

Abstract
Rhabdomyosarcoma (RMS) is a highly malignant tumor and is one of the few life-threatening diseases that present first to the ophthalmologist. It is the most common soft-tissue sarcoma of the head and neck in childhood with 10% of all cases occurring in the orbit. RMS has been reported from birth to the seventh decade, with the majority of cases presenting in early childhood. Survival has changed drastically over the years, from 30% in the 1960’s to 90% presently, with the advent of new diagnostic and therapeutic modalities. The purpose of this review is to provide a general overview of primary orbital RMS derived from a literature search of material published over the last 10 years, as well as to present two representative cases of patients that have been managed at our institute.
Keywords: Rhabdomyosarcoma, Tumor, Orbit, Eye, AMORE, Genetics, Pediatrics, Brachytherapy
Introduction
Rhabdomyosarcoma (RMS) is a highly malignant tumor in which the tissue of origin is pluripotent mesenchyme.1 It is the most common soft-tissue sarcoma of the head and neck in childhood and comprises 4% of all pediatric malignancies, with 10% of all cases occurring in the orbit.1,2 Most of these tumors occur in the first decade of life,3 however RMS has been reported from birth to the eighth decade.1,4,5
The incidence of malignant orbital tumors in general has been increasing.6 Turner et al. in a study of population based incidence and survival in head and neck RMS reported an annual percentage increase of 1.16% and a statistically unchanged 5-year survival over the past 30 years despite advances in treatment modalities; although other studies have reported improved survival.3,7 Relative survival (RS) was more dependent on the extent of disease rather than primary site, and it was noted that most orbital RMS tumors (60.6%) presented with localized disease and consequently had a 5-year survival of about 84.3% which is more favorable when compared with other sites in the head and neck.3,8 Other factors positively influencing survival were younger age (<10 years), female sex and embryonal histology (the most common in the orbit).3,4,8–10
Ocular RMS comprises tumors that occur in the orbit, or rarely in other ocular adnexal structures or within the eye.2 The purpose of this review is to provide a general overview of primary orbital RMS derived from a literature search of material published over the last 10 years, as well as to present two representative cases of patients that have been managed at our institute.
Orbital RMS is one of the few life-threatening diseases that present first to the ophthalmologist; therefore prompt diagnosis and treatment is a life-saving issue.2 Hence, knowledge of the clinical, histopathological, and radiographic features as well as the more recent advances in the management of this entity is essential.
Clinical features
Patients with orbital RMS usually present with proptosis developing rapidly over weeks (80–100%), or globe displacement (80%) which is usually downward and outward because two-thirds of these tumors are supero-nasal.1,2,4
Metastatic spread of orbital RMS is uncommon, however if left untreated RMS has a propensity to metastasize to the lung, bone and bone marrow mainly via hematogenous spread (because orbital lymphatics are scarce).2 Locally, orbital RMS can invade the orbital bones and can extend intracranially.2 Metastatic orbital RMS has an unfavorable prognosis when compared to localized disease; however in a joint European-North American pooled analysis orbital site proved to be favorable.2,11,12
Diagnosis
A detailed history is essential in any child suspected to have orbital RMS (i.e. under 2 years of age and presenting with an orbital mass).2 The ophthalmologist should specifically ask about pain, visual loss, and signs of sinusitis, and should also keep in mind possible misleading presentations such as rapidly progressing alterations of the lid, conjunctiva or caruncle.2,13
The history will help exclude other differential diagnoses such as orbital cellulitis, lymphangioma, idiopathic orbital inflammation, dermoid cyst, hemangioma, Langerhans cell histiocytosis (eosinophilic granuloma), sarcomas, metastatic neuroblastoma, and lymphoma.2 Imaging also plays an important role in the differentiation of these conditions. Biopsy should be performed if RMS is suspected based on clinical and radiological findings.2
Imaging
Orbital RMS is usually extraconal (37–87%) or both intra- and extraconal (13–47%), and more commonly superonasal in location especially for embryonal RMS (inferior location is more common for alveolar).4,9,14–16 The mass is usually close to extraocular muscles, but there is no enlargement of the muscle belly.9 In early stages the tumor is well circumscribed, but in later stages, where there is pseudocapsular invasion, the borders are irregular.2 There may be some bone deformity, but frank bone destruction with bone involvement is rare, and the diagnosis in this case will change from orbital to parameningeal RMS.15 The tumor may show hemorrhages and cyst formation.2
Ultrasound (US)
US findings are neither specific for orbital RMS nor sensitive for detecting parameningeal spread, and US penetration into deeper tissues is limited; hence reports on US findings in orbital tumors are limited.9,15,17 It mainly appears as a well-circumscribed, heterogenous, irregular mass with low to medium echogenicity on US and a variable intratumoral vascular flow pattern on Doppler US.9,17 A “pseudocystic” appearance has been reported as characteristic, and that is thought to be due to spindle cells in embryonal RMS which have abundant cytoplasm and are separated loosely by edematous fluid.18
Computed tomography (CT) and magnetic resonance imaging (MRI)
CT and MRI are important for the preoperative evaluation, staging, and follow-up of orbital RMS.6 CT is especially important for the detection of bone involvement, and MR is important for better spatial resolution, soft-tissue contrast, and detection of intracranial spread.9,14 Both are also used for post-treatment follow up: CT to show change in bone involvement, and MR to show residual or recurrent disease.6 Hence, CT and MR are complementary.4,14,15 However, when using CT one must be aware that exposure to ionizing radiation may theoretically induce a second potentially fatal cancer in children who have already manifested a tendency to harbor cancer; therefore, especially for follow up purposes MRI is the preferred imaging modality.19
CT
Orbital RMS appears as a well-circumscribed, homogenous, soft tissue mass that is isodense as compared to muscles without bone destruction in earlier stages, but with invasion of surrounding structures and calcification in more advanced cases where there is destruction of the adjacent bone.2,4,9,14,16,20 A heterogeneous appearance is possible in the event of focal hemorrhage or necrosis, and there is moderate to marked general enhancement with intravenous contrast.1,2,4,9,16 A common finding is eyelid thickening, regardless of lid involvement, and a less common finding is a cavitated mass with ring-like enhancement.9,14
MRI
On MRI, the tumor appears isointense with respect to extraocular muscles and hypointense with respect to orbital fat on T1-weighted images, and hyperintense (with respect to both orbital fat and extraocular muscles) on T2-weighted images.4,16,20 Increased signal on T1 and T2 is seen in focal areas of chronic hemorrhage.1 The globe may be distorted or displaced but rarely invaded.9 Visualization is best with gadolinium, (moderate to marked enhancement), and fat suppression for better delineation.2 Pre- and postcontrast comparison is useful to best detect intracranial and adjacent paranasal sinus invasion.14
Diffusion-weighted imaging (DWI) is an MR imaging technique that gives information about normality of proton and water diffusion through tissue characterized by DWI and ADC (Apparent Diffusion Coefficient) values, respectively.21 Because of high cellular density, malignant tumors usually have restricted diffusion. Lope et al. studied the usefulness of DWI in identifying characteristics of orbital tumors in order to aid radiographic diagnosis.21 They found that RMS was predominantly restricted on DWI/ADC, which was especially helpful in differentiating it from hemangioma, (which has increased DWI/ADC) since the two exhibit a similar appearance on T1 and T2.21
Nuclear imaging
Bone scintigraphy is being used in the work-up particularly for the detection of osteoblastic metastases.14
Some studies have demonstrated that PET-CT is better at the detecting bone and lymph node metastasis than CT and US; however whole body MR is being advocated (sensitivity 97.5%, specificity 99.4%) as superior to PET-CT (sensitivity 90%, specificity 100%).14,15,22 It is still unclear whether PET-CT or whole body MR should be used routinely because of lack of clinical trials aimed at RMS, however, they can be contemplated in cases with increased metastatic risk.14
Biopsy
RMS is the most common biopsied malignant orbital tumor in a child, and it is important in terms of establishing diagnosis and determining prognosis.2,23 The biopsy can be incisional or excisional based on clinical and imaging findings, however fine needle aspiration is less useful because the limited amount of tissue obtained is usually insufficient for pathological diagnosis.
We advise incisional biopsy, as opposed to excisional, in order to avoid dissemination of tumor cells into the adjacent healthy tissue.
Pathology
Previously, RMS was believed to arise from extraocular muscles, but now it is thought that it originates from pluripotent mesenchymal cells that have the ability to differentiate into skeletal muscle.2,9,20 Some cases may be related to prior radiation.2
RMS is divided into three histological subgroups: pleomorphic, embryonal, and alveolar; with the majority of orbital RMS being of the embryonal type, and the pleomorphic subtype occurring almost exclusively in adults (median age: sixth decade).2
The embyonal type (Fig. 1) comprises 50–70% of orbital RMS. Frequently seen cells are bipolar cells with tapered cytoplasmic processes, and less commonly seen cells are “tadpole-like” with long cytoplasmic extensions.9 The cells are usually arranged in a pattern of interlacing fascicle.9 Cross-striations (which are bundles of actin and myosin filaments) in the cytoplasm may be visible with Masson trichrome or phosphotungstic acid–hematoxylin stains.2,9,20
Figure 1.
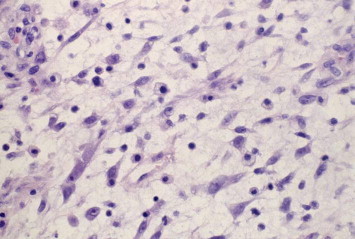
Embryonal orbital RMS. Elongated to round spindle cells with features of skeletal muscle in different stages of embryogenesis with a highly eosiniphilic cytoplasm and hyperchromatic nuclei.
The alveolar type (Fig. 2) (20–30% of orbital RMS) is characterized by ill-defined aggregates of poorly differentiated malignant cells that are loosely arranged and separated into irregular ovoid spaces by thin fibrovascular septa in an alveolar pattern which is absent in the “solid” form.2,9,20,24 This type has a poor prognosis, and any focus of alveolar morphology is sufficient to classify the tumor as alveolar RMS.24
Figure 2.
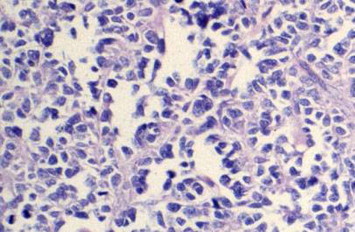
Alveolar orbital RMS. The cells are large, with abundant eosinophilic cytoplasm and round to polygonal in shape with vesicular nuclei which are a diagnostic feature of alveolar type as opposed to embryonal.
The botryoid seems to be a variant of the embryonal type that assumes a papillary configuration.2
Distinction of the different types of RMS is important and can be aided by immunohistochemical staining and cytogenetic studies.24
Immunohistochemistry
Immunohistochemical studies constitute the main approach to diagnosis. The markers typically found in RMS include antibodies against desmin (90%), muscle-specific actin, myoD1 (71–91%) and myoglobin.2,23,25–28 Myogenin (90%) is expressed more in the alveolar than in the embryonal type.24–26,28,29 Vimentin and desmin are usually positive but less specific since they can be positive in other tumors with skeletal muscle differentiation.2,25,30
Myogenin and MyoD1 are myogenic transcriptional regulators that are expressed early in skeletal muscle differentiation (earlier than desmin, actin, myoglobin and myosin).25
Electron microscopy
Not usually used for diagnosis of RMS, but may be helpful if histopathologic diagnosis is uncertain by demonstrating parallel arrays of thick myosin filaments.2
Genetics
RMS has been noted to occur more commonly in certain familial syndromes like Li-Fraumeni familial cancer syndrome (associated with p53 mutation), neurofibromatosis, Noonan, Beckwith–Wiedemann and Costello syndromes which suggest a possible genetic contribution to RMS.2,24,31–33
The most common cytogenetic finding in alveolar RMS is a t(2;13)(q35–37;q14) translocation resulting in a transcript composed of 5′ PAX3 sequences fused to 3′ FKHR sequences, and another translocation found in a subset of alveolar RMS is t(1;13)(p36;q14).24,26 One of these two fusion genes is usually found in the majority of alveolar RMS.24,26 It is of note that the detection of PAX3-FKHR fusion protein indicates an unfavorable variant of alveolar RMS, whereas the PAX7-FKHR fusion protein indicates a relatively favorable variant.10,34
p53 in RMS varies in a different series ranging from 5% to 44% of cases; CDK4 (cyclin-dependent kinase 4) is also a cell cycle control gene reported to be positive in about 80% of alveolar RMS.25,26 C-erbB2 expression is reported to be positive in 60% of cases of RMS.25 In a study by Andrade et al., the investigators found p53 to be positive in 13.8%, CDK4 in 34.5%, C-erbB2 in 70.4% and FAS in 31% of cases.25
Chan et al. reported hypermethylation of three tumor suppressor genes (HIN-1, RB1, and CDX-1) in a patient with alveolar RMS, in addition to HIC-1 (present in 100% of studied alveolar RMS cases).35 This is important especially in light of the absence of MyoD1 methylation in this patient, knowing that it is present in 91% of embryonal RMS.35 This can thus play a role in differentiating embryonal and alveolar RMS; however the sample size was too small to make definitive conclusions and further studies are warranted.35
Staibano et al. studied DNA ploidy and immunohistochemical expression of p53 (tumor suppressor gene), bcl-2 (protooncogene), MDR-1 (multidrug resistance protein) and MIB1 (Ki67; cell proliferation associated protein) in the prognostic evaluation of orbital RMS.7,26 They divided their findings into two groups:
-
-
Good prognosis i.e. relapse free.
-
-
Poor prognosis i.e. relapsing RMS requiring more aggressive therapeutic protocols.
Their results showed that the cases of tetraploid and/or multiploid RMS overexpressing p53 and MDR-1 were characterized by a worse prognosis; whereas tumors with a good prognosis showed hyperexpression of MIB1 and absence of mutated p53 expression.7,36 No difference was found with respect to bcl-2 expression.7
It is worth noting that the point mutations of p53 as related to recurrence/progression/worse prognosis is still controversial in the literature.25,31
These findings, and further studies, can prove useful for subtyping RMS and tailoring treatment protocols.7
Management
Guidelines regarding management of RMS since the early seventies have been intoduced mostly by the North American Intergroup Rhabdomyosarcoma Study Group (IRSG) and European cooperative groups: International Society of Pediatric Oncology-Malignant Mesenchymal Tumor Committee (SIOP-MMT), Cooperative Weichteilsarkom Study Group (CWS) and Italian Cooperative Group (ICG).2 Prior to that, overall survival (OS) was about 25–30% (with the historically recommended treatment being exenteration prior to the late 1960’s), and with the introduction of surgery combined with adjuvant chemotherapy as well as radiotherapy in cooperative group trials OS improved to around 90%.1,4,10,15,37–40
Although the IRSG is directed at RMS in general, some investigators have retrieved data that pertains only to orbital RMS.2,41 The IRSG conclusions represent the American school of thought, whereas the findings SIOP-MMT, CWS and ICG represent the European school of thought.2,42,40
The diagnosis of orbital RMS is made based on histopathologic findings following excisional or incisional biopsy.2 Some surgeons believe that an incisional biopsy is sufficient (European protocol) since adequate margins cannot be reached and orbital RMS has a good prognosis following radiation and chemotherapy regardless of amount of tissue excised.43 Others believe that complete excision or maximal debulking decreases the tumor burden and facilitates subsequent medical treatment.2 In a pooled analysis of 306 patients with orbital RMS treated according to international protocols (IRSG, SIOP-MMT, CWS, and ICG) in the period 1978–1992, 222/306 (72%) of patients underwent an initial inicisional biopsy only and had tumors that were considered unresectable.40 A partial excision was achieved by an initial surgery in 75/306 patients (25%), and in only 9/306 patients (3%) an initial oncological radical excision could be performed. Although the initial surgical approach differed between cooperative groups, with the percentage of patients starting therapy after biopsy only varying from 82% (IRSG) to 46% (ICG), this had no impact on prognosis.
Following biopsy, staging for orbital RMS is internationally uniformly done according to the IRS post-surgical staging system.22,44–46
Group I: localized disease, completely resected (excisional biopsy).
Group II: microscopic disease remaining after biopsy.
Group III: gross residual disease remaining after biopsy.
Group IV: distant metastasis present at onset.
This classification is useful in terms of treatment, stratification and prognosis prediction.2,44
Decision-making for management is based on histopathologic confirmation as well as staging of orbital RMS, which is done by reviewing imaging (MRI of primary tumor, chest-CT, Tc bone scan), pathology, and further work-up for metastases (bilateral bone marrow punctures and trephines).1
Current management includes surgery, irradiation and chemotherapy depending on the stage.2,47–49
-
-
Group I are treated with chemotherapy only: VA (vincristine and actinomycin).
-
-
Group II are treated with a combination of chemotherapy (VA and cyclophosphamide; VAC) and radiotherapy (36 Gy).
-
-
Group III are treated with a combination of chemotherapy (VAC) and radiotherapy (45 Gy).
-
-
Group IV are treated with a combination of intensive chemotherapy and radiotherapy.
Patients having a higher risk of relapse also receive cyclophosphamide based on the IRSGIII and IV.12,49 Details regarding the different possible chemotherapeutic regimens depending on the risk is beyond the scope of this article but have been reported in the literature.49,50,47,51
The current European pediatric Soft tissue sarcoma Study Group (EpSSG) protocol (EpSSG-RMS-2005) proposes the following treatment strategy.42,52
Group I: chemotherapy consisting of VA.
Groups II and III: chemotherapy VA with Ifosfamide added in the first four courses; when in complete remission after three chemotherapy courses policy is to either leave out radiotherapy but add more ifosfamide, or add radiotherapy (36 Gy) and leave out further ifosfamide. Those not in complete remission after induction chemotherapy get radiotherapy (45 Gy) without further ifosfamide.
Group IV: intensified chemotherapy regimen (IVA and doxorubicin) followed by one year of maintenance chemotherapy and radiotherapy to all involved sites (when possible).
A pooled analysis on 306 orbital RMS patients according to European and North American protocols showed 10 year EFS to be significantly better for patients receiving RT as part of their initial treatment compared to those who did not (82 v 53%), however confirmed no statistical difference in OS (87 v 86%), taking advantage of a favorable ‘salvage gap’ with rescue utilizing further treatment.40 SIOP group data from that analysis showed that up to 40% of patients with an orbital localization could be treated successfully without the use of RT and without disadvantage to the survival of the whole group. However, the total burden of therapy must be taken into account as those who relapsed not only received RT as part of their second therapy but also needed additional chemotherapy.40
Breneman et al. reported no change in prognosis with reduced doses of radiotherapy with the addition of a moderate cumulative dose of cyclophosphamide.31 No specific guidelines concerning radiotherapy were specified apart from the use of brachytherapy whenever possible.31,53
Brachytherapy has been used for local treatment at our center since 1991.15 The AMORE protocol is a novel technique established at our institute for the treatment of non-resectable head and neck RMS.55 This protocol has since been followed for orbital RMS with the only remark that reconstruction is often not needed at the orbital site.54,53 It consists of Ablative surgery, MOulage technique brachytherapy and surgical REconstruction; it targets residual tumor mass.15,55 After diagnosis and staging, chemotherapy is given according to the European protocol, after which imaging is repeated to look for residual disease.15 The aim of this protocol is to maximize local treatment and to avoid external beam radiotherapy (EBRT). Ablative surgery is performed as conservatively as possible with an effort not to sacrifice important tissue at the expense of possible microscopic remnants- which will be amenable to the brachytherapy which is initiated directly post surgery.15,55 Brachytherapy has advantages over EBRT including a reduction in the treatment time and a focused dose delivery to the tumor bed and rapid fall-off of the dose beyond the treatment volume, this way sparing surrounding tissues and reducing morbidity, allowing organ preservation and bone growth, potentially improving the functional and cosmetic outcome.55
Radiotherapy
Radiation is usually in the range of 3600–5040 cGy over 4–5 weeks, and in the case of parameningeal extension the region of extension is irradiated + 2 cm.1,2,52
The main goal in novel management options is to maintain excellent survival while reducing the late side effects of treatment.58 All of this has prompted enthusiasm for no or reduced-dose radiotherapy in low-risk RMS2,14,41,48,49,31,56 and for new technologies in radiation oncology including proton beam radiotherapy, intensity modulated radiotherapy (IMRT), 3-D conformational radiotherapy and implant brachytherapy all with the goal to minimize dose to normal tissue.15,22,43,57,58
IMRT
Wolden et al. studied IMRT for head-and-neck RMS where they used a smaller margin (1.5 cm as opposed to the standard 2 cm in IRSG protocols) made possible by using imaging (CT, PET, MR) in radiotherapy planning.59 They concluded that IMRT with image fusion achieved excellent local control (in cases that did not have alveolar histology) with minimal dose reaching adjacent tissue when dose-limiting structures are adjacent to the tumor.10,36,59
Hein et al. focused on the possibility of organ-sparing (particularly lens-sparing) irradiation in children with orbital RMS by using IMRT as opposed to 3D conformational photon radiotherapy.60 They concluded that although IMRT resulted in a reduced dose to the lens (in treating tumors in retrobulbar and lateral positions) and ipsilateral lacrimal gland, no significant difference was noted for the ipsilateral retina and optic nerve, and more importantly, it resulted in an expanded volume of the brain receiving low dose radiation as compared to 3D conformational radiotherapy.60
Fractionated proton radiotherapy
Yock et al. reported results on using fractionated proton radiotherapy as superior over 3D conformational photon radiotherapy for orbital RMS; they concluded that proton therapy achieves excellent tumor coverage at the same time as it reduces radiation to the adjacent normal tissue like the brain, pituitary, hypothalamus, temporal lobes, and ipsilateral and contralateral orbital structures.10,36,54,61 Proton irradiation has excellent potential for conformal treatment to normal tissue that is better than 3D conformal radiation and perhaps even IMRT- which although advanced in tumor targeting as compared to 3D conformational photon irradiation- does distribute low dose to normal tissue: a disadvantage in a pediatric population with excellent survival that is more prone to secondary malignancies and growth retardation.60,61
Intensity-modulated brachytherapy (IMBT)
Perioperative IMBT has also been studied, and in cases of refractory orbital RMS was found to improve local tumor control and quality of life.62
Brachytherapy
Brachytherapy offers a high dose to the tumor bed with a low dose to the surrounding tissues and can be considered in cases where radiotherapy is necessary and brachytherapy characteristics fit; and although there are no conclusive clinical data available yet, the findings after IMRT might seem to be less favorable than those of brachytherapy.54,53
Following SIOP guidelines, Blank et al. have recently reported the use of brachytherapy instead of EBRT as part of the treatment of orbital RMS.54 The side effects were found to be very low and not clinically significant. They thus concluded that surgery followed by brachytherapy can be considered when there is no residual intracranial extension of the tumor after chemotherapy, macroscopic tumor removal is feasible, and when EBRT is expected to be harmful to eye structures.54
Side effects of radiotherapy
Many complications can arise from treatment with EBRT, including cataract (55%), dry eyes (36%), orbital hypoplasia (24%), ptosis (9%), radiation retinopathy (90%), facial asymmetry secondary to bone hypoplasia, keratoconjunctivitis, lacrimal duct stenosis, dental defects, growth retardation (in case of incidental irradiation of the pituitary) and secondary neoplasms such as osteogenic sarcoma, lymphoblastic leukemia and melanoma.1,2,41,57,58,63
Although brachytherapy is not free of side effects, it is less toxic than EBRT especially with respect to orbital hypoplasia.54,53,57 The side effects of brachytherapy can include cataract, radiation retinopathy, dryness, ptosis; all of which are relatively mild and/or amenable to treatment.54
Evaluation of metastatic disease
Work-up for metastatic disease should include chest-CT, Tc bone scan, and bone marrow punctures and trephines. Staging combines TNM (tumor, nodes, metastasis), which includes clinical features, histological grade, and the clinical grouping classification for RMS by the IRSG.14,25 A pooled analysis of 788 metastatic patients treated according to international cooperative group protocols (IRSG/COG, SIOP-MMT, ICG) in the time period 1984–2000 revealed that 3-year event free survival (EFS) was significantly and adversely influenced by age (⩽1 or ⩾10 years), location of the primary tumor at an unfavorable site (defined as extremity and “other” sites, the latter predominantly meaning trunk, excluding bladder and prostate), presence of three or more sites of metastatic disease, and the presence of bone or bone marrow involvement. EFS was 50% for patients with zero adverse factors and was respectively 42%, 18%, 12%, and 5% in patients with one, two, three, or four factors.12
Recurrence
Recurrence of orbital RMS can occur in about 17% of cases at a median time of 18 months, with 92% of these cases being local and 8% at distant sites.14,64 There are no clear guidelines for treatment in cases of recurrence. Possible options include debulking in addition to chemotherapy, additional EBRT, brachytherapy; and if all fails, exenteration is the remaining option.2,37,47
Follow-up
Follow-up of patients post treatment for orbital RMS is essential in order to look for late side-effects of radiotherapy, recurrence and secondary tumors. Radiological follow up with MRI is specified in cooperative group protocols and will generally start at 3 month intervals, with decreasing frequency until 5 years after treatment. Initially, ocular examination has to be done every 3–4 months, and after the first year every 4–6 months for several years and then yearly; and depending on the clinical findings periodic imaging can be done.2,15,63
Prognosis
Orbital RMS has a good prognosis because of the favorable anatomic location, (symptoms apparent at an early stage allowing early diagnosis), favorable histology and biology (80% embryonal) and possibly patient age.2,37 Embryonal RMS has a 94% 5-year survival (versus alveolar 74%) (2). Overall survival is excellent for groups I, II and III (92% at 5 years and 87% at 10 years).1,14,25 Survival after relapse is dependent on age at initial presentation, histology, IRS clinical group, nodal-stage, and previous chemotherapy and radiotherapy.65,66
Clinical cases
We present two cases that were treated at our institute following the above-mentioned AMORE protocol.
Case 1
A 1 year old male boy was referred to our center with proptosis OS. MR revealed a large intraorbital mass with local invasion of the lamina papyracea and no intracranial extension (Fig. 3). A biopsy of the tumor revealed an alveolar RMS on histology (positive for PAX3/FKHFR). There were no metastases. The patient was treated with six courses of IVA chemotherapy (ifosfamide, vincristine, dactinomycin) to which the tumor responded very well, but there was some residual tumor. External beam radiotherapy in such a young child would lead to severe late effects and therefore the delayed local treatment of choice was surgery followed by brachytherapy (AMORE protocol). The residual tumor was macroscopically removed and in the same session a silicon mold with three parallel plastic tubes was constructed and positioned in the medial part of the orbit. The wound was closed between the plastic tubes. A dose of 40 Gy in 23 pulses of 1,25 Gy was applied to treat the residual tumor area with a high active 192Iridium source during a three-day period (Fig. 4).
Figure 3.
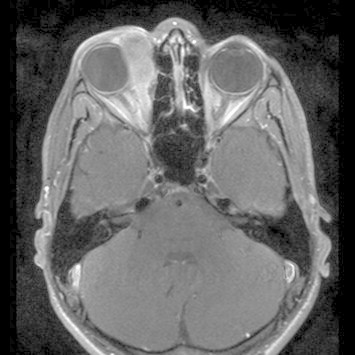
MR orbits and brain. Large intraorbital mass with local invasion of the lamina papyracea and no intracranial extension.
Figure 4.
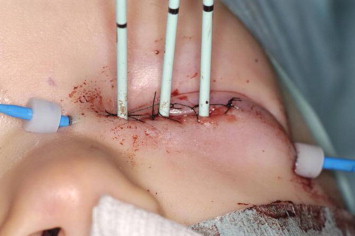
A silicon mold with three parallel plastic tubes positioned in the medial part of the orbit.
The mold was removed directly afterward and the skin closed. Following the total procedure there was good movement of the eye and no strabismus. The patient is now in persistent complete remission 7 years post treatment and without any side effects related to brachytherapy.
Case 2
A 12 year old female patient presented to our clinic with a rapidly growing mass in the left orbital/forehead area. MRI revealed a solid soft tissue tumor, from the left ethmoidal sinus with expansion into the maxillary sinus and the left orbit with dural enhancement in the anterior fossa along a bone defect. No metastasis was found on further work-up. Biopsy revealed an embryonal RMS. The patient was started on IVA chemotherapy (five cycles). Due to the existence of a residual tumor it was decided to perform a macroscopic radical resection (Fig. 4) followed by brachytherapy and reconstruction. The treatment consisted of 36 pulses of 1.25 Gy per pulse (total dose 45 Gy), after which the patient received four additional cycles of IVA chemotherapy. Reconstructive surgery was performed consisting of Galea flap attached to a calvarian bone, which were brought to the orbit to reconstruct the medial wall and orbital floor (Figs. 5 and 6). Complications secondary to brachytherapy included nasolacrimal duct obstruction which was subsequently treated successfully with endoscopic dacryocystorhinostomy, after which the patient complained of dry eyes. The patient is still in persistent complete remission nine years after completion of treatment.
Figure 5.
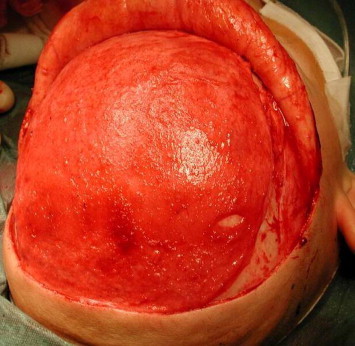
Galea flap attached to a calvarian bone.
Figure 6.
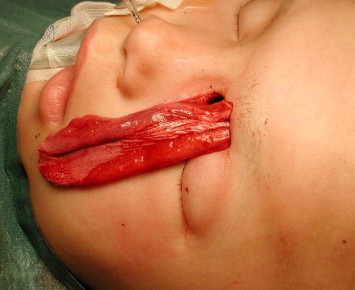
Galea flap attached to a calvarian bone, brought together to the orbit to reconstruct the medial wall and orbital floor.
Conclusion
Orbital RMS is one of the few life-threatening diseases that presents first to the ophthalmologist; therefore prompt diagnosis and treatment is a life-saving issue. Hence, knowledge of the clinical, histopathological, and radiographic features as well as the more recent advances in the management of this entity is necessary. A multidisciplinary approach for the treatment of orbital RMS is essential.
Conflict of interest
The authors declared that there is no conflict of interest.
Footnotes
Peer review under responsibility of Saudi Ophthalmological Society, King Saud University.

References
- 1.Rootman J., editor. Neoplasia. Vol. 54. Lippincott Williams and Wilkins; Philadelphia: 2003. pp. 262–268. (Diseases of the orbit: a multidisciplinary approach). [Google Scholar]
- 2.Shields J.A., Shields C.L. Rhabdomyosarcoma: review for the ophthalmologist. Surv Ophthalmol. 2003;48:39–57. doi: 10.1016/s0039-6257(02)00415-0. [DOI] [PubMed] [Google Scholar]
- 3.Turner J.H., Richmon J.D. Head and neck rhabdomyosarcoma: a critical analysis of population-based incidence and survival data. Otolaryngol Head Neck Surg. 2011;145:967–973. doi: 10.1177/0194599811417063. [DOI] [PubMed] [Google Scholar]
- 4.Conneely M.F., Mafee M.F. Orbital rhabdomyosarcoma and simulating lesions. Neuroimaging Clin N Am. 2005;15:121–136. doi: 10.1016/j.nic.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 5.Koopman J.H., van der Heiden-van der Loo M., van Dijk M.R., Bijlsma W.R. Incidence of primary malignant orbital tumours in The Netherlands. Eye (Lond) 2011;25:461–465. doi: 10.1038/eye.2011.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Staibano S., Franco R., Tranfa F., Mezza E., Lo Muzio L., Strianese D. Orbital rhabdomyosarcoma: relationship between DNA ploidy, p53, bcl-2, MDR-1 and Ki67 (MIB1) expression and clinical behavior. Anticancer Res. 2004;24:249–257. [PubMed] [Google Scholar]
- 7.Crist W.M., Garnsey L., Beltangady M.S., Gehan E., Ruymann F., Webber B. Prognosis in children with rhabdomyosarcoma: a report of the intergroup rhabdomyosarcoma studies I and II. Intergroup rhabdomyosarcoma committee. J Clin Oncol. 1990;8:443–452. doi: 10.1200/JCO.1990.8.3.443. [DOI] [PubMed] [Google Scholar]
- 8.Chung E.M., Smirniotopoulos J.G., Specht C.S., Schroeder J.W., Cube R. From the archives of the AFIP: pediatric orbit tumors and tumorlike lesions: nonosseous lesions of the extraocular orbit. Radiographics. 2007;27:1777–1799. doi: 10.1148/rg.276075138. [DOI] [PubMed] [Google Scholar]
- 9.Terezakis S.A., Wharam M.D. Radiotherapy for rhabdomyosarcoma: indications and outcome. Clin Oncol (R Coll Radiol) 2013;25:27–35. doi: 10.1016/j.clon.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 10.Breneman J.C., Lyden E., Pappo A.S., Link M.P., Anderson J.R., Parham D.M. Prognostic factors and clinical outcomes in children and adolescents with metastatic rhabdomyosarcoma – a report from the intergroup rhabdomyosarcoma study IV. J Clin Oncol. 2003;21:78–84. doi: 10.1200/JCO.2003.06.129. [DOI] [PubMed] [Google Scholar]
- 11.Schworm H.D., Boergen K.P., Stefani F.H. The initial clinical manifestations of rhabdomyosarcoma. Ophthalmologe. 1995;92:362–365. [PubMed] [Google Scholar]
- 12.Rao A.A., Naheedy, Chen J.Y., Robbins S.L., Ramkumar H.L. A clinical update and radiologic review of pediatric orbital and ocular tumors. J Oncol. 2013;2013:975908. doi: 10.1155/2013/975908. doi: 10.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freling N.J., Merks J.H., Saeed P., Balm A.J., Bras J., Pieters B.R. Imaging findings in craniofacial childhood rhabdomyosarcoma. Pediatr Radiol. 2010;40:1723–1738. doi: 10.1007/s00247-010-1787-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sohaib S.A., Moseley I., Wright J.E. Orbital rhabdomyosarcoma-the radiological characteristics. Clin Radiol. 1998;53:357–362. doi: 10.1016/s0009-9260(98)80009-3. [DOI] [PubMed] [Google Scholar]
- 15.Neudorfer M., Leibovitch I., Stolovitch C., Dray J.P., Hermush V., Nagar H. Intraorbital and periorbital tumors in children-value of ultrasound and color Doppler imaging in the differential diagnosis. Am J Ophthalmol. 2004;137:1065–1072. doi: 10.1016/j.ajo.2004.01.050. [DOI] [PubMed] [Google Scholar]
- 16.Boparai M.S., Dash R.G. Clinical, ultrasonographic and CT evaluation of orbital rhabdomyosarcomas with management. Indian J Ophthalmol. 1991;39:129–131. [PubMed] [Google Scholar]
- 17.Karcioglu Z.A., Hadjistilianou D., Rozans M., DeFrancesco S. Orbital rhabdomyosarcoma. Cancer Control. 2004;11:328–333. doi: 10.1177/107327480401100507. [DOI] [PubMed] [Google Scholar]
- 18.Lope L.A., Hutcheson K.A., Khademian Z.P. Magnetic resonance imaging in the analysis of pediatric orbital tumors: utility of diffusion-weighted imaging. J AAPOS. 2010;14:257–262. doi: 10.1016/j.jaapos.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 19.Malempati S., Hawkins D.S. Rhabdomyosarcoma: review of the Children’s Oncology Group (COG) soft-tissue sarcoma committee experience and rationale for current COG studies. Pediatr Blood Cancer. 2012;59:5–10. doi: 10.1002/pbc.24118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Viswanathan S., George S., Ramadwar M., Shet T., Arora B., Laskar S. Extraconal orbital tumors in children – a spectrum. Virchows Arch. 2009;454:703–713. doi: 10.1007/s00428-009-0775-1. [DOI] [PubMed] [Google Scholar]
- 21.Passmore L.M., Myers P., Gilbert-Barness E. Pathology teach and tell: solid variant alveolar rhabdomyosarcoma of the orbit. Fetal Pediatr Pathol. 2006;25:51–57. doi: 10.1080/15227950600701602. [DOI] [PubMed] [Google Scholar]
- 22.Andrade C.R., Takahama Junior A., Nishimoto I.N., Kowalski L.P., Lopes M.A. Rhabdomyosarcoma of the head and neck: a clinicopathological and immunohistochemical analysis of 29 cases. Braz Dent J. 2010;21:68–73. doi: 10.1590/s0103-64402010000100011. [DOI] [PubMed] [Google Scholar]
- 23.Constine L.S., Marcus R.B., Jr., Halperin E.C. The future of therapy for childhood rhabdomyosarcoma: clues from molecular biology. Int J Radiat Oncol Biol Phys. 1995;32:1245–1249. doi: 10.1016/0360-3016(95)00237-s. [DOI] [PubMed] [Google Scholar]
- 24.Tapscott S.J., Thayer M.J., Weintraub H. Deficiency in rhabdomyosarcomas of a factor required for MyoD activity and myogenesis. Science. 1993;259:1450–1453. doi: 10.1126/science.8383879. [DOI] [PubMed] [Google Scholar]
- 25.Sun X.L., Zheng B.H., Li B., Li L.Q., Soejima K., Kanda M. Orbital rhabdomyosarcoma. Immunohistochemical studies of seven cases. Chin Med J (Engl) 1990;103:485–488. [PubMed] [Google Scholar]
- 26.Cintorino M., Vindigni C., Del Vecchio M.T., Tosi P., Frezzotti R., Hadjistilianou T. Expression of actin isoforms and intermediate filament proteins in childhood orbital rhabdomyosarcomas. J Submicrosc Cytol Pathol. 1989;21:409–419. [PubMed] [Google Scholar]
- 27.Felix C.A., Kappel C.C., Mitsudomi T., Nau M.M., Tsokos M., Crouch G.D. Frequency and diversity of p53 mutations in childhood rhabdomyosarcoma. Cancer Res. 1992;52:2243–2247. [PubMed] [Google Scholar]
- 28.Jung A., Bechthold S., Pfluger T., Renner C., Ehrt O. Orbital rhabdomyosarcoma in Noonan syndrome. J Pediatr Hematol Oncol. 2003;25:330–332. doi: 10.1097/00043426-200304000-00014. [DOI] [PubMed] [Google Scholar]
- 29.Thavaraj V., Sethi A., Arya L.S. Incomplete Beckwith–Wiedemann syndrome in a child with orbital rhabdomyosarcoma. Indian Pediatr. 2002;39:299–304. [PubMed] [Google Scholar]
- 30.Chan W.M., Liu D.T., Pang C.P., Lam D.S., To K.F., Choi P.C. Pediatric malignancies. Case 1: hypermethylation in orbital alveolar rhabdomyosarcoma. J Clin Oncol. 2005;23:4790–4791. doi: 10.1200/JCO.2005.06.037. [DOI] [PubMed] [Google Scholar]
- 31.Wolden S.L., Wexler L.H., Kraus D.H., Laquaglia M.P., Lis E., Meyers P.A. Intensity-modulated radiotherapy for head-and-neck rhabdomyosarcoma. Int J Radiat Oncol Biol Phys. 2005;61:1432–1438. doi: 10.1016/j.ijrobp.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 32.Raney B., Huh W., Hawkins D., Hayes-Jordan A., Million L., Rodeberg D. Outcome of patients with localized orbital sarcoma who relapsed following treatment on Intergroup Rhabdomyosarcoma Study Group (IRSG) protocols-III and -IV, 1984–1997: a report from the children’s oncology g. Pediatr Blood Cancer. 2013;60:371–376. doi: 10.1002/pbc.24289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ashton N., Morgan G. Embryonal sarcoma and embryonal rhabdomyosarcoma of the orbit. J Clin Pathol. 1965;18:699–714. doi: 10.1136/jcp.18.6.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Rijn R.R., Wilde J.C., Bras J. Imaging findings in noncraniofacial childhood rhabdomyosarcoma. Pediatr Radiol. 2008;38:617–634. doi: 10.1007/s00247-008-0751-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raney R.B., Anderson J.R., Kollath J., Vassilopoulou-Sellin R., Klein M.J., Heyn R. Late effects of therapy in 94 patients with localized rhabdomyosarcoma of the orbit: report from the Intergroup Rhabdomyosarcoma Study (IRS)-III, 1984–1991. Med Pediatr Oncol. 2000;34:413–420. doi: 10.1002/(sici)1096-911x(200006)34:6<413::aid-mpo6>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 36.Orbach D., Rey A., Oberlin O., Sanchez de Toledo J., Terrier-Lacombe MJ., van Unnik A. Soft tissue sarcoma or malignant mesenchymal tumors in the first year of life: experience of the International Society of Pediatric Oncology (SIOP) malignant mesenchymal tumor committee. J Clin Oncol. 2005;23:4363–4371. doi: 10.1200/JCO.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 37.Olivier Pascual N., Calvo J.M., Abelairas Gómez J.M. Orbital rhabdomyosarcoma: difficulties with European treatment protocol. Arch Soc Esp Oftalmol. 2005;80:331–338. doi: 10.4321/s0365-66912005000600006. [DOI] [PubMed] [Google Scholar]
- 38.Raney R.B., Maurer H.M., Anderson J.R., Andrassy R.J., Donaldson S.S., Qualman S.J. The Intergroup Rhabdomyosarcoma Study Group (IRSG): major lessons from the IRS-I through IRS-IV studies as background for the current IRS-V treatment protocols. Sarcoma. 2001;5:9–15. doi: 10.1080/13577140120048890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wharam M.D., Hanfelt J.J., Tefft M.C., Johnston J., Ensign L.G., Breneman J. Radiation therapy for rhabdomyosarcoma: local failure risk for clinical group III patients on intergroup rhabdomyosarcoma study II. Int J Radiat Oncol Biol Phys. 1997;38:797–804. doi: 10.1016/s0360-3016(97)00120-x. [DOI] [PubMed] [Google Scholar]
- 40.Shields C.L., Shields J.A., Honavar S.G., Demirci H. Primary ophthalmic rhabdomyosarcoma in 33 patients. Trans Am Ophthalmol Soc. 2001;99:133–143. [PMC free article] [PubMed] [Google Scholar]
- 41.Wolden S.L., Anderson J.R., Crist W.M., Breneman J.C., Wharam M.D., Jr, Wiener E.S. Indications for radiotherapy and chemotherapy after complete resection in rhabdomyosarcoma: a report from the intergroup rhabdomyosarcoma studies I to III. J Clin Oncol. 1999;17:3468–3475. doi: 10.1200/JCO.1999.17.11.3468. [DOI] [PubMed] [Google Scholar]
- 42.Orbach D., Brisse H., Helfre S., Freneaux P., Husseini K.I. Effectiveness of chemotherapy in rhabdomyosarcoma: example of orbital primary. Expert Opin Pharmacother. 2003;4:2165–2174. doi: 10.1517/14656566.4.12.2165. [DOI] [PubMed] [Google Scholar]
- 43.Raney R.B., Walterhouse D.O., Meza J.L., Andrassy R.J., Breneman J.C., Crist W.M. Results of the intergroup rhabdomyosarcoma study group D9602 protocol, using vincristine and dactinomycin with or without cyclophosphamide and radiation therapy, for newly diagnosed patients with low-risk embryonal rhabdomyosarcoma: a report from the soft tissue sarcoma committee of the children’s oncology group. J Clin Oncol. 2011;29:1312–1318. doi: 10.1200/JCO.2010.30.4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ruymann F.B., Vietti T., Gehan E. Cyclophosphamide dose escalation in combination with vincristine and actinomycin D (VAC) in gross residual sarcoma: a pilot study without hematopoietic growth factor support evaluating toxicity and response. J Pediatr Hematol Oncol. 1995;17:331–337. doi: 10.1097/00043426-199511000-00009. [DOI] [PubMed] [Google Scholar]
- 45.Arndt C., Tefft M., Gehan E. A feasibility, toxicity, and early response study of etoposide, ifosfamide, and vincristine for the treatment of children with rhabdomyosarcoma: a report from the intergroup rhabdomyosarcoma study (IRS) IV pilot study. J Pediatr Hematol Oncol. 1997;19:124–129. doi: 10.1097/00043426-199703000-00005. [DOI] [PubMed] [Google Scholar]
- 46.Blank L.E., Koedooder K., van der Grient H.N., Wolffs N.A., van de Kar M., Merks J.H. Brachytherapy as part of the multidisciplinary treatment of childhood rhabdomyosarcomas of the orbit. Int J Radiat Oncol Biol Phys. 2010;77:1463–1469. doi: 10.1016/j.ijrobp.2009.06.015. [DOI] [PubMed] [Google Scholar]
- 47.Breneman J., Meza J., Donaldson S.S., Raney R.B., Wolden S., Michalski J. Local control with reduced-dose radiotherapy for low-risk rhabdomyosarcoma: a report from the children’s oncology group D9602 study. Int J Radiat Oncol Biol Phys. 2012;83:720–726. doi: 10.1016/j.ijrobp.2011.06.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tyl J.W., Blank L.E., Koornneef L. Brachytherapy in orbital tumors. Ophthalmology. 1997;104:1475–1479. doi: 10.1016/s0161-6420(97)30113-4. [DOI] [PubMed] [Google Scholar]
- 49.Buwalda J., Schouwenburg P.F., Blank L.E., Merks J.H., Copper M.P., Strackee S.D. A novel local treatment strategy for advanced stage head and neck rhabdomyosarcomas in children: results of the AMORE protocol. Eur J Cancer. 2003;39:1594–1602. doi: 10.1016/s0959-8049(03)00363-0. [DOI] [PubMed] [Google Scholar]
- 50.Forstner D., Borg M., Saxon B. Orbital rhabdomyosarcoma: multidisciplinary treatment experience. Australas Radiol. 2006;50:41–45. doi: 10.1111/j.1440-1673.2005.01526.x. [DOI] [PubMed] [Google Scholar]
- 51.Regine W.F., Fontanesi J., Kumar P., Zeitzer K., Greenwald C., Bowman L. A phase II trial evaluating selective use of altered radiation dose and fractionation in patients with unresectable rhabdomyosarcoma. Int J Radiat Oncol Biol Phys. 1995;31:799–805. doi: 10.1016/0360-3016(94)00459-5. [DOI] [PubMed] [Google Scholar]
- 52.Abramson D.H., Fass D., McCormick B., Servodidio C.A., Piro J.D., Anderson L.L. Implant brachytherapy: a novel treatment for recurrent orbital rhabdomyosarcoma. J AAPOS. 1997;1:154–157. doi: 10.1016/s1091-8531(97)90058-6. [DOI] [PubMed] [Google Scholar]
- 53.Hein P.A., Gladstone D.J., Bellerive M.R., Hug E.B. Importance of protocol target definition on the ability to spare normal tissue: an IMRT and 3D-CRT planning comparison for intraorbital tumors. Int J Radiat Oncol Biol Phys. 2005;62:1540–1548. doi: 10.1016/j.ijrobp.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 54.Yock T., Schneider R., Friedmann A., Adams J., Fullerton B., Tarbell N. Proton radiotherapy for orbital rhabdomyosarcoma: clinical outcome and a dosimetric comparison with photons. Int J Radiat Oncol Biol Phys. 2005;63:1161–1168. doi: 10.1016/j.ijrobp.2005.03.052. [DOI] [PubMed] [Google Scholar]
- 55.Strege R.J., Kovács G., Meyer J.E., Holland D., Claviez A., Mehdorn M.H. Perioperative intensity-modulated brachytherapy for refractory orbital rhabdomyosarcomas in children. Strahlenther Onkol. 2009;185:789–798. doi: 10.1007/s00066-009-2012-x. [DOI] [PubMed] [Google Scholar]
- 56.Yip C.C., Kersten R.C., McCulley T.J., Ballard E.T., Kulwin D.R. Osteogenic sarcoma after orbital radiation rhabdomyosarcoma. Ophthalmology. 2003;110:1996–1999. doi: 10.1016/S0161-6420(03)00478-0. [DOI] [PubMed] [Google Scholar]
- 57.Shields C.L., Shields J.A., Honavar S.G., Demirci H. Clinical spectrum of primary ophthalmic rhabdomyosarcoma. Ophthalmology. 2001;108:2284–2292. doi: 10.1016/s0161-6420(01)00840-5. [DOI] [PubMed] [Google Scholar]
- 58.Warrier A.R., Syriac S., Rathnam K.K. Late recurrence in orbital rhabdomyosarcoma: complete remission after multimodality management. J Cancer Res Ther. 2010;6:307–309. doi: 10.4103/0973-1482.73326. [DOI] [PubMed] [Google Scholar]
- 59.Sultan I., Qaddoumi I., Yaser S., Rodriguez-Galindo C., Ferrari A. Comparing adult and pediatric rhabdomyosarcoma in the surveillance, epidemiology and end results program, 1973 to 2005: an analysis of 2,600 patients. J Clin Oncol. 2009;27:3391–3397. doi: 10.1200/JCO.2008.19.7483. [DOI] [PubMed] [Google Scholar]
- 60.Oberlin O., Rey A., Lyden E., Bisogno G., Stevens M.C., Meyer W.H. Prognostic factors in metastatic rhabdomyosarcomas: results of a pooled analysis from United States and European cooperative groups. J Clin Oncol. 2008;26:2384–2389. doi: 10.1200/JCO.2007.14.7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Paterson A., Frush D.P., Donnelly L.F. Helical CT of the body: are settings adjusted for pediatric patients? AJR Am J Roentgenol. 2001;176:297–301. doi: 10.2214/ajr.176.2.1760297. [DOI] [PubMed] [Google Scholar]
- 62.Missiaglia E., Williamson D., Chisholm J., Wirapati P., Pierron G., Petel F. PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J Clin Oncol. 2012;30:1670–1677. doi: 10.1200/JCO.2011.38.5591. [DOI] [PubMed] [Google Scholar]
- 63.Oberlin O., Rey A., Anderson J., Carli M., Raney R.B., Treuner J. Treatment of orbital rhabdomyosarcoma: survival and late effects of treatment–results of an international workshop. J Clin Oncol. 2001;19:197–204. doi: 10.1200/JCO.2001.19.1.197. [DOI] [PubMed] [Google Scholar]
- 64.Maurer H.M., Beltangady M., Gehan E.A., Crist W., Hammond D., Hays D.M. The intergroup rhabdomyosarcoma study-IA final report. Cancer. 1988;61:209–220. doi: 10.1002/1097-0142(19880115)61:2<209::aid-cncr2820610202>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 65.Chisholm J.C., Marandet J., Rey A., Scopinaro M., de Toledo J.S., Merks J.H. Prognostic factors after relapse in nonmetastatic rhabdomyosarcoma: a nomogram to better define patients who can be salvaged with further therapy. J Clin Oncol. 2011;29:1319–1325. doi: 10.1200/JCO.2010.32.1984. [DOI] [PubMed] [Google Scholar]
- 66.Mazzoleni S., Bisogno G., Garaventa A., Cecchetto G., Ferrari A., Sotti G. Outcomes and prognostic factors after recurrence in children and adolescents with nonmetastatic rhabdomyosarcoma. Cancer. 2005;104:183–190. doi: 10.1002/cncr.21138. [DOI] [PubMed] [Google Scholar]