider C., Porter N. A., Brash A. R. 2008. Routes to 4-hydroxynonenal: fundamental issues in the mechanisms of lipid peroxidation. J. Biol. Chem. 283: 15539–15543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sayre L. M., Lin D., Yuan Q., Zhu X., Tang X. 2006. Protein adducts generated from products of lipid oxidation: focus on HNE and ONE*. Drug Metab. Rev. 38: 651–675 [DOI] [PubMed] [Google Scholar]
- 21.Vila A., Tallman K. A., Jacobs A. T., Liebler D. C., Porter N. A., Marnett L. J. 2008. Identification of protein targets of 4-hydroxynonenal using click chemistry for ex vivo biotinylation of azido and alkynyl derivatives. Chem. Res. Toxicol. 21: 432–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Codreanu S. G., Zhang B., Sobecki S. M., Billheimer D. D., Liebler D. C. 2009. Global analysis of protein damage by the lipid electrophile 4-hydroxy-2-nonenal. Mol. Cell. Proteomics. 8: 670–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perluigi M., Coccia R., Butterfield D. A. 2012. 4-Hydroxy-2-nonenal, a reactive product of lipid peroxidation, and neurodegenerative diseases: a toxic combination illuminated by redox proteomics studies. Antioxid. Redox Signal. 17: 1590–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zarkovic K. 2003. 4-Hydroxynonenal and neurodegenerative diseases. Mol. Aspects Med. 24: 293–303 [DOI] [PubMed] [Google Scholar]
- 25.Leonarduzzi G., Chiarpotto E., Biasi F., Poli G. 2005. 4-Hydroxynonenal and cholesterol oxidation products in atherosclerosis. Mol. Nutr. Food Res. 49: 1044–1049 [DOI] [PubMed] [Google Scholar]
- 26.Jacobs A. T., Marnett L. J. 2010. Systems analysis of protein modification and cellular responses induced by electrophile stress. Acc. Chem. Res. 43: 673–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim H-Y. H., Tallman K. A., Liebler D. C., Porter N. A. 2009. An azido-biotin reagent for use in the isolation of protein adducts of lipid-derived electrophiles by streptavidin catch and photorelease. Mol. Cell. Proteomics. 8: 2080–2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Milne S. B., Tallman K. A., Serwa R., Rouzer C. A., Armstrong M. D., Marnett L. J., Lukehart C. M., Porter N. A., Brown H. A. 2010. Capture and release of alkyne-derivatized glycerophospholipids using cobalt chemistry. Nat. Chem. Biol. 6: 205–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tallman K. A., Armstrong M. D., Milne S. B., Marnett L. J., Brown H. A., Porter N. A. 2013. Cobalt carbonyl complexes as probes for alkyne-tagged lipids. J. Lipid Res. 54: 859–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rostovtsev V. V., Green L. G., Fokin V. V., Sharpless K. B. 2002. A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. Engl. 41: 2596–2599 [DOI] [PubMed] [Google Scholar]
- 31.Thirumurugan P., Matosiuk D., Jozwiak K. 2013. Click chemistry for drug development and diverse chemical-biology applications. Chem. Rev. 113: 4905–4979 [DOI] [PubMed] [Google Scholar]
- 32.Liu W., Xu L., Lamberson C. R., Merkens L. S., Steiner R. D., Elias E. R., Haas D., Porter N. A. 2013. Assays of plasma dehydrocholesteryl esters and oxysterols from Smith-Lemli-Opitz syndrome patients. J. Lipid Res. 54: 244–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hong V., Presolski S. I., Ma C., Finn M. G. 2009. Analysis and optimization of copper-catalyzed azide-alkyne cycloaddition for bioconjugation. Angew. Chem. Int. Ed. Engl. 48: 9879–9883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McComsey D. F., Reitz A. B., Maryanoff C. A., Maryanoff B. E. 1986. Deoxygenation of acetylenic carbinols. reduction of cobalt carbonyl adducts with borane-methyl sulfide and trifluoroacetic acid. Synth. Commun. 16: 1535–1549 [Google Scholar]
- 35.Aluise C. D., Rose K., Boiani M., Reyzer M. L., Manna J. D., Tallman K., Porter N. A., Marnett L. J. 2013. Peptidyl-prolyl cis/trans-isomerase A1 (Pin1) Is a target for modification by lipid electrophiles. Chem. Res. Toxicol. 26: 270–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Witsch-Baumgartner M., Löffler J., Utermann G. 2001. Mutations in the human DHCR7 gene. Hum. Mutat. 17: 172–182 [DOI] [PubMed] [Google Scholar]
- 37.Korade Z., Kenworthy A. K., Mirnics K. 2009. Molecular consequences of altered neuronal cholesterol biosynthesis. J. Neurosci. Res. 87: 866–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liang X., Nazarian A., Erdjument-Bromage H., Bornmann W., Tempst P., Resh M. D. 2001. Heterogeneous fatty acylation of Src family kinases with polyunsaturated fatty acids regulates raft localization and signal transduction. J. Biol. Chem. 276: 30987–30994 [DOI] [PubMed] [Google Scholar]
- 39.Ekkebus R., van Kasteren S. I., Kulathu Y., Scholten A., Berlin I., Geurink P. P., de Jong A., Goerdayal S., Neefjes J., Heck A. J. R., et al. 2013. On terminal alkynes that can react with active-site cysteine nucleophiles in proteases. J. Am. Chem. Soc. 135: 2867–2870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cygan N. K., Scheinost J. C., Butters T. D., Wentworth P. 2011. Adduction of cholesterol 5,6-secosterol aldehyde to membrane-bound myelin basic protein exposes an immunodominant epitope. Biochemistry. 50: 2092–2100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nieva J., Shafton A., Altobell L. J., Tripuraneni S., Rogel J. K., Wentworth A. D., Lerner R. A., Wentworth P. 2008. Lipid-derived aldehydes accelerate light chain amyloid and amorphous aggregation. Biochemistry. 47: 7695–7705 [DOI] [PubMed] [Google Scholar]
- 42.Zhang Q., Powers E. T., Nieva J., Huff M. E., Dendle M. A., Bieschke J., Glabe C. G., Eschenmoser A., Wentworth P., Lerner R. A., et al. 2004. Metabolite-initiated protein misfolding may trigger Alzheimer's disease. Proc. Natl. Acad. Sci. USA. 101: 4752–4757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bosco D. A., Fowler D. M., Zhang Q., Nieva J., Powers E. T., Wentworth P., Jr, Lerner R. A., Kelly J. W. 2006. Elevated levels of oxidized cholesterol metabolites in Lewy body disease brains accelerate alpha-synuclein fibrilization. Nat. Chem. Biol. 2: 249–253 [DOI] [PubMed] [Google Scholar]
- 44.Korade Z., Xu L., Shelton R., Porter N. A. 2010. Biological activities of 7-dehydrocholesterol-derived oxysterols: implications for Smith-Lemli-Opitz syndrome. J. Lipid Res. 51: 3259–3269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu L., Mirnics K., Bowman A. B., Liu W., Da J., Porter N. A., Korade Z. 2012. DHCEO accumulation is a critical mediator of pathophysiology in a Smith-Lemli-Opitz syndrome model. Neurobiol. Dis. 45: 923–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Payré B., de Medina P., Boubekeur N., Mhamdi L., Bertrand-Michel J., Tercé F., Fourquaux I., Goudounèche D., Record M., Poirot M., et al. 2008. Microsomal antiestrogen-binding site ligands induce growth control and differentiation of human breast cancer cells through the modulation of cholesterol metabolism. Mol. Cancer Ther. 7: 3707–3718 [DOI] [PubMed] [Google Scholar]
- 47.de Sain-van der Velden M. G. M., Verrips A., Prinsen B. H. C. M. T., de Barse M., Berger R., Visser G. 2008. Elevated cholesterol precursors other than cholestanol can also be a hallmark for CTX. J. Inherit. Metab. Dis. 31(Suppl. 2): S387–S393 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.