

Abstract
Apoptosis is a form of programmed cell death that is controlled at the mitochondrion by the BCL-2 family of proteins. While much has been learned about the structure and function of these proteins over the past two decades, the important goal of predicting cell fate decisions in response to toxic stimuli is largely unrealized. BH3 profiling is a functional approach that can be used to predict cellular responses to stimuli based on measuring the response of mitochondria to perturbation by a panel of BH3 domain peptides. BH3 profiling has proven useful in identifying and understanding cellular dependence on individual anti-apoptotic proteins like BCL-2 or MCL-1. Consequently, it can also be used to predict cellular response to chemotherapy agents such as ABT-737 that target these individual proteins.
Background on BCL-2 proteins
The B cell leukemia/lymphoma-2 (BCL-2) family of proteins interact at the crux of the mitochondrial pathway of apoptosis. Initially identified as a participant in the t(14;18) translocation of follicular lymphomas, BCL-2 was the first oncogene described that inhibited cell death [1-3]. Over the two decades since its discovery, multiple proteins with structural and functional similarities have been identified to comprise what is called the BCL-2 family of proteins. BCL-2 proteins are categorized broadly as anti- or pro-apoptotic. Homo- and heterodimerization of these proteins regulates apoptosis at the mitochondrion [4-8]. In very simplified terms, when the abundance of active pro-apoptotic proteins exceeds the binding capacity of anti-apoptotic proteins, apoptosis proceeds to permeabilization of the mitochondrial outer membrane, the hallmark of the mitochondrial pathway of apoptosis.
All family members share homology in one or more BCL-2 homology domains (called BH domains, numbered BH1 - BH4) that are essential for their function [6, 9]. Anti-apoptotic members such as BCL-2 and BCL-XL share homology in all four BH regions. A surface-exposed hydrophobic cleft formed by BH 1-3 participates in hetero- and homodimerization with the BH3 domain of other family members [10]. Multi-domain pro-apoptotic members BAX and BAK have conserved homology in BH1 – BH3 as well as a C-terminal membrane anchor and are the effectors of mitochondrial outer membrane permeabilization (MOMP) [11-12]. While they are monomers in healthy cells, BAX and BAK can be triggered to homo-oligomerize and participate in pores that cause MOMP, allowing the release of cytochrome c and other pro-apoptotic molecules that commit a cell to apoptosis [12-13]. Either multidomain pro-apoptotic protein is sufficient for release of cytochrome c, but at least one of the two is required for this function. The anti-apoptotic proteins, including BCL-2, MCL-1, BCL-w, BCL-XL and BFL1/A1, can inhibit apoptosis by sequestering monomeric activated BAX and BAK before they can oligomerize [14] (Figure 1).
Figure 1. Model of apoptotic control by BCL-2 family proteins.

Cellular damage induces upregulation of activator and/or sensitizer proteins. BAX and BAK oligomerization is then induced by direct binding of activators like BID and BIM leading to MOMP then cytochrome c release and subsequent cell death. Anti-apoptotic proteins inhibit apoptosis by binding and sequestering activators or activated BAX or BAK. Sensitizers can bind to anti-apoptotic proteins and displace pre-bound activators, or previously activated BAX or BAK, inducing BAX/BAK oligomerization and mitochondrial permeabilization. (adapted from Certo et al. [15]).
Other pro-apoptotic family members that share homology only in the BH3 domain, called BH3-only proteins, are subdivided as either activators or sensitizers. “Activators'”, such as BIM, BID and possibly PUMA, exert their pro-death function by interacting directly with BAX and BAK and induce the allosteric changes that lead to BAX and BAK homo-oligomerization [15-23]. Another way that anti-apoptotic proteins inhibit apoptosis is by binding and sequestering activators, preventing their activation of BAX and BAK. BH3-only proteins that lack activator function, called “sensitizers”, exert their pro-death function by competitive displacement of activator BH3-only proteins or activated BAX or BAK monomers from inhibitor anti-apoptotic proteins like BCL-2 [16] (Figure 1). These binding interactions involve the binding of the hydrophobic face of the amphipathic alpha-helical BH3 domain to the hydrophobic BH3-binding cleft in anti-apoptotic proteins. It is clear that the ability of an antiapoptotic protein like BCL-2 to inhibit subsequent death signaling in a cellular context will depend entirely on its not already being occupied by any one of many pro-apoptotic molecules. This probably explains why measurements of levels of single anti-apoptotic proteins are generally insufficient to predict response to toxic stimuli. As a result, some have employed systems biology approaches to understand cell fate decisions based on initial conditions of BCL-2 family proteins [24].
Early experiments examining the interactions among BCL-2 proteins revealed differential binding patterns. In the course of studying whether the BH3 domain of BH3-only proteins separated from the context of the whole protein could interact with multi-domain anti- and pro-apoptotic proteins, it was found that individual BH3 domains interacted differently [25]. For example BAD was found to bind to BCL-2 but not MCL-1. NOXA displayed an opposite binding pattern and BID, BIM or PUMA interacted with all anti-apoptotic proteins as well as BAX and BAK [15, 21, 26]. Experiments utilizing recombinant anti-apoptotic proteins and fluoroscein-tagged BH3 domain peptides in fluorescence polarization assays gave rise to the “binding code” [15] (Figure 2). The binding code revealed a selectivity of interaction among BCL-2 proteins. It may be that cells exploit this selectivity of interaction to finely tune individual cells to select stimuli.
Figure 2. The binding code.

Summary of the selective binding between BH3-only peptides and different antiapoptotic BCL-2 family members. + indicates moderate binding, ++ tight binding, and – no binding interaction based on fluorescent polarization binding studies (adapted from Certo et al. [15]).
BCL-2 proteins in cancer
A common observation that is nevertheless often overlooked is that many cancer cells that express large amounts of anti-apoptotic proteins are exquisitely chemosensitive [17-19]. Examples of this apparent paradox include acute lymphoblastic leukemia (ALL) and chronic lymphocytic leukemia (CLL), both of which consistently express large amounts of BCL-2 [16-17]. While cure is frequently obtained only in the case of ALL, both diseases show a very high rate of initial response to chemotherapy. If BCL-2 acts to inhibit cell death, how can this be? The answer lies in the understanding that commitment to apoptotic death depends up on levels of many BCL-2 family proteins, both pro- and anti-apoptotic. One can imagine, therefore, that if the BCL-2 in these cancers is largely occupied by pro-apoptotic proteins, there would be a negligible amount of protection against apoptosis afforded by the BCL-2 present. In the case of ALL and CLL, it is clear that these cancers simultaneously express large amounts of the pro-apoptotic activator BH3-only protein BIM which is sequestered by BCL-2 [16-17].
During oncogenesis, oncogene activation, metastasis and genomic instability are examples of common phenotypes which can induce pro-apoptotic signaling, including up-regulation of activator BH3-only proteins [15, 27-28]. In cancer cells such as ALL and CLL, which concomitantly overexpress anti-apoptotic proteins, apoptosis can be avoided because the highly expressed BCL-2 binds and sequesters the excess BIM thereby preventing BAX/BAK activation [16-17, 28]. These cells therefore depend on continued function of their anti-apoptotic BCL-2 to avoid apoptosis. Such cancer cells could thus be described as addicted to BCL-2. When substantial activators, or perhaps activated BAX or BAK, are present in cells but they are bound to anti-apoptotic proteins such as BCL-2 or MCL-1, and addiction to BCL-2 or MCL-1 ensues, we refer to these cells as being “primed for death” [4, 15, 18, 29-31]. If activators could be displaced from anti-apoptotic proteins in these primed cells, they would be predicted to cause apoptotic cell death. Indeed, as predicted by this model, the primed cancer cells in ALL and CLL very readily die via apoptosis when treated with a BCL-2 antagonist ABT-737 [16-17].
BH3 profiling
BH3 profiling was designed as a tool to predict cellular fate decisions and understand addiction to BCL-2 family proteins. The basis of BH3 profiling is to expose mitochondria to known concentrations of BH3 peptides and measure the resulting permeabilization of the outer mitochondrial membrane. As long as conditions are well-controlled, comparisons can then be made between the mitochondria of different cell lines, different tissues or subject to different perturbations. Initially, BH3 profiling was conducted with isolated mitochondria [15-18]. The read-out of results was cytochrome c release (Figure 3A). In intact mitochondria, cytochrome c is contained in the intermembrane space. When BAX/BAK are engaged and oligomerize, MOMP occurs and cytochrome c is released, which can be detected by ELISA or Western blot. Understanding the binding code provided the opportunity to selectively identify dependence on individual BCL-2 family members. For instance, a mitochondrion that was sensitive to the NOXA BH3 peptide would indicate a dependence on MCL-1, while one that was sensitive to BAD BH3 would be dependent on BCL-2 (or perhaps BCL-XL or BCL-w, which also interact with BAD BH3).
Figure 3. BH3 Profiling Assay Protocols.

Flow charts representing the various steps for BH3 profiling using cytochrome c ELISA (A), JC-1 in a plate reader format(B), and JC-1 with flow cytometry (C) (adapted from Ryan et al [33]).
An initial test venue for BH3 profiling was in cell lines overexpressing BCL-2 or MCL-1. We found that the technique could accurately identify when the cell was dependent on either individual protein [15]. Protein analysis showed that cellular dependence occurred only when upregulated BIM was sequestered by the relevant anti-apoptotic protein. Extending BH3 profiling to in vivo systems, a murine leukemia model that was demonstrated to be BCL-2 dependent in vivo via a conditional BCL-2 allele was subjected to BH3 profiling, revealing a BCL-2 dependent BH3 profile [15, 28]. The BCL-2 in this case was found to be in complex with BIM and PUMA BH3 only-proteins. A related MCL-1 dependent leukemia model demonstrates a distinct BH3 profile, with high NOXA activity and low BAD activity, consistent with MCL-1 dependence [29].
Other work has tested a series of lymphoma cell lines, and demonstrated that BH3 profiling correctly identifies those cell lines that are BCL-2 dependent based on correlation with response to the BCL-2 antagonist ABT-737 [18]. Throughout our work on many cell types, we have found an excellent correlation between a BH3 profile that indicates BCL-2 dependence and sensitivity to ABT-737. This is perhaps not surprising, since ABT-737, and its clinically-tested derivative, ABT-263, are essentially small molecule mimetics of the BAD BH3 domain, and the BAD BH3 is the key peptide indicating BCL-2 dependence in the BH3 profiling assay. We have also performed BH3 profiling of primary cancer cells, including ALL and CLL, and similarly found that a BH3 profile indicative of BCL-2 dependence was predictive of ABT-737 sensitivity [16-17]. These experiments were the first instance that suggested that BH3 profiling could be a useful diagnostic for clinical treatment. Since BH3 profiling is rapid, taking less than 4 hours from blood draw to profiling results, as well as accurate, it could potentially be used to predict clinical response to an antagonist of an anti-apoptotic protein.
Whole cell assay
BH3 profiling using isolated mitochondria can require between 107 to 109 cells depending on the number of mitochondria per cell. To make BH3 profiling clinically useful in situations where fewer cells are obtained, to improve upon the time it takes from patient biopsy to results, and allow use of using mixed cell populations, a whole cell assay was developed. First, a readout of cytochrome c release other than western blot or ELISA was sought. Testing revealed that the dual emission fluorescent probe JC-1 would be ideal. Reading a JC-1 fluorescent signal at A595, healthy mitochondria with an intact membrane potential and mitochondria where BAX/BAK have oligomerized causing the decline and eventual abolishment of the membrane potential could be distinguished. Since JC-1 is cell permeant and fluorescence is detectable using a fluorimeter or by flow cytometry the next obstacle to attack was delivery of the activator and sensitizer peptides to the cells. Since the binding of peptides to anti-apoptotic proteins is dependent on structure interactions, attachment to a cell-penetrating signal, such as TAT or Antennapedia domain, could change binding or induce off-target effects [32]. Therefore an alternative way to introduce peptides into the cell was sought. Since long term cell viability was not a concern, we turned to detergents. After evaluating multiple detergents and concentrations, ultra low concentrations of digitonin (0.001 – 0.005%) were found to permeabilize the plasma membrane without affecting the mitochondrial membrane potential in both cell lines and primary samples for up to 3 hours (Figure 3B) [33]. Further stabilization of the cells was then achieved by using a trehalose-based buffer for experiments [34] and the assay was then adapted to flow cytometry (Figure 3C). Overall, BH3 profiling results using isolated mitochondria and whole cells produce similar results, indicating that utilizing the trehalose and JC-1 does not affect the results (Figure 4). By adapting the assay to flow cytometry not only was the procedure time decreased but mixed populations of cells could then be used because cell surface markers could allow definition of subpopulations of interest. One example of its use is to distinguish the BH3 profiles of distinct T-cell subsets of the thymus, explaining the great sensitivity of CD4+ CD8+ lymphocytes to apoptotic stimuli [33]. In the laboratory we now find that we can perform BH3 profiling on any normal or malignant tissue once it is reduced to a single cell suspension, and have had success in analyzing cell subsets even when they represent a very small fraction of the total cells, as long as we can identify cell surface markers to distinguish them.
Figure 4. BH3 profiling using isolated mitochondria and whole cell assays produce comparable results.
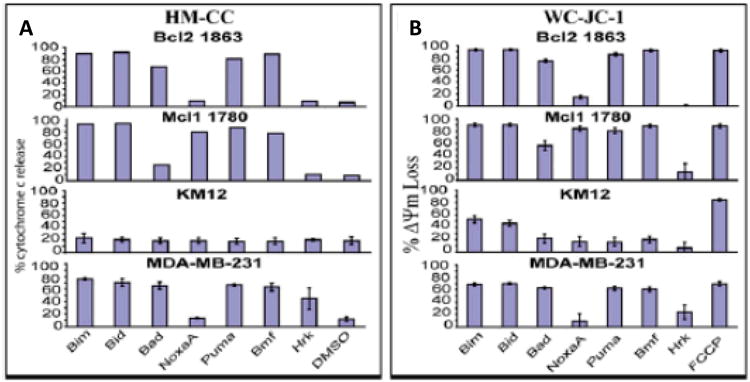
BCL-2 1863, MCL-1 1780 and MDA-MB-231 cell lines were used to either isolate mitochondria via a heavy membrane preparation and profiled with 100μM peptides either using a cytochrome c ELISA (HM-CC) (A) or whole cells used in the JC-1 (WC-JC-1) profiling assay (B). Two Upper panels in A represent data from a single experiment while all the rest triplicates; means shown with bars for SD (adapted from Ryan et al [33]).
Conclusions
Apoptosis transpires via the dynamic interaction of BCL-2 proteins at the mitochondrion. Dependence, or addiction, to an anti-apoptotic protein can lead to a cancer phenotype and priming of the mitochondria can allow initial chemosensitivity. Both situations can be monitored conveniently by BH3 profiling, the novel technique that assesses the aggregate state of BCL-2 proteins at the mitochondrion (Figure 4). After multiple iterations, BH3 profiling is now conducted with whole cells and with heterogenous samples. The data from this assay not only is informative from a basic science standpoint but also is a step toward personalized medicine where response to a drug can be evaluated prior to administration.
References
- 1.Bakhshi A, Jensen JP, Goldman P, Wright JJ, McBride OW, Epstein AL, Korsmeyer SJ. Cloning the chromosomal breakpoint of t(14;18) human lymphomas: clustering around JH on chromosome 14 and near a transcriptional unit on 18. Cell. 1985;41:899–906. doi: 10.1016/s0092-8674(85)80070-2. [DOI] [PubMed] [Google Scholar]
- 2.Cleary ML ML, Sklar J. Nucleotide sequence of a t(14;18) chromosomal breakpoint in follicular lymphoma and demonstration of a breakpoint-cluster region near a transcriptionally active locus on chromosome 18. Proceedings of the National Academy of Sciences of the United States of America. 1985;82:7439–7443. doi: 10.1073/pnas.82.21.7439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsujimoto Y, Gorham J, Cossman J, Jaffe E, Croce CM. The t(14;18) chromosome translocations involved in B-cell neoplasms result from mistakes in VDJ joining. Science. 1985;229:1390–1393. doi: 10.1126/science.3929382. [DOI] [PubMed] [Google Scholar]
- 4.Brunelle JK, Letai A. Control of mitochondrial apoptosis by the Bcl-2 family. J Cell Sci. 2009;122:437–441. doi: 10.1242/jcs.031682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Green DR, Evan GI GI. A matter of life and death. Cancer cell. 2002;1:19–30. doi: 10.1016/s1535-6108(02)00024-7. [DOI] [PubMed] [Google Scholar]
- 6.Cory D, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nature reviews Cancer. 2002;2:647–656. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 7.Liang H, Fesik SW. Three-dimensional structures of proteins involved in programmed cell death. Journal of molecular biology. 1997;274:291–302. doi: 10.1006/jmbi.1997.1415. [DOI] [PubMed] [Google Scholar]
- 8.Rutledge SE, Chin JW, Schepartz A. A view to a kill: ligands for Bcl-2 family proteins. Current opinion in chemical biology. 2002;6:479–485. doi: 10.1016/s1367-5931(02)00352-6. [DOI] [PubMed] [Google Scholar]
- 9.Petros AM, Olejniczak ET, Fesik SW. Structural biology of the Bcl-2 family of proteins. Biochimica et biophysica acta. 2004;1644:83–94. doi: 10.1016/j.bbamcr.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 10.Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M, Yoon HS, Shuker SB, Chang BS, Minn AJ, Thompson CB, Fresik SW. Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science. 1997;275:983–986. doi: 10.1126/science.275.5302.983. [DOI] [PubMed] [Google Scholar]
- 11.Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, Chen Y, Wei M, Eng VM, Adelman DM, Simon MC, Ma A, Golden JA, Evan G, Korsmeyer SJ, MacGregor GR, Thompson CB. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Molecular Cell. 2000;6:1389–1399. doi: 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng EH, Wei MC, A HY, Weiler S, Flavell RA, Mak TW, Lindsten T, Korsmeyer SJ. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Molecular Cell. 2001;8:705–711. doi: 10.1016/s1097-2765(01)00320-3. [DOI] [PubMed] [Google Scholar]
- 14.Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 15.Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer SJ, Armstrong SA, Letai A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–365. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 16.Del Gaizo Moore V, Brown JR, Certo M, Love TM, Novina CD, Letai A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest. 2007;117:112–121. doi: 10.1172/JCI28281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Del Gaizo Moore V, Schlis KD, Sallan SE, Armstrong SA, Letai A. BCL-2 dependence and ABT-737 sensitivity in acute lymphoblastic leukemia. Blood. 2008;111:2300–2309. doi: 10.1182/blood-2007-06-098012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp MA, Letai A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell. 2007;12:171–185. doi: 10.1016/j.ccr.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 19.Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler SS, Koresmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–192. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- 20.Cartron PF, Gallenne T, Bougras G, Gautier F, Manero F, Vusio P, Meflah K, Vallette FM, Juin P. The first alpha helix of Bax plays a necessary role in its ligand-induced activation by the BH3-only proteins Bid and PUMA. Molecular Cell. 2004;16:807–818. doi: 10.1016/j.molcel.2004.10.028. [DOI] [PubMed] [Google Scholar]
- 21.Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, Newmeyer DD. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Molecular Cell. 2005;17:525–535. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 22.Zong WX, Lindsten T, Ross AJ, MacGregor GR, Thompson CB. BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes & Development. 2001;15:1481–1486. doi: 10.1101/gad.897601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gavathiotis E, Suzuki M, Davis ML, Pitter K, Bird GH, Katz SG, Tu H, Kim H, Cheng E, Tjandra N, Walensky LD. BAX activation is initiated at a novel interaction site. Nature. 2008;455:1076–1081. doi: 10.1038/nature07396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Albeck JG, Burke JM, Aldridge BB, Zhang M, Lauffenburger DA, Sorger PK. Quantitative analysis of pathways controlling extrinsic apoptosis in single cells. Mol Cell. 2008;30:11–25. doi: 10.1016/j.molcel.2008.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature. 2003;426:671–676. doi: 10.1038/nature02067. [DOI] [PubMed] [Google Scholar]
- 26.Chen L, Willis SA, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DCS. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 27.Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, Waters CM, Penn LZ, Hancock DC. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- 28.Letai A, Beard C, Sorcinelli M, Korsmeyer SJ. Anti-apoptotic BCL-2 is required for maintenance of a model leukemia. Cancer Cell. 2004;6:241–249. doi: 10.1016/j.ccr.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 29.Brunelle JK, Ryan J, Yecies D, Opferman JT, Letai A. MCL-1-dependent leukemia cells are more sensitive to chemotherapy than BCL-2-dependent counterparts. J Cell Biol. 2009;187:429–442. doi: 10.1083/jcb.200904049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Del Gaizo Moore V, Letai A. Rational design of therapeutics targeting the BCL-2 family: are some cancer cells primed for death but waiting for a final push? Adv Exp Med Biol. 2008;615:159–175. doi: 10.1007/978-1-4020-6554-5_8. [DOI] [PubMed] [Google Scholar]
- 31.Letai AG. Diagnosing and exploiting cancer's addiction to blocks in apoptosis. Nat Rev Cancer. 2008;8:21–132. doi: 10.1038/nrc2297. [DOI] [PubMed] [Google Scholar]
- 32.Schimmer AD, Hedley DW, Chow S, Pham NA, Chakrabartty A, Bouchard D, Mak TW, Trus MR, Minden MD. The BH3 domain of BAD fused to the Antennapedia peptide induces apoptosis via its alpha helical structure and independent of Bcl-2. Cell Death Differ. 2001;8:725–733. doi: 10.1038/sj.cdd.4400870. [DOI] [PubMed] [Google Scholar]
- 33.Ryan JA, Brunelle JK, Letai A. Heightened mitochondrial priming is the basis for apoptotic hypersensitivity of CD4+ CD8+ thymocytes. Proc Natl Acad Sci U S A. 2010;107:12895–12900. doi: 10.1073/pnas.0914878107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamaguchi R, Andreyev A, Murphy AN, Perkins GA, Ellisman MH, Newmeyer DD. Mitochondria frozen with trehalose retain a number of biological functions and preserve outer membrane integrity. Cell Death Differ. 2007;14:616–624. doi: 10.1038/sj.cdd.4402035. [DOI] [PubMed] [Google Scholar]