

Abstract
Emerging evidence supports an inhibitory role for vitamin D in colorectal carcinogenesis, but the mechanism remains unclear. The APC/β-catenin pathway plays a critical role in colorectal carcinogenesis. The purpose is this study is to explore the interactions of vitamin D and APC/β-catenin pathways in intestinal tumor development. APCmin/+ mice with genetic inactivation of the vitamin D receptor (VDR) were generated through breeding. Intestinal tumorigenesis was compared between APCmin/+ and APCmin/+VDR−/− mice at different ages. No differences were seen in the number of small intestinal and colonic tumors between APCmin/+ and APCmin/+VDR−/− mice at 3, 4 and 6–7 months of age. The size of the tumors, however, was significantly increased in APCmin/+VDR−/− mice in all age groups. Immunostaining showed a significant increases in β-catenin, cyclin D1, phosphorylated Stat-3 and MSH-2 levels and decreases in Stat-1 in APCmin/+VDR−/− tumors compared to APCmin/+ tumors. These observations suggest that VDR signaling inhibits tumor growth rather than tumor initiation in the intestine. Thus, the increased tumor burden in APCmin/+VDR−/− mice is likely due to the loss of the growth-inhibiting effect of VDR. This study provides strong evidence for the in vivo relevance of the interaction demonstrated in vitro between the vitamin D and β-catenin signaling pathways in intestinal tumorigenesis.
Keywords: vitamin D receptor, beta-catenin, intestinal tumor, colon cancer, Apc(min/+)
Introduction
Colorectal cancer is the second leading cause of cancer-related deaths in men and women in the United States. The APC/β-catenin pathway plays a central role in colorectal carcinogenesis, as nearly all colon tumors have dysregulated β-catenin signaling, with either inactivating mutations in the adenomatous polyposis coli (APC) protein or activating mutations in β-catenin protein 1. Beta-catenin is a proto-oncogene that is predominately bound to E-cadherin in the adherence junctions. Tyrosine phosphorylation of β-catenin leads to its disassociation from E-cadherin and transfer to the cytosol, where it is either degraded by the 26S proteasome or translocated into the nucleus 1. In the nucleus β-catenin forms a complex with members of the TCF/LEF family of transcription factors and activates genes encoding c-Myc and cyclin D1, among others, to promote cell proliferation. In the cytosol, β-catenin degradation requires the involvement of GSK-3β, AXIN and APC 1. Mutations in the APC gene prevent APC binding to β-catenin and thereby stabilize β-catenin and increase its nuclear translocation, ultimately leading to tumorigenesis. APCmin/+ mice are the first reported genetic mouse model of intestinal tumorigenesis, which was originally derived from an Apc germ-line mutation induced by ethylnitrosourea treatment 2. Heterozygous (APCmin/+) mice develop multiple intestinal polyps after 3–4 months of age that are predominantly in the small intestine, as a result of spontaneous inactivation of the remaining wild-type Apc allele (loss of heterozygosity).
1,25-dihydroxyvitamin D3 (1,25(OH)2D3) is a secosteroid hormone whose actions are mostly mediated by VDR, a member of nuclear receptor superfamily. A large body of literature has suggested a suppressive role for vitamin D in colorectal cancer development. Epidemiological data, for example, showed an inverse relationship between sunlight exposure or vitamin D intake and human colon cancer prevalence 3, 4. Low circulating vitamin D levels are associated with increased polyp formation in the distal colon in women 5, and diets deficient in vitamin D increase hyperplasia and proliferation of colonic crypt cells 6. On the other hand, vitamin D supplementation alone or with calcium can inhibit experimental colonic carcinogenesis induced by high-fat diets or intrarectal instillation of lithocholic acid, a tumor-promoting bile acid 7, 8. Although vitamin D is known to inhibit colon cancer cell proliferation and induce colon caner cell apoptosis 9, 10, the mechanisms involved in vitamin D suppression of colonic carcinogenesis remain elusive.
Several studies have examined VDR haplotypes in an effort to identify risk alleles that could modulate the effects of vitamin D on colon cancer prevention. While some studies have reported an association of VDR polymorphisms and colonic cancer risk 11, 12, others have not 13, 14. Thus this remains a controversial area that needs more investigations.
Prior in vitro and in vivo studies have suggested a potentially important relationship between the vitamin D and APC/β-catenin signaling pathways. It has been reported that treatment with vitamin D or its synthetic analogs decreases tumor burden in APCmin/+ mice 15. In SW480 cells 1,25(OH)2D3 induces E-cadherin expression, promotes VDR-β-catenin interaction and prevents β-catenin nuclear translocation, leading to inhibition of TCF-4 responsive genes such as c-myc 16, a proto-oncogene required for tumor formation in APCmin/+ mice 17. The molecular basis underlying the protein-to-protein interaction between liganded VDR and β-catenin has also been established 18. These observations suggest that vitamin D may inhibit colon cancer cell proliferation by antagonizing the APC/β-catenin pathway. The relevance of this hypothesis, however, has not been tested in an in vivo setting. In the present study we compared tumorigenesis in VDR-null and wild-type APCmin/+ mice to study the relationship between the VDR and APC/β-catenin pathways in intestinal neoplastic transformation.
Materials and Methods
Animal studies
APCmin/+ mice on C57BL/6 background were purchased from Jackson Laboratory (Bar Harbor, Maine). VDR+/− and VDR−/− mice on C57BL/6 background have been reported previously 19. APCmin/+VDR−/− mice were produced through APCmin/+ × VDR+/− cross. Mouse genotyping was performed by genomic PCR. APCmin/+ and APCmin/+VDR−/− mice were fed standard rodent chow, and sacrificed at 3, 4, 6 and 7 months of age for analysis. We did not feed the mice the high calcium rescue diet 20, because dietary calcium is well known to affect intestinal carcinogenesis 21, 22 independent of vitamin D. Dietary calcium in the intestinal lumen could activate membrane calcium sensing receptors to directly inhibit β-catenin signaling 23. Two hours before sacrifice, mice were injected i.p. with 50 mg/kg BrdU to label proliferating crypt cells. After sacrifice, the entire intestine was dissected, cut longitudinally, placed onto a filter paper with luminal side facing up, and then fixed flat in 4% formaldehyde (made in PBS, pH 7.2). The number of visible polyps in the intestine was counted, and the size (the diameter) of each tumor was measured using an electronic digital calipers (Fisher Scientific). The Institutional Animal Care and Use Committee at the University of Chicago approved the animal protocol used in the study.
Histology and immunostaining
Paraffin embedded sections were cut at 4-μm with a Leica microtone 2030. Slides were deparaffinized, hydrated and stained with hematoxylin and eosin. For immunostaining, antigens were retrieved by 10–15 minute boiling in 10 mM citrate (pH 6.0). The slides were stained with primary antibodies, peroxidase-conjugated secondary antibodies, and visualized with a DAB peroxidase substrate kit (Vector Laboratories). Antibodies against β-catenin, E-cadherin, cyclin D1, p-Stat-3 and VDR were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), antibodies against BCL-2 and vimentin-1 from Dako (Carpinteria, CA), Stat-1 antibody from BD Biosciences (San Jose, CA), and MSH-2 antibody from Zymed (South San Francisco, CA). Immunostained slides were quantified using a quantitative computer assisted imaging system or Image J software with color deconvolution as described 24. Basically, we performed color deconvolution on digitized images of immunostained tumors, and examined at least 3 tumors in each group and quantified at least 5 fields that were representative of protein staining. Collectively at least 50,000 cells were quantified for each genotype and protein. Means and SEM were calculated based on these multiple measurements.
Western blot
Tissue samples were homogenized in Laemmli buffer, followed by 5 min boiling and centrifugation to obtain a soluble fraction. Protein concentrations were determined using a BioRad Protein Assay kit. Proteins were separated by SDS-PAGE and transferred onto Immobilon P membranes. Western blotting was carried out as previously described 25.
Co-immunoprecipitation (Co-IP) assays
SW480 cells (human colonic carcinoma cell line) and CCD-18Co (human colonic fibroblast cell line) were cultured in DMEM supplemented with 10% FBS. Cells at 80% confluence were treated with ethanol or 2×10−8 M 1,25(OH)2D3 for 4 hours. The cells were lysed with cold immunoprecipitation buffer (1% Triton X-100, 150 mM NaCl, 10 mM Tris -HCl, pH 7.4, 1 mM EDTA, 1 mM EGTA, pH 8.0, 0.2 mM sodium orthovanadate) containing protease inhibitor cocktail (Boehringer Mannheim), and the lysate was precipitated with anti-β-catenin or anti-VDR antibodies. The immunoprecipitated proteins were resolved by SDS-PAGE, and transferred to Immobilon-P membranes that were blotted with anti-VDR or anti-β-catenin antibodies. In some cases, the cells were fractionated into cytosolic and nuclear fractions for Co-IP experiments or Western blot analyses.
Real time RT-PCR
First-strand cDNAs were synthesized from total RNAs using MML-V reverse transcriptase (Invitrogen) and hexanucleotide random primers. Real time PCR was performed in an Applied Biosystems 7900 Real Time PCR System using aSYBR green PCR reagent kit (Applied Biosystems). The PCR primers used in this study are: p21waf1/cip1, 5′AGACCAGCCTGACAGATTTCT3′ (forward), and 5′ACACACAGAGTGAGGGCTAA3′ (reverse); p27kip1, 5′ACCTGCTGCAGAAGATT-CTTCT3′ (forward), and 5′CAGATGGGGTGTCAGTTTTGT3′ (reverse); Snail-1, 5′AGTCGCGGAAGATCTTCAACT3′ (forward), and 5′AGAATGGCTTCTCACCAGTGT3′ (reverse).
Statistical analysis
Data are expressed as means ± SEM. Statistical comparisons between groups were made using two-tailed unpaired Student’s t-test, with P< 0.05 being considered significant.
Results
To assess the relationship between the vitamin D and β-catenin pathways we first examined the co-association of VDR and β-catenin proteins in SW480 and CCD-18Co cells. Consistent with a previous report 16, co-immunoprecipitation assays using SW480 cells demonstrated that the VDR protein in the presence of 1,25(OH)2D3 was precipitated by anti-β-catenin antibody, whereas unliganded VDR did not interact with β-catenin (Fig. 1A). A similar result was seen in CCD-18Co cells, but in these cells, β-catenin was precipitated by antibody against VDR, and the association of VDR and β-catenin was markedly increased in the presence of 1,25(OH)2D3 (Fig. 1B). Increased VDR and β-catenin association was observed in the nuclear fraction compared to the cytosolic fraction after 1,25(OH)2D3 treatment (Fig. 1C), which may reflect VDR nuclear translocation upon 1,25(OH)2D3 stimulation. These data suggest that 1,25(OH)2D3 may interfere with the APC/β-catenin signaling pathway by promoting the physical interaction of VDR and β-catenin.
Figure 1.
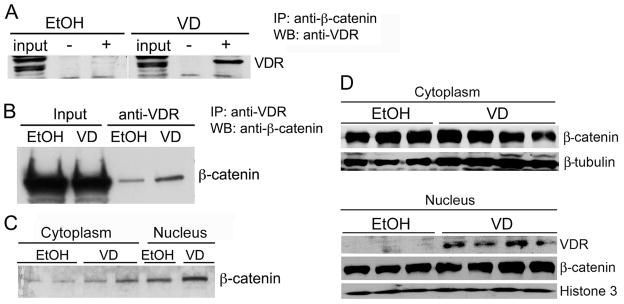
Interaction of liganded VDR with β-catenin. (A) Co-IP assay in SW480 cells. Cells were treated with ethanol (EtOH) or 2×10−8 M 1,25(OH)2D3 (VD) for 4 hours and cell lysates were immunoprecipitated (IP) with (+) or without (−) antibody against β-catenin. The precipitated proteins were analyzed by Western blot (WB) with anti-VDR antibody. (B) Co-IP assays in CCD-18Co cells. Cells were treated with EtOH or 2×10−8 M 1,25(OH)2D3. Total cell lysates were precipitated with anti-VDR antibody, following by WB analysis with anti-β-catenin antibody. (C) Co-IP of cytoplasmic and nuclear extracts from CCD-18Co cells. Cytoplasmic and nuclear extracts were prepared from EtOH- or VD-treated CCD-18Co cells, and analyzed by Co-IP assays as in (B). (D) Effect of 1,25(OH)2D3 treatment on β-catenin nuclear translocation. CCD-18Co cells were treated with EtOH or 1,25(OH)2D3 for 24 hours, and the cytoplasmic and nuclear extracts were prepared for Western blot analyses with antibodies against β-catenin, VDR, β-tubulin or histone-3 as indicated. Upper panel: Cytoplasmic fractions; Lower panel: Nuclear fractions. Each lane represents an independent sample.
We further investigated whether 1,25(OH)2D3 treatment altered β-catenin nuclear translocation in CCD-18Co cells. As shown in Figure 1D, β-catenin was abundant in both the cytoplasmic and nuclear compartments, and 1,25(OH)2D3 treatment did not appear to change the distribution of β-catenin in cytoplasm and nucleus (Fig. 1D); however, 1,25(OH)2D3 promoted VDR nuclear translocation as expected (Fig. 1D). This result suggests that VDR-β-catenin interaction does not block β-catenin nuclear translocation, but may inhibit the trans-activating activity of β-catenin in the nucleus.
To explore the in vivo relevance of the interaction between the VDR and APC/β-catenin pathways, we set out to compare intestinal tumor development in APCmin/+ mice with or without a functional VDR. We generated APCmin/+VDR−/− mice through breeding, and these double mutant mice developed normally and were viable for our studies. Although mice with the VDR−/− allele were known to develop hypocalcemia that can be corrected by a high calcium-containing diet 20, we did not use the high calcium diet in order to avoid the confounding effect of calcium, because calcium is well known to suppress colonic tumorigenesis 21, 22. APCmin/+ and APCmin/+VDR−/− mice were studied in parallel, and the number and size of intestinal polyps were assessed at 3, 4, and 6–7 months of age.
Consistent with previously published observations 2, APCmin/+ mice developed intestinal adenomatous polyps beginning at about 3 months of age. The number and size of the polyps peaked at 6–7 months of age, which was often followed by rectal bleeding and mortality in the next few weeks. APCmin/+VDR−/− double mutant mice showed a similar tumor phenotype, with the majority of tumors seen in the small intestine. As shown in Table 1, the total number of tumors in the intestine was approximately the same in APCmin/+ and APCmin/+VDR−/− mice at all ages. Interestingly, however, APCmin/+VDR−/− mice developed significantly (p<0.05) more large tumors at 4 months of age compared to APCmin/+ mice. At this time point, APCmin/+VDR−/− mice had 6.4±2.4 tumors in 3–4 mm size range and 4.2±1.3 tumors larger than 4 mm, whereas APCmin/+ mice had only 0.7±0.5 in the 3–4 mm size range and none >4 mm (Table 1). At 6–7 months, tumor sizes were no longer significantly different between APCmin/+ and APCmin/+VDR−/− mice (p=0.10), most likely reflecting the fact that most of these tumors had already achieved their maximal size. These data suggest that VDR has little effect on tumor initiation but inhibits tumor growth.
Table 1.
Intestinal tumor number (per mouse) and size in APCmin/+ and APCmin/+VDR−/− mice
| APCmin/+ | APCmin/+VDR−/− | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Tumor size | 3 months n=3 |
4 months n=8 |
6–7 months n=9 |
3 months n=3 |
4 months n=7 |
6–7 months n=7 |
| <2 mm | 4.3±1.5 | 9.2±1.8 | 5.1±1.8 | 2.7±1.5 | 3.1±1.8 | 4.6±1.8 |
| 2–3 mm | 0.7±1.2 | 4.7±3.5 | 11.1±2.3 | 0.3±0.5 | 8.5±1.6 | 11.3±2.3 |
| 3–4 mm | 0 | 0.7±0.5 | 7.9±1.9 | 0.3±0.5 | 6.4±2.4* | 7.8±1.8 |
| >4 mm | 0 | 0 | 3.1±0.9 | 0 | 4.2±1.3* | 5.0±1.0# |
| Total number | 5.0±1.0 | 13.6±3.2 | 33.6±3.2 | 3.3±1.5 | 17.7±4.2 | 34.6±4.3 |
Note: Data are presented as mean ± SEM.
P<0.05;
P=0.10 vs. the corresponding APCmin/+ value.
Figure 2 shows representative H&E sections of adenomas from the small intestine (Fig. 2A and D) and colon (Fig. 2C) and colonic aberrant crypt foci (ACF) (Fig. 2B) in APCmin/+ and APCmin/+VDR−/− mice. Histologically the tumors from APCmin/+ and APCmin/+VDR−/− mice were similar and most were adenomas.
Figure 2.

H&E staining of representative intestinal tumors. (A) Small intestinal adenoma from 3-month old APCmin/+ mouse (Magnification: 50x); (B) Colonic aberrant crypt focus (ACF) from 3-month old APCmin/+VDR−/− mouse (50x); (C) Colonic adenoma from 4-month old APCmin/+VDR−/− mouse (100x). (D) Small intestinal adenoma from 4-month old APCmin/+VDR−/− mouse (50x).
The tumors from APCmin/+ and APCmin/+VDR−/− mice were examined for VDR and β-catenin expression and proliferation by immunostaining. As expected and in contrast to APCmin/+ mice, no VDR staining was seen in APCmin/+VDR−/− tumors (Fig. 3A, a and b). Both APCmin/+ and APCmin/+VDR−/− tumors showed strong staining for β-catenin (Fig. 3A, c and d) and BrdU (Fig. 3A, e and f). Interestingly, β-catenin staining appeared more intense in both nuclei and cytosol in APCmin/+VDR−/− tumors than in APCmin/+ tumors (see insets in Fig. 3A, c and d for higher magnification). Western blot analyses of tissue lysates confirmed that β-catenin expression was higher in tumors from APCmin/+VDR−/− mice than from APCmin/+ mice (Fig. 3B). No clear difference was detected in BrdU labeling of the tumors from these two mouse lines (Fig. 3A, e and f).
Figure 3.

Levels of VDR, β-catenin, cyclin D1 and phospho-Stat-3 in the tumors. (A) VDR, β-catenin and BrdU immunostaining. Tumors from 4-month old APCmin/+ (a, c and e) and APCmin/+VDR−/− (b, d and f) mice were immunostained with antibodies against VDR (a and b), β-catenin (c and d) and BrdU (e and f). Magnification 200x. The insets in panels c and d are in higher magnification (400x). (B) Western blot analysis for β-catenin. Cell lysates from tumors and neighboring normal intestinal tissues from APCmin/+ and APCmin/+VDR−/− mice were subject to Western blotting analysis with anti-β-catenin antibody. Fold-increase of β-catenin for each group is presented below the blot. Each lane represents one mouse. (C) Cyclin D1 and phospho-Stat-3 immunostaining analysis. Tumors from 4-month old APCmin/+ (a and c) or APCmin/+VDR−/− (b and d) mice were immunostained with antibodies against cyclin D1 (a and b) or p-Stat-3 (c and d). Magnification, 200x. (D) Quantitative data from immunostained slides shown in (C) for relative levels of cyclin D1 and p-Stat-3, using computer assisted imaging system. * P<0.05, ** P<0.01 vs. APCmin/+.
We next determined the levels of cyclin D1 and phosphorylated (p) Stat-3 in these tumors by immunostaining. Cyclin D1, a cell cycle regulator, and Stat-3, a transcription factor, are known to be elevated or activated in colon cancer to promote colonic tumorigenesis 26, 27. As shown in Figure 3C, APCmin/+VDR−/− tumors (Fig 3C, b and d) showed markedly higher levels of cyclin D1 and p-Stat-3 compared to APCmin/+ tumors (Fig. 3C, a and c). Semi-quantitative data showed a 1.8- and 8.4-fold increase in cyclin D1- and p-Stat-3-positive cells, respectively, in APCmin/+VDR−/− tumors (Fig. 3D). All these data are consistent with the larger tumor size seen in the double mutant mice.
We further examined tumors for several other proto-oncogenes and tumor suppressor genes, including BCL-2, Stat-1, MSH-2 and vimentin-1 (Fig. 4). Both APCmin/+ and APCmin/+VDR−/− tumors showed similar low levels of BCL-2 staining (Fig. 4A and B). Stat-1, a transcription factor activated in response to external stimuli, showed decreased staining in APCmin/+VDR−/− tumors compared to APCmin/+ tumors (Fig. 4C, D and I). On the other hand, APCmin/+VDR−/− tumors exhibited a marked increase in the staining of MSH-2, a DNA mismatch repair protein (Fig. 4E and F), and a moderate increase in the staining for vimentin-1, a marker for mesenchymal cells (Fig. 4G and H), compared to APCmin/+ tumors. By semi-quantitative analysis we determined that Stat-1 staining was about 40% decreased, nuclear MSH-2 in epithelial cells was approximately 9-fold increased, and vimentin-1 staining, seen in both stromal and epithelial cells, was about 2.5-fold higher in APCmin/+VDR−/− tumors (Fig. 4I).
Figure 4.

Levels of BCL-2, Stat-1, MSH-2 and vimentin in tumors. Tumors from 4-month old APCmin/+ (A, C, E and G) and APCmin/+VDR−/− (B, D, F and H) mice were immunostained with antibodies against BCL-2 (A and B), Stat-1 (C and D), MSH-2 (E and F), and vimentin-1 (G and H). Magnification 200x. (I) Quantitative results of Stat-1, vimentin-1 and MSH-2 immunostaining using the Image J software. * P<0.05; ** P<0.01 vs. APCmin/+. Note the decreased Stat-1 and increased MSH-2 and vimentin-1 staining in APCmin/+VDR−/− tumors compared to APCmin/+ tumors.
We also compared mRNA transcript levels of p21waf1/cip1, p27kip1 and Snail-1 in these two types of tumors by real time RT-PCR quantification. The mRNA levels of these three genes were lower in APCmin/+VDR−/− tumors compared to APCmin/+ tumors, and p21waf1/cip1 and p27kip1 reduction reached statistical significance (Fig. 5A–C).
Figure 5.
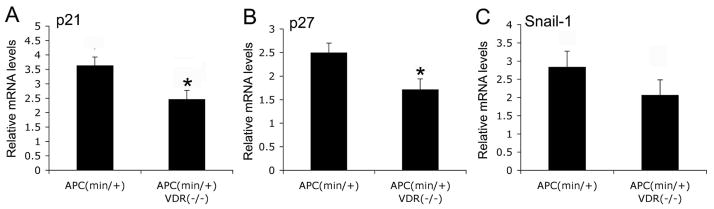
Quantification of p21, p27 and snail-1 mRNA levels. Total RNAs were extracted from intestinal tumors from APCmin/+ and APCmin/+VDR−/− mice at 4-month of age, and levels of p21waf1/cip1 (A), p27kip1 (B) and snail-1 (C) were quantified by real time RT-PCR. *, P ≤0.05 vs. APCmin/+.
Discussion
A large body of literature has shown that vitamin D and its analogues have potent anti-proliferative activity against tumor cells of different origins, including colon, breast, prostate and the hematopoietic system. Several mechanisms, including suppression of cell cycle progression and induction of apoptosis, have been proposed to explain vitamin D inhibition of tumor cell proliferation 9, 28. It was previously showed that a vitamin D analog significantly inhibited aberrant crypt foci (ACF) formation and colorectal tumor initiation and progression in azoxymethane-treated animals 29. In mice lacking the VDR, increased crypt cell proliferation and oxidative DNA damage was reported in colonic epithelia 30, suggesting that VDR knockout mice are predisposed to colon tumors.
In the present study we used a genetic approach to explore the role of the VDR in intestinal tumorigenesis. We chose the well-established APCmin/+ model based on the interaction between the VDR and APC/β-catenin signaling pathways observed in vitro. Our data showed that APCmin/+ mice lacking VDR developed increased tumor burden compared to APCmin/+ mice with wild-type VDR, consistent with the anti-cancer property of the VDR. In fact, the number of intestinal tumors was not altered in APCmin/+VDR−/− mice, indicating that VDR inactivation does not have a significant impact on tumor initiation in the APCmin/+ background. The difference seen in APCmin/+VDR−/− mice is the significant increase in tumor size at the time (4 months of age) when tumor growth is in an exponential phase. At a later stage of tumor growth (6–7 month of age), the difference in tumor burden between APCmin/+ and APCmin/+VDR−/− mice was no longer apparent, probably because the tumor size had reached a plateau. These data suggest that the VDR inhibits tumor growth rather than tumor initiation. These observations are in agreement with an earlier report that treatment with a vitamin D analog decreased tumor size, but not tumor numbers in APCmin/+ mice 15. Together, the data from the genetic models suggest that in vivo vitamin D and vitamin D analogs are more likely to inhibit tumor growth rather than to suppress tumor initiation in colorectal carcinogenesis.
The physical interaction seen in vitro between liganded VDR and β-catenin suggests that VDR binding might prevent β-catenin translocation to the nucleus, or block the transactivating activity of β-catenin in the nucleus. Our data do not support the inhibition of β-catenin nuclear translocation, but show an increased interaction between liganded VDR and β-catenin in the nucleus. This is not surprising, as 1,25(OH)2D3 induces VDR nuclear translocation (Figures 1). While blocking β-catenin activity, VDR-β-catenin interaction may also increase the degradation of β-catenin by 26S proteasome. The latter predicts an increased β-catenin level in VDR-negative cells, consistent with our immunostaining and Western blotting data that revealed higher levels of β-catenin in APCmin/+VDR−/− tumors than in APCmin/+ tumors (Figure 3A). These data suggest that the increased activation of the β-catenin pathway in the absence of the VDR is the basis for the increased tumor growth seen in APCmin/+VDR−/− mice. Consistent with this conjecture, recent studies have also shown that β-catenin activity increases with adenoma growth 31.
Immunostaining analysis revealed varying degrees of increases in the expression of cyclin D1, p-Stat-3, MSH2 and vimentin-1, and decreases in Stat-1 in tumors from APCmin/+VDR−/− mice. Given the heterogeneity of tumors and intrinsic semi-quantitative nature of immunostaining, the observed difference in staining signals, however, may be inaccurate or potentially an artifact of the method. Nevertheless, as discussed below, the differences in immunostaining that we observed between the two genotypes are consistent with the increased size in APCmin/+VDR−/− tumors.
Cyclin D1 is a key cell cycle regulator promoting G1 progression and is increased in human and experimental colonic tumors 27, 32. Cyclin D1 is a direct target of APC/β-catenin signals and has been shown to regulate APCmin/+ tumor growth, although its role in more advanced human colon cancer is less clear 33.
Stat-3 is activated in colon cancer and associated with enhanced cell proliferation and tumor growth 26. Stat-3 can up-regulate cyclin D1 and c-Myc, both of which are known to control proliferation. Phospho-Stat-3 was identified as a prognostic factor for invasive human colonic tumors, although in APCmin/+ mice Stat-3 may control tumor initiation rather than tumor progression 34, 35. Loss of VDR, however, may have uncovered an important role for Stat-3 in tumor growth. While Stat-3 has oncogenic properties, Stat-1 is known to induce pro-apoptotic and anti-proliferative effects and to mediate tumor suppressor signals in colon cancer cells 36. Thus, lower Stat-1 levels in APCmin/+VDR−/− tumors are consistent with increased growth of these tumors. In agreement with these anti-tumor effects, Stat-1 was not required for APCmin/+ adenoma formation 37.
MSH2, a subunit of DNA mismatch repair complex, is frequently mutated in hereditary non-polyposis colorectal cancer syndrome (Lynch Syndrome) 38. The implication of higher MSH2 expression in the VDR-null tumors is unclear, but may reflect a secondary response to increased oxidative DNA damage in the intestine of VDR-null mice that was previously reported 39. Alternatively, MSH2 up-regulation might reflect increased colonocyte cell cycling since E2F transcription factors regulate MSH2 expression 40.
Vimentin-1, a marker of epithelial-to-mesenchymal transition, has been reported to be increased in intestinal adenomas in APCmin/+ mice 41. Vimentin-1 appears to be further up-regulated in APCmin/+VDR−/− adenoma. Whereas positive staining appears mainly in stromal cells in the APCmin/+ tumor, both the epithelial and stromal cells show stronger vimentin-1 staining in the APCmin/+VDR−/− adenoma. As epithelial-to-mesenchymal transition is recognized as a key step in tumor progression and metastasis and to be closely related to cancer stem cells 42, the seemingly increased vimentin-1 staining in the APCmin/+VDR−/− tumors might reflect a phenotype of enhanced tumor progression and/or more “stemness” of the tumor cells. The roles of vimentin-1 in stromal versus epithelial cells, however, remain uncertain and will require further investigations.
Vitamin D and its analogs have been shown to suppress the expression of β-catenin targets cyclin D1, c-Myc and COX-2 10, 43 and induces p21waf1/cip1 and p27kip1 28, 44, inhibitors of cell cycle progression. These growth-inhibiting alterations are thought to be involved in vitamin D inhibition of carcinogenesis. Furthermore, loss of p21waf1/cip1 and p27kip1 was shown to increase APCmin/+ tumorigenesis 45, 46. Thus, increased tumor size is consistent with lower p21waf1/cip1 and p27kip1 expression in APCmin/+VDR−/− tumors. Stat-1 has been implicated in stabilizing p27kip1 and the observed lower Stat-1 in APCmin/+VDR−/− adenomas might contribute to p27kip1 reduction in these tumors. Taken together, increased cyclin D1 and reduced cyclin dependent kinase inhibitors p21waf1/cip1 and p27kip1 are expected to accelerate the cell cycle and enhance proliferation in agreement with larger APCmin/+VDR−/− tumors.
While the transcription factor Snail-1 is known to suppress VDR expression in colon cancer cells 47, potential VDR regulation of Snail-1 has not been examined. We observed that Snail-1 was reduced but not significantly in APCmin/+VDR−/− adenomas. A larger study is required to further examine whether this trend in decreased Snail-1 expression in APCmin/+VDR−/− tumors is significant.
In summary, the genetic approach that we employed in the present study reveals a suppressive role for the VDR in intestinal tumor growth rather than an effect on tumor initiation in the APCmin/+ mouse model. The possible molecular basis for this suppression involves in part blockade of the β-catenin signaling pathway (via inhibition of β-catenin activity and promotion of its degradation) in colonic epithelial cells through liganded VDR-β-catenin interaction. This finding advances our understanding of the molecular mechanisms underlying the chemopreventive actions of vitamin D against colon cancer. Development of vitamin D analogs that induce preferential binding of VDR to β-catenin might provide a useful strategy for colon cancer chemoprevention.
Acknowledgments
This work was supported in part by National Institutes of Health grants T32DK07074 and R03CA117472, and National Natural Science Foundation of China grant No. 20972046.
Abbreviations
- VDR
vitamin D receptor
- 1,25(OH)2D3
1,25-dihydroxyvitamin D3
- APC
adenomatous polyposis coli
References
- 1.Morin PJ. beta-catenin signaling and cancer. Bioessays. 1999;21:1021–30. doi: 10.1002/(SICI)1521-1878(199912)22:1<1021::AID-BIES6>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 2.Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, Gould KA, Dove WF. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science. 1992;256:668–70. doi: 10.1126/science.1350108. [DOI] [PubMed] [Google Scholar]
- 3.Newmark HL, Lipkin M. Calcium, vitamin D, and colon cancer. Cancer Res. 1992;52:2067s–70s. [PubMed] [Google Scholar]
- 4.Garland CF, Comstock GW, Garland FC, Helsing KJ, Shaw EK, Gorham ED. Serum 25-hydroxyvitamin D and colon cancer: eight-year prospective study. Lancet. 1989;2:1176–8. doi: 10.1016/s0140-6736(89)91789-3. [DOI] [PubMed] [Google Scholar]
- 5.Platz EA, Hankinson SE, Hollis BW, Colditz GA, Hunter DJ, Speizer FE, Giovannucci E. Plasma 1,25-dihydroxy- and 25-hydroxyvitamin D and adenomatous polyps of the distal colorectum. Cancer Epidemiol Biomarkers Prev. 2000;9:1059–65. [PubMed] [Google Scholar]
- 6.Sadava D, Remer T, Petersen K. Hyperplasia, hyperproliferation and decreased migration rate of colonic epithelial cells in mice fed a diet deficient in vitamin D. Biol Cell. 1996;87:113–5. [PubMed] [Google Scholar]
- 7.Kawaura A, Tanida N, Sawada K, Oda M, Shimoyama T. Supplemental administration of 1 alpha-hydroxyvitamin D3 inhibits promotion by intrarectal instillation of lithocholic acid in N-methyl-N-nitrosourea-induced colonic tumorigenesis in rats. Carcinogenesis. 1989;10:647–9. doi: 10.1093/carcin/10.4.647. [DOI] [PubMed] [Google Scholar]
- 8.Yang K, Lamprecht SA, Shinozaki H, Fan K, Yang W, Newmark HL, Kopelovich L, Edelmann W, Jin B, Gravaghi C, Augenlicht L, Kucherlapati R, et al. Dietary calcium and cholecalciferol modulate cyclin D1 expression, apoptosis, and tumorigenesis in intestine of adenomatous polyposis coli1638N/+ mice. J Nutr. 2008;138:1658–63. doi: 10.1093/jn/138.9.1658. [DOI] [PubMed] [Google Scholar]
- 9.Diaz GD, Paraskeva C, Thomas MG, Binderup L, Hague A. Apoptosis is induced by the active metabolite of vitamin D3 and its analogue EB1089 in colorectal adenoma and carcinoma cells: possible implications for prevention and therapy. Cancer Res. 2000;60:2304–12. [PubMed] [Google Scholar]
- 10.Kumagai T, O’Kelly J, Said JW, Koeffler HP. Vitamin D2 analog 19-nor-1,25-dihydroxyvitamin D2: antitumor activity against leukemia, myeloma, and colon cancer cells. J Natl Cancer Inst. 2003;95:896–905. doi: 10.1093/jnci/95.12.896. [DOI] [PubMed] [Google Scholar]
- 11.Ochs-Balcom HM, Cicek MS, Thompson CL, Tucker TC, Elston RC, SJP, Casey G, Li L. Association of vitamin D receptor gene variants, adiposity and colon cancer. Carcinogenesis. 2008;29:1788–93. doi: 10.1093/carcin/bgn166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sweeney C, Curtin K, Murtaugh MA, Caan BJ, Potter JD, Slattery ML. Haplotype analysis of common vitamin D receptor variants and colon and rectal cancers. Cancer Epidemiol Biomarkers Prev. 2006;15:744–9. doi: 10.1158/1055-9965.EPI-05-0814. [DOI] [PubMed] [Google Scholar]
- 13.Egan JB, Thompson PA, Ashbeck EL, Conti DV, Duggan D, Hibler E, Jurutka PW, Leroy EC, Martinez ME, Mount D, Jacobs ET. Genetic polymorphisms in vitamin D receptor VDR/RXRA influence the likelihood of colon adenoma recurrence. Cancer Res. 2010;70:1496–504. doi: 10.1158/0008-5472.CAN-09-3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poynter JN, Jacobs ET, Figueiredo JC, Lee WH, Conti DV, Campbell PT, Levine AJ, Limburg P, Le Marchand L, Cotterchio M, Newcomb PA, Potter JD, et al. Genetic variation in the vitamin D receptor (VDR) and the vitamin D-binding protein (GC) and risk for colorectal cancer: results from the Colon Cancer Family Registry. Cancer Epidemiol Biomarkers Prev. 2010;19:525–36. doi: 10.1158/1055-9965.EPI-09-0662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huerta S, Irwin RW, Heber D, Go VL, Koeffler HP, Uskokovic MR, Harris DM. 1alpha,25-(OH)(2)-D(3) and its synthetic analogue decrease tumor load in the Apc(min) Mouse. Cancer Res. 2002;62:741–6. [PubMed] [Google Scholar]
- 16.Palmer HG, Gonzalez-Sancho JM, Espada J, Berciano MT, Puig I, Baulida J, Quintanilla M, Cano A, de Herreros AG, Lafarga M, Munoz A. Vitamin D(3) promotes the differentiation of colon carcinoma cells by the induction of E-cadherin and the inhibition of beta-catenin signaling. J Cell Biol. 2001;154:369–87. doi: 10.1083/jcb.200102028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sansom OJ, Meniel VS, Muncan V, Phesse TJ, Wilkins JA, Reed KR, Vass JK, Athineos D, Clevers H, Clarke AR. Myc deletion rescues Apc deficiency in the small intestine. Nature. 2007;446:676–9. doi: 10.1038/nature05674. [DOI] [PubMed] [Google Scholar]
- 18.Shah S, Islam MN, Dakshanamurthy S, Rizvi I, Rao M, Herrell R, Zinser G, Valrance M, Aranda A, Moras D, Norman A, Welsh J, et al. The molecular basis of vitamin D receptor and beta-catenin crossregulation. Mol Cell. 2006;21:799–809. doi: 10.1016/j.molcel.2006.01.037. [DOI] [PubMed] [Google Scholar]
- 19.Li YC, Pirro AE, Amling M, Delling G, Baron R, Bronson R, Demay MB. Targeted ablation of the vitamin D receptor: an animal model of vitamin D-dependent rickets type II with alopecia. Proc Natl Acad Sci U S A. 1997;94:9831–5. doi: 10.1073/pnas.94.18.9831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li YC, Amling M, Pirro AE, Priemel M, Meuse J, Baron R, Delling G, Demay MB. Normalization of mineral ion homeostasis by dietary means prevents hyperparathyroidism, rickets, and osteomalacia, but not alopecia in vitamin D receptor-ablated mice. Endocrinology. 1998;139:4391–6. doi: 10.1210/endo.139.10.6262. [DOI] [PubMed] [Google Scholar]
- 21.Sitrin MD, Halline AG, Abrahams C, Brasitus TA. Dietary calcium and vitamin D modulate 1,2-dimethylhydrazine-induced colonic carcinogenesis in the rat. Cancer Res. 1991;51:5608–13. [PubMed] [Google Scholar]
- 22.Pence BC, Dunn DM, Zhao C, Landers M, Wargovich MJ. Chemopreventive effects of calcium but not aspirin supplementation in cholic acid-promoted colon carcinogenesis: correlation with intermediate endpoints. Carcinogenesis. 1995;16:757–65. doi: 10.1093/carcin/16.4.757. [DOI] [PubMed] [Google Scholar]
- 23.Chakrabarty S, Radjendirane V, Appelman H, Varani J. Extracellular calcium and calcium sensing receptor function in human colon carcinomas: promotion of E-cadherin expression and suppression of beta-catenin/TCF activation. Cancer Res. 2003;63:67–71. [PubMed] [Google Scholar]
- 24.Ruifrok AC, Johnston DA. Quantification of histochemical staining by color deconvolution. Analytical and quantitative cytology and histology/the International Academy of Cytology [and] American Society of Cytology. 2001;23:291–9. [PubMed] [Google Scholar]
- 25.Li YC, Bolt MJG, Cao L-P, Sitrin MD. Effects of vitamin D receptor inactivation on the expression of calbindins and calcium metabolism. Am J Physiol Endocrinol Metab. 2001;281:E558–E64. doi: 10.1152/ajpendo.2001.281.3.E558. [DOI] [PubMed] [Google Scholar]
- 26.Corvinus FM, Orth C, Moriggl R, Tsareva SA, Wagner S, Pfitzner EB, Baus D, Kaufmann R, Huber LA, Zatloukal K, Beug H, Ohlschlager P, et al. Persistent STAT3 activation in colon cancer is associated with enhanced cell proliferation and tumor growth. Neoplasia. 2005;7:545–55. doi: 10.1593/neo.04571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hulit J, Wang C, Li Z, Albanese C, Rao M, Di Vizio D, Shah S, Byers SW, Mahmood R, Augenlicht LH, Russell R, Pestell RG. Cyclin D1 genetic heterozygosity regulates colonic epithelial cell differentiation and tumor number in ApcMin mice. Molecular and cellular biology. 2004;24:7598–611. doi: 10.1128/MCB.24.17.7598-7611.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park WH, Seol JG, Kim ES, Jung CW, Lee CC, Binderup L, Koeffler HP, Kim BK, Lee YY. Cell cycle arrest induced by the vitamin D(3) analog EB1089 in NCI-H929 myeloma cells is associated with induction of the cyclin-dependent kinase inhibitor p27. Exp Cell Res. 2000;254:279–86. doi: 10.1006/excr.1999.4735. [DOI] [PubMed] [Google Scholar]
- 29.Wali RK, Bissonnette M, Khare S, Hart J, Sitrin MD, Brasitus TA. 1 alpha,25-Dihydroxy-16-ene-23-yne-26,27-hexafluorocholecalciferol, a noncalcemic analogue of 1 alpha,25-dihydroxyvitamin D3, inhibits azoxymethane-induced colonic tumorigenesis. Cancer Res. 1995;55:3050–4. [PubMed] [Google Scholar]
- 30.Kallay E, Pietschmann P, Toyokuni S, Bajna E, Hahn P, Mazzucco K, Bieglmayer C, Kato S, Cross HS. Characterization of a vitamin D receptor knockout mouse as a model of colorectal hyperproliferation and DNA damage. Carcinogenesis. 2001;22:1429–35. doi: 10.1093/carcin/22.9.1429. [DOI] [PubMed] [Google Scholar]
- 31.Oyama T, Yamada Y, Hata K, Tomita H, Hirata A, Sheng H, Hara A, Aoki H, Kunisada T, Yamashita S, Mori H. Further upregulation of beta-catenin/Tcf transcription is involved in the development of macroscopic tumors in the colon of ApcMin/+ mice. Carcinogenesis. 2008;29:666–72. doi: 10.1093/carcin/bgn001. [DOI] [PubMed] [Google Scholar]
- 32.Bissonnette M, Khare S, von Lintig FC, Wali RK, Nguyen L, Zhang Y, Hart J, Skarosi S, Varki N, Boss GR, Brasitus TA. Mutational and nonmutational activation of p21ras in rat colonic azoxymethane-induced tumors: effects on mitogen-activated protein kinase, cyclooxygenase-2, and cyclin D1. Cancer Res. 2000;60:4602–9. [PubMed] [Google Scholar]
- 33.Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–6. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 34.Kusaba T, Nakayama T, Yamazumi K, Yakata Y, Yoshizaki A, Nagayasu T, Sekine I. Expression of p-STAT3 in human colorectal adenocarcinoma and adenoma; correlation with clinicopathological factors. Journal of clinical pathology. 2005;58:833–8. doi: 10.1136/jcp.2004.023416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Musteanu M, Blaas L, Mair M, Schlederer M, Bilban M, Tauber S, Esterbauer H, Mueller M, Casanova E, Kenner L, Poli V, Eferl R. Stat3 is a negative regulator of intestinal tumor progression in Apc(Min) mice. Gastroenterology. 2010;138:1003–11. e1–5. doi: 10.1053/j.gastro.2009.11.049. [DOI] [PubMed] [Google Scholar]
- 36.Stephanou A, Latchman DS. Opposing actions of STAT-1 and STAT-3. Growth Factors. 2005;23:177–82. doi: 10.1080/08977190500178745. [DOI] [PubMed] [Google Scholar]
- 37.Liddle FJ, Frank DA. STAT1 expression is not required for polyp formation in Min mice. Molecular carcinogenesis. 2008;47:75–9. doi: 10.1002/mc.20371. [DOI] [PubMed] [Google Scholar]
- 38.Peltomaki P. Lynch syndrome genes. Fam Cancer. 2005;4:227–32. doi: 10.1007/s10689-004-7993-0. [DOI] [PubMed] [Google Scholar]
- 39.Kallay E, Bareis P, Bajna E, Kriwanek S, Bonner E, Toyokuni S, Cross HS. Vitamin D receptor activity and prevention of colonic hyperproliferation and oxidative stress. Food Chem Toxicol. 2002;40:1191–6. doi: 10.1016/s0278-6915(02)00030-3. [DOI] [PubMed] [Google Scholar]
- 40.Polager S, Kalma Y, Berkovich E, Ginsberg D. E2Fs up-regulate expression of genes involved in DNA replication, DNA repair and mitosis. Oncogene. 2002;21:437–46. doi: 10.1038/sj.onc.1205102. [DOI] [PubMed] [Google Scholar]
- 41.Chen X, Halberg RB, Burch RP, Dove WF. Intestinal adenomagenesis involves core molecular signatures of the epithelial-mesenchymal transition. J Mol Histol. 2008;39:283–94. doi: 10.1007/s10735-008-9164-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741–51. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jensen SS, Madsen MW, Lukas J, Binderup L, Bartek J. Inhibitory effects of 1alpha,25-dihydroxyvitamin D(3) on the G(1)-S phase-controlling machinery. Mol Endocrinol. 2001;15:1370–80. doi: 10.1210/mend.15.8.0673. [DOI] [PubMed] [Google Scholar]
- 44.Liu M, Lee M-H, Cohen M, Bommakanti M, Freedman LP. Transcriptional activation of the Cdk inhibitor p21 by vitamin D3 leads to the induced differentiation of the myelomonocytic cell line U937. Genes Dev. 1996;10:142–53. doi: 10.1101/gad.10.2.142. [DOI] [PubMed] [Google Scholar]
- 45.Philipp-Staheli J, Kim KH, Payne SR, Gurley KE, Liggitt D, Longton G, Kemp CJ. Pathway-specific tumor suppression. Reduction of p27 accelerates gastrointestinal tumorigenesis in Apc mutant mice, but not in Smad3 mutant mice. Cancer Cell. 2002;1:355–68. doi: 10.1016/s1535-6108(02)00054-5. [DOI] [PubMed] [Google Scholar]
- 46.Yang WC, Mathew J, Velcich A, Edelmann W, Kucherlapati R, Lipkin M, Yang K, Augenlicht LH. Targeted inactivation of the p21(WAF1/cip1) gene enhances Apc-initiated tumor formation and the tumor-promoting activity of a Western-style high-risk diet by altering cell maturation in the intestinal mucosal. Cancer Res. 2001;61:565–9. [PubMed] [Google Scholar]
- 47.Palmer HG, Larriba MJ, Garcia JM, Ordonez-Moran P, Pena C, Peiro S, Puig I, Rodriguez R, de la Fuente R, Bernad A, Pollan M, Bonilla F, et al. The transcription factor SNAIL represses vitamin D receptor expression and responsiveness in human colon cancer. Nat Med. 2004;10:917–9. doi: 10.1038/nm1095. [DOI] [PubMed] [Google Scholar]