

Abstract
Neuromyelitis optica (NMO) is an autoimmune demyelinating disease associated with recurrent episodes of optic neuritis and transverse myelitis, often resulting in permanent blindness and/or paralysis. The discovery of autoantibodies (AQP4-IgG) that target aquaporin-4 (AQP4) has accelerated our understanding of the cellular mechanisms driving NMO pathogenesis. AQP4 is a bidirectional water channel expressed on the plasma membranes of astrocytes, retinal Müller cells, skeletal muscle, and some epithelial cells in kidney, lung and the gastrointestinal tract. AQP4 tetramers form regular supramolecular assemblies at the cell plasma membrane called orthogonal arrays of particles. The pathological features of NMO include perivascular deposition of immunoglobulin and activated complement, loss of astrocytic AQP4, inflammatory infiltration with granulocyte and macrophage accumulation, and demyelination with axon loss. Current evidence supports a causative role of AQP4-IgG in NMO, in which binding of AQP4-IgG to AQP4 orthogonal arrays on astrocytes initiates complement-dependent and antibody-dependent cell-mediated cytotoxicity and inflammation. Immunosuppression and plasma exchange are the mainstays of therapy for NMO optic neuritis. Novel therapeutics targeting specific steps in NMO pathogenesis are entering the development pipeline, including blockers of AQP4-IgG binding to AQP4 and inhibitors of granulocyte function. However, much work remains in understanding the unique susceptibility of the optic nerves in NMO, in developing animal models of NMO optic neuritis, and in improving therapies to preserve vision.
Keywords: NMO, optic nerve, aquaporin-4, Devic’s disease, neuroinflammation
1. Introduction
1.1 Overview
NMO is a multifocal, inflammatory demyelinating disorder of the central nervous system that preferentially affects the optic nerves and spinal cord. Isolated optic neuritis (ON) is a common presenting sign in NMO as well as in the more common demyelinating disorder multiple sclerosis (MS). However, while most MS patients recover significant visual acuity following ON, NMO patients often manifest severe visual loss. The mechanisms producing these distinct clinical outcomes are beginning to be clarified. The discovery of a highly specific serum immunoglobulin G autoantibody (AQP4-IgG) that targets AQP4 (Lennon et al., 2004, 2005), the major plasma membrane water channel on astrocytes, has established NMO as a distinct clinical entity from MS and suggested AQP4 as a specific immunologic target in NMO ON. Recent research has demonstrated a pathogenic role of AQP4-IgG and led to the development of new NMO treatment strategies aimed at preventing AQP4-IgG binding to AQP4 and downstream mechanisms of neuropathology. Herein, we review the major clinical aspects of NMO ON, recent progress in elucidating NMO pathogenesis mechanisms, and emerging therapeutics.
1.2 History
In 1870, Sir Thomas Albutt described the syndrome now known as NMO. Albutt observed that some patients with co-existent transverse myelitis (TM) and ON tended to have a severe disease course. In 1894, Eugène Devic and his graduate student, Fernand Gault, summarized 16 patients with unilateral or bilateral vision loss, who within weeks developed spastic weakness of the limbs, loss of sensation, and incontinence (Devic, 1894; Gault, 1894). Initially described as a monophasic disorder of bilateral ON and TM, NMO is now recognized as a relapsing disorder with protean manifestations (O’Riordan et al., 1996; Wingerchuk et al., 1999; Popescu et al., 2011; Iorio et al., 2011).
2. Optic nerve anatomy
The human optic nerve consists of 1.2 million, parallel cell axons that extend from ganglion cells in the inner retina. The ganglion cells synapse mostly in the lateral geniculate nucleus (Figure 1), and to a lesser extent in pretectum and superior colliculus of the midbrain and the suprachiasmic nuclei of the hypothalamus. The intraorbital portion of the optic nerve is surrounded by dura, arachnoid, and pia mater. The dura (the outmost sheath) is continuous with the sclera anteriorly and fuses with the periosteum and annulus of Zinn at the orbital apex. In the orbit, the optic nerve tissue is perfused by centripetally penetrating capillaries that are contained within connective tissue invested by the pia that divides the intraorbital optic nerve into septae. The blood supply of the optic nerve may be of particular importance in NMO because major pathology occurs in the perivascular space.
Figure 1.
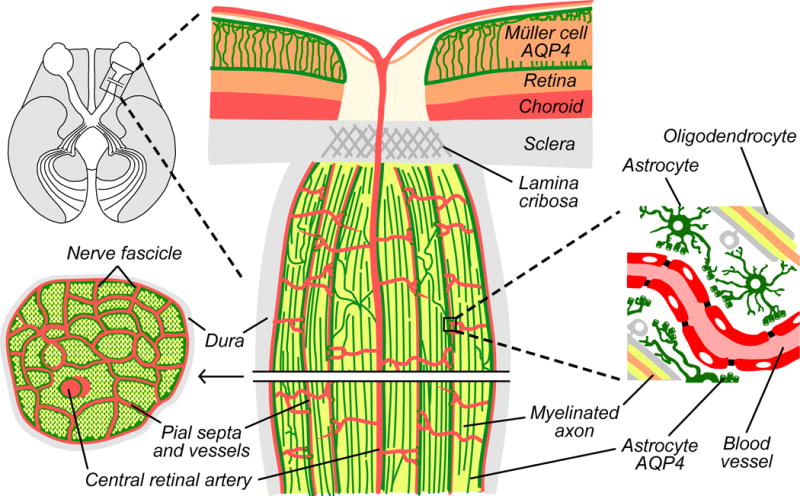
Schematic of optic nerve anatomy. Top left, axial view of the brain through the visual pathway. The majority of ganglion cells synapse in the lateral geniculate nucleus, as illustrated. Center, longitudinal section through the posterior pole of the globe and optic nerve. Inner retinal Müller cells express AQP4 (green), whereas astrocytes of the unmyelinated optic nerve head do not. Behind the lamina cribosa, astrocytes express AQP4, especially around branches of pial vessels and the central retinal artery. Astrocytes separate myelinated axons from pial surfaces and from one another. Bottom left, cross-section through retrolaminar optic nerve, showing fascicles of parallel myelinated axons (yellow) surrounded by AQP4-expressing astrocytes (green). Right, retrolaminar astrocytes protect myelinated axons by forming end-feet (with high AQP4 expression) over blood vessels.
Posterior to the collagenous lamina cribrosa, oligodendrocytes wrap optic nerve axons concentrically with myelin, similar to white matter tracts in the brain and spinal cord. Astrocytes are abundant throughout the optic nerve and their characteristics vary by region. Filamentous astrocytes of the unmyelinated optic nerve head have weak AQP4 expression, whereas fibrous astrocytes, which extend posteriorly from the lamina, strongly express AQP4 (Triviño et al., 1996; Nagelhus et al., 1998; Oyama et al., 2006). Retrolaminar astrocytes serve as the scaffolding that protects myelinated axons by forming end-feet over blood vessels and preventing contact with connective pial septae. Some astrocytes extend transverse processes across the full width of the optic nerve, while others project longitudinal processes in parallel with axons (Sun et al., 2009). Astrocytes express AQP4, the target of AQP4-IgG. Astrocyte disruption precedes the profound demyelination and neuronal damage observed in some NMO lesions (Parratt and Prineas, 2010).
3. Clinical features of NMO
3.1 Epidemiology
NMO has a low prevalence, estimated at 0.3–4.4 per 100,000. NMO accounts for a small proportion of demyelinating disease in Caucasians (1–2%), but a much larger percentage in Asians (20–48%) (Kira, 2003; Cabre et al., 2005; Cabrera-Gomez et al., 2009; Asgari et al., 2011; Cossburn et al., 2012). In the United States, patients of African descent are also overrepresented, making up over one-third of cases (Mealy et al., 2012). Women are preferentially affected with a female: male ratio of nearly 10:1. The age of onset varies widely, with 1/6 of AQP4-IgG seropositive cases occurring in pediatric and elderly patients (Quek et al., 2012). The median age of onset of NMO is about 40 years (Wingerchuk et al., 2007; Mealy et al., 2012). As with other autoimmune disorders, there are potential complex genetic contributions to NMO. Several HLA variants and other polymorphisms have been identified in association with NMO (Zéphir et al., 2009; Wang et al., 2011; Asgari et al., 2012), and about 3% of patients have affected relatives (Matiello et al., 2010). NMO is associated 10–40% of the time with other autoimmune disorders, including myasthenia gravis, systemic lupus erythematous, Sjögren’s syndrome, and celiac disease (Pittock et al., 2008; Bergamaschi et al., 2009; Wandinger et al., 2010; Jarius et al., 2011; Leite et al., 2012).
3.2 Neuro-ophthalmic manifestations
Approximately one-half of NMO patients present with isolated ON, of which about 20% is bilateral (Wingerchuk et al., 1999; Papais-Alvarenga et al., 2008). Profound and persistent visual loss is a hallmark of ON in NMO, but not in MS (Beck et al., 1992). Eighty percent of NMO eyes experience severe loss of visual acuity (< 20/200) during an acute attack, compared with 36% in MS. Sixty percent of NMO patients experience unilateral or bilateral blindness at a median of 7.7 years after disease onset, compared with 4% of MS ON patients at 15-year follow up (Wingerchuk et al., 1999; Optic Neuritis Study Group, 2008). In NMO, the median time from the onset of ON to ipsilateral blindness is 2 years, and to contralateral ON is 3 years (Merle et al., 2007). Deterioration of visual acuity in pediatric patients may be more rapid than in adult NMO patients (Collongues et al., 2010).
NMO ON may be distinguished from MS ON by imaging and functional measures. While magnetic resonance imaging (MRI) shows changes typical of acute ON, such as optic nerve enlargement, T2 hyperintensity, and gadolinium enhancement, the lesions are often more extensive and likely to involve the optic chiasm or adjacent hypothalamus (Figure 2A; Li et al., 2008). NMO ON generally causes more severe visual field defects than MS ON (Fernandes et al., 2012a), and, given its potential to involve the optic chiasm and tracts, may manifest with bitemporal or homonymous visual field defects (Raz et al., 2010; Costa et al., 2007; Romero et al., 2012). With the more routine serologic testing of individuals with acute optic neuritis, the spectrum of visual symptoms associated with NMO ON may expand over time.
Figure 2.
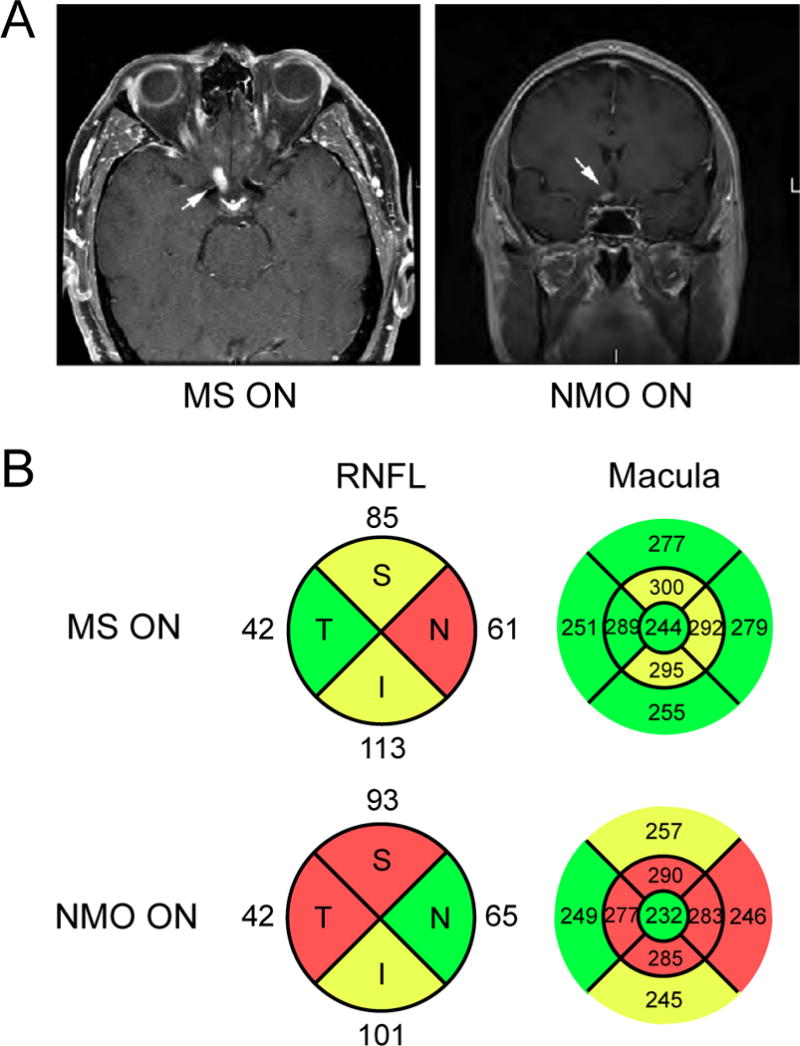
MRI and OCT imaging of NMO optic neuritis. A. Axial and coronal post-contrast T1-weighted images of acute NMO optic neuritis demonstrate acute inflammation of the prechiasmatic right optic nerve (arrows). B. Spectral domain OCT of the peripapillary RNFL and macula of eyes affected by MS- and NMO-associated ON. Note the increased retinal thinning in the macula affected by NMO ON despite a comparable amount of peripapillary RNFL loss (red: < 1st percentile; yellow: < 5th percentile; green: 5th–95th percentile). Thickness shown in microns.
Optical coherence tomography (OCT) shows more substantial peripapillary retinal nerve fiber layer (RNFL) loss in NMO than MS in both monophasic and recurrent ON (Figure 2B; Merle et al., 2008; Ratchford et al., 2009; Naismith et al., 2009; Green and Cree, 2009). In contrast to the temporal RNFL loss that is typical of MS, substantial atrophy is seen in the superior and inferior quadrants in NMO (Naismith et al., 2009). More recent OCT studies have evaluated macular thinning as an additional indirect measure of axonal loss. The macular nerve fiber and retinal ganglion cell layers show similar atrophy in NMO and MS, consistent with the loss of temporal RNFL fibers seen in both disorders. However, the inner nuclear layer manifests significant thickening in NMO compared to MS (Fernandes et al., 2012b; Monteiro et al., 2012). This finding, together with the recognition of inner retinal microcystic changes in 25% of NMO patients (Gelfand et al., 2012; Sotirchos et al., 2012), may suggest concurrent retinal pathology. Similar findings have been observed in MS ON and other non-inflammatory optic neuropathies; therefore further characterization of this pathology will be needed to identify an underlying mechanism (Gelfand et al., 2012; Saidha et al., 2012; Barboni et al., 2013).
3.3 Diagnostic criteria and testing
According to the current criteria (Wingerchuk et al., 2006), definite NMO requires the presence of ON and TM, and at least two of three supportive criteria that enhance diagnostic specificity (MRI evidence of a contiguous spinal cord lesion in at least 3 segments; brain MRI not diagnostic of MS; and AQP4-IgG seropositivity). These criteria allow the diagnosis of definite NMO without detectable AQP4-IgG. Current flow cytometry and cell-based immunofluorescence assays for AQP4-IgG have high sensitivity (73–77%) and specificity (approaching 100%) (Waters et al., 2012). The existence of seronegative NMO may be due to the limited sensitivity of currently available antibody detection assays and/or the presence of alternative disease-causing autoantibodies (Jarius et al., 2012). Autoantibodies to myelin oligodendrocyte glycoprotein have been identified in some pediatric patients with recurrent ON, and in some NMO patients that are seronegative for AQP4-IgG (Mader et al., 2011; Rostasy et al., 2012).
Seropositive patients with isolated ON or TM are currently classified as having NMO spectrum disorder (NMOSD), which carries a high risk of eventual conversion into definite NMO (Wingerchuk et al., 2007). In one series, more than 50% of seropositive patients who initially presented with TM developed either ON (and thus definite NMO) or recurrent TM within one year (Weinshenker et al., 2006). Several studies report a short interval between the sentinel and subsequent clinical attacks in AQP4-IgG seropositive NMO patients (O’Riordan et al., 1996; Wingerchuk et al., 1999; Merle et al., 2007; Rivera et al., 2008).
Debate exists about the role for routine AQP4-IgG serum testing in patients with acute monosymptomatic ON given the low frequency (3–5%) of AQP-IgG positivity in this group (Jarius et al., 2010a; Petzold et al., 2010; Costa et al., 2012). Because NMO carries a poor prognosis and warrants aggressive immunotherapy, identifying patients with NMOSD could potentially enable earlier treatment. Furthermore, standard MS therapeutics such as interferons, fingolimod and natalizumab might be deleterious in NMO (Palace et al., 2010; Kleiter et al., 2012; Min et al., 2012). It is generally recommended that serologic testing be performed at minimum in those ON patients with MRI changes atypical of MS, with bilateral or recurrent ON, with poor visual recovery, or with ON associated with autoimmune disease (Galetta and Cornblath, 2010). Additional characteristics of acute ON attacks, include the presence of longitudinally extensive enhancement on MRI and the absence of pain, may provide additional rationale for obtaining AQP4-IgG testing (Papais-Alvarenga et al., 2008; Daily et al., 2013).
3.4 Current therapy
To date, no acute therapy has demonstrated significant benefit in improving visual outcome in NMO or preventing optic nerve atrophy. Most OCT studies have demonstrated that RNFL loss in NMO accrues from discrete attacks of ON in the absence of additional demyelination in the optic radiations or visual cortex, whereas in MS there may be gradual visual decline due to repeated subclinical injury in these regions during the relapsing and secondary progressive phases of disease (Ratchford et al., 2009; Popescu et al., 2010; Syc et al., 2012). These distinct patterns of retinal neuronal loss suggest that effective treatment of acute NMO ON might ameliorate vision loss and improve outcomes.
Current treatment regimens for acute attacks of ON include corticosteroids and plasma exchange (PE). In the Optic Neuritis Treatment Trial, high dose intravenous methylprednisolone (IVMP) (1000 mg daily for 3 days) resulted in faster recovery, enhanced contrast sensitivity, and improved color vision in patients with idiopathic acute ON at 6 months; however, the improvement was not maintained at 1 year. While high dose IVMP has shown no benefit in reducing post-inflammatory optic atrophy following idiopathic ON (Hickman et al., 2003), one study suggested that early administration of IVMP might limit optic atrophy following NMO ON (Nakamura et al., 2010).
Clinical observations have suggested benefit for PE following high-dose IVMP in cases of refractory vision loss or following NMO exacerbations (Ruprecht et al., 2004; Watanabe et al., 2007). A recent nonrandomized study of NMO/NMOSD patients treated early after onset of inaugural ON demonstrated long-term visual acuity gains and preservation of peripapillary RNFL in patients treated with sequential IVMP and PE compared to IVMP alone (Merle et al., 2012). The benefit of PE may be due in part to reduced circulating AQP4-IgG, though seropositivity is not critical to PE effectiveness (Jarius et al., 2008; Merle et al., 2012). Anecdotal evidence suggests that PE might stabilize the clinical course in patients with steroid-refractory NMO (Khatri et al., 2012).
Intravenous immunoglobulin, which is frequently used in other antibody-mediated neurological diseases, has not demonstrated clear benefit in treating acute or chronic vision loss associated with ON (Noseworthy et al., 2001; Roed et al., 2005; Magraner et al., 2012). Immunosuppressive drugs such as azathioprine, mycophenolate mofetil, and rituximab are used as long-term preventative NMO therapies, but are generally not used to treat acute attacks (Jacob et al., 2008, 2009; Costanzi et al., 2011; Kim et al., 2011).
4. Pathogenesis mechanisms
4.1 Overview
An expanding body of evidence supports a pathogenic role of AQP4-IgG in NMO. NMO lesions show perivascular deposition of immunoglobulin and complement, corresponding to areas of high AQP4 expression. Binding of AQP4-IgG to AQP4 on astrocytes activates antibody effector function, resulting in astrocyte destruction. Early human NMO lesions show loss of AQP4 and glial fibrillary acidic protein (GFAP) in the absence of significant myelin loss, suggesting early and isolated astrocyte damage. Release of inflammatory factors, including cytokines and chemokines, results in disruption of the blood-brain barrier and infiltration by neutrophils, eosinophils, and macrophages. Secondary injury to oligodendrocytes results in demyelination, axonal injury, and, ultimately, cavitation, necrosis, and gliosis (Mandler et al., 1993; Lucchinetti et al., 2002; Misu et al., 2007; Parratt and Prineas, 2010). The chronology of astrocyte injury leading to secondary myelin loss is supported by data from mouse models (Saadoun et al., 2010). Though complement dependent cytotoxicity (CDC) is probably the major initiating mechanism in NMO, other pathogenic mechanisms triggered by AQP4-IgG have been proposed, as discussed below.
4.2 AQP4, the target of AQP4-IgG
AQP4 is a water-transporting integral membrane protein that was cloned in 1994 from rat lung (Hasegawa et al., 2004) and deleted in mice in 1997 (Ma et al., 1997) (Figure 3). X-ray structure analysis shows that each AQP4 monomer contains 6-helical, membrane-spanning domains and two short helical segments surrounding a narrow aqueous pore (Ho et al., 2009). AQP4 monomers form stable tetramers, which further aggregate in cell plasma membranes to form supramolecular assembles called orthogonal arrays of particles (OAPs) (Yang et al., 1996; Rossi et al., 2012a). Human AQP4 is expressed in two isoforms: a long (M1) isoform with translation initiation at Met-1, and a short (M23) isoform with translation initiation at Met-23 (Yang et al., 1995). Astrocytes express both isoforms, which coassemble in heterotetramers (Tajima et al., 2010). AQP4 assembly into OAPs is driven by intermolecular N-terminus interactions between M23-AQP4 monomers involving residues just downstream of Met-23 (Crane and Verkman, 2009). AQP4 OAP assembly appears to be crucial in NMO pathogenesis, as AQP4-IgG generally binds to OAPs with much greater affinity than to individual AQP4 tetramers (Crane et al., 2011), and AQP4 OAPs greatly enhance CDC through multivalent binding of complement protein C1q to clustered AQP4-IgG bound to OAPs (Phuan et al., 2012).
Figure 3.
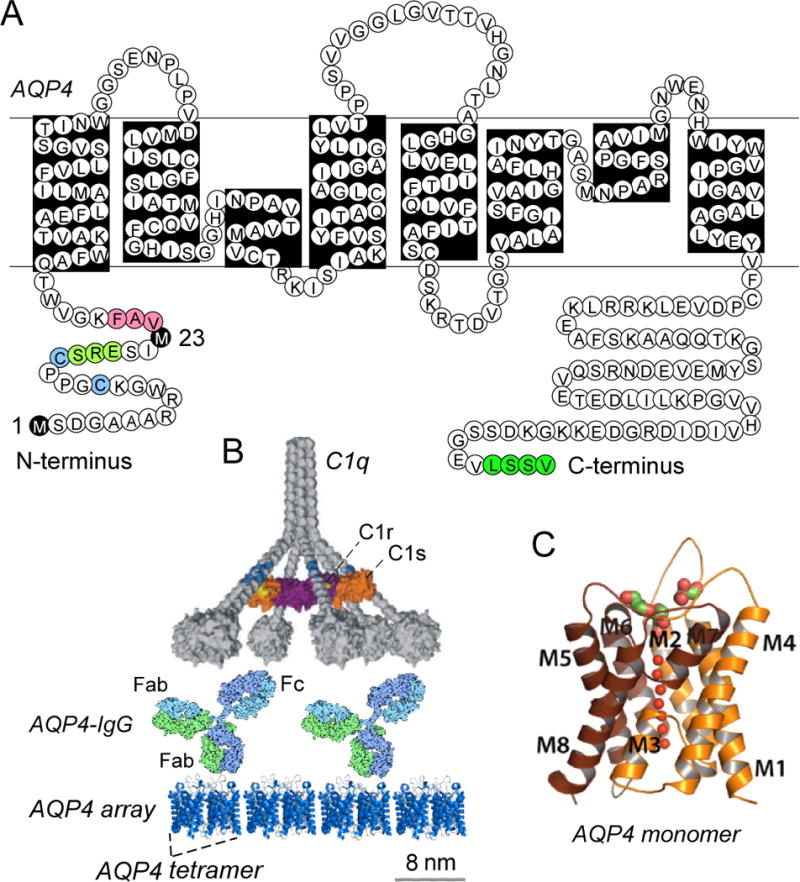
AQP4, the target of AQP4-IgG autoantibodies. A. Amino acid sequence of human AQP4 showing Met-1 and Met-23 translation inhibition sites (black), residues involved in intermolecular N-terminus associations to form OAPs (pink); residues preventing OAP formation by M1-AQP4 (light green); cysteines involved in palmitoylation-regulated OAP assembly (blue); and C-terminus PDZ domain (dark green). B. CDC requires AQP4 assembly in OAPs. Multivalent binding of C1q to Fc regions of clustered AQP4-IgG on AQP4 OAPs. AQP4 tetramers shown with a generic IgG antibody and C1 on the same size scale. C. Crystal structure of human AQP4 (PDB, 3GD8).
AQP4 is expressed in astrocytes throughout the CNS, including brain, spinal cord and optic nerve, as well as in skeletal muscle and in epithelial cells in kidney (collecting duct), stomach (parietal cells), airways (surface epithelia) and glands (acinar epithelia) (Frigeri et al., 1995). In brain, AQP4 is concentrated in astrocyte end-feet at pial and ependymal surfaces in contact with the CSF and blood vessels (Nielsen et al., 1997). AQP4 is weakly expressed in the non-myelinated optic nerve head compared with the myelinated retrolaminar optic nerve (Li et al., 2002; Mizokami et al., 2011). AQP4 is also highly expressed in astrocyte-like Müller cells in the inner retina and in the ciliary epithelium (Hamann et al., 1998).
The biological roles of AQP4 have been elucidated from phenotype analysis of transgenic mice lacking AQP4. Though AQP4 knockout mice have normal appearance, survival, growth and neuroanatomy, they manifest interesting phenotypes when stressed. Outside of the CNS they manifest a mild defect in maximum urinary concentrating ability (Ma et al., 1997), but normal skeletal muscle, lung and gastric functions (reviewed in Verkman, 2011). AQP4 knockout mice also manifest electrophysiological evidence of defective auditory (Li et al., 2001) and olfactory (Lu et al., 2008) signal transduction. In brain, AQP4 is involved in water movement and brain edema (Manley et al., 2000; Papadopoulos et al., 2004), neuroexcitation (Binder et al., 2006), astrocyte migration (Saadoun et al., 2005; Auguste et al., 2007) and neuroinflammation (Li et al., 2011). The water transporting function of AQP4 in astrocytes is probably responsible for each of these functions. AQP4 knockout mice show reduced b-wave amplitude and latency by electroretinography, without demonstrable abnormalities in retinal or optic nerve morphology (Li et al., 2002).
4.3 Cellular consequences of AQP4-IgG binding to AQP4
In general, antibody binding to its cellular target can cause: (i) altered target function; (ii) target internalization; (iii) CDC; and/or (iv) antibody-dependent cell-mediated cytotoxicity (ADCC). AQP4 functions as a bidirectional, water-selective transporter. A recent study reported reduced AQP4 water permeability following AQP4-IgG binding (Hinson et al., 2012); however, data from our lab (Rossi et al., 2012b) and others (Nicchia et al., 2009; Melamud et al., 2012;) showed no inhibition of AQP4 water permeability. AQP4-IgG can induce AQP4 internalization in transfected cell cultures and some astrocyte culture models (Hinson et al., 2008), but does not cause internalization in mouse brain in vivo (Ratelade et al., 2011a); AQP4 internalization, if it occurred, would be a protective during lesion formation.
Though CDC is probably a major mechanism in NMO pathogenesis, the importance of ADCC has become apparent. AQP4-IgG together with NK cells cause death of AQP4-transfected cells and astrocyte cultures through ADCC. In ADCC, NK or other effector cells bind to the Fc region of AQP4-IgG, resulting in release of toxic perforins and granzymes. Intracerebral injection of AQP4-IgG and NK-cells in mice produced NMO-like lesions with astrocyte injury, but without myelin loss (Ratelade et al., 2012). Though NK-cells are rarely seen in human NMO lesions (Saadoun et al., 2012a), neutrophils, eosinophils and macrophages are abundant in NMO lesions. Each of these cell types can cause ADCC, as well as participate in complement-dependent cell-mediated cytotoxicity (CDCC) through enhanced phagocytosis and activated complement (anaphalotoxin)-induced degranulation. Anaphalotoxins produced in NMO lesions by CDCC are potent chemoattractants for circulating granulocytes, monocytes, and macrophages. Studies in mouse models and spinal cord slice cultures have implicated the involvement of neutrophils (Zhang et al., 2011; Saadoun et al., 2012b) and eosinophils (Zhang and Verkman, 2013) in exacerbating NMO pathology through the release of neutrophil proteases and eosinophil granule toxins. These studies support an important role for ADCC in NMO pathogenesis, suggesting that therapies targeting CDC exclusively may have limited efficacy.
Microglia/macrophages are activated in the optic nerve and retina following optic nerve inflammation or injury (Wohl et al., 2010; Fairless et al., 2012; Roh et al., 2012), producing a variety of potentially destructive cytokines and trophic factors (reviewed in Cui et al., 2009). In mouse experimental autoimmune encephalomyelitis (EAE), for example, marked microglial activation in optic nerve coincides with onset of visual dysfunction (Matsunaga et al., 2012). In spinal cord slice cultures, microglia are not essential for lesion formation; however, addition of macrophages or TNF-α (a cytokine that is synthesized and released from astrocytes and microglia in the CNS), or pre-treatment with LPS (which activates endogenous microglia), greatly potentiated NMO lesions (Zhang et al., 2011). Microglial activation might thus represent another cellular mechanism of optic nerve damage in NMO.
4.4 Functional consequences of astrocyte cytotoxicity
While the antibody- and complement-dependent cellular mechanisms described above can produce secondary oligodendrocyte and axonal toxicity in NMO lesions, disrupted astrocyte function might also contribute to neuronal and oligodendrocyte damage. Astrocytes support neurotransmission by clearing extracellular potassium and water, especially near the non-myelinated nodes of Ranvier, where electrical excitation is concentrated. Alexander disease, the prototype genetic disorder of astrocytes caused by mutations in GFAP and associated with intracellular accumulation of GFAP (known as Rosenthal fibers), is remarkable for diffuse demyelination with relative axonal preservation (Brenner et al., 2001; Mignot et al., 2004; Sawaishi, 2009). Interestingly, aged mice that lack GFAP manifest impaired optic nerve and spinal cord myelination (Liedtke et al., 1996), suggesting an important role for astrocytes in the maintenance of myelination. Astrocytes are involved in an extensive glial network involving astrocyte-astrocyte and astrocyte-oligodendrocyte coupling through homotypic and heterotypic connexin (Cx) gap junction proteins, respectively.
Gap junctions might represent important mediators of myelin and axonal damage in NMO and other astrocyte-mediated diseases (reviewed in Cotrina and Nedergaard, 2012). Astrocyte Cx30 and Cx43, which are responsible for astrocyte-astrocyte and astrocyte-oligodendrocyte coupling, are lost at the glia limitans in NMO-like white matter lesions in animal models (Sharma et al., 2010). Still-myelinated human NMO optic nerve lesions also manifest vacuolated edema within myelin (Parratt and Prineas, 2010), similar to the gross myelin vacuolization in double-knockout mice lacking the heterotypic astrocyte-oligodendrocyte gap junction proteins Cx32 and Cx47 (Menichella et al., 2003). In these mice, neuronal activity was implicated in the development of myelin vacuoles. Increasing retinal ganglion cell activity (with intraocular cholera toxin) exacerbated vacuole formation, whereas inhibiting activity (with intraocular tetrodotoxin) reduced vacuole formation (Menichella et al., 2006). The high metabolic demands of ganglion cells resulting from their constant activity, high surface area-to-volume ratio, and their need for efficient transport along long myelinated optic nerve fibers, might exacerbate lesion pathology in NMO ON. NMO lesions are notably prevalent in other highly active white matter regions, including the descending respiratory motor tracts of the spinal cord, the corpus callosum, and axon bundles that lie adjacent to circumventricular organs.
Several studies have demonstrated the vulnerability of optic nerve oligodendrocytes and myelination to glutamate excitotoxic mechanisms (Micu et al., 2006; Domercq et al., 2005; Wilke et al., 2004; Matule, 1998). Of relevance to NMO, one study concluded that AQP4-IgG indirectly promotes internalization of glutamate transporter EAAT2 and hence glutamate excitotoxicity (Hinson et al., 2008); however, a subsequent report failed to observe this mechanism (Ratelade et al., 2011a). Nonetheless, astrocyte damage may produce a toxic bystander effect on oligodendrocytes given the critical role of astrocytes in extracellular glutamate removal. Finally, the generation of free radical oxygen species appears to represent an important downstream mechanism limiting remyelination of injured axons. Oxidative stress has been shown to promote astrogliosis and inhibit oligodendrocyte regeneration by altering transcription through the bone morphogeneic protein, Wnt, and Notch development signaling pathways (John et al., 2002; Ara et al., 2008; Fancy et al., 2011; Reid et al., 2012).
4.5 Sensitivity of the optic nerve to NMO pathology
As mentioned above and diagrammed in Figure 4, it is thought that complement-mediated astrocyte damage is an initiating event in NMO, followed by granulocyte infiltration, oligodendrocyte death, and ultimately neuron death. It remains unclear why NMO lesions localize to the central nervous system, and the optic nerve in particular, and why peripheral AQP4-expressing organs are spared. AQP4-IgG concentration in serum is more than 500-fold greater than in CSF (Jarius et al., 2010b). Regional differences in blood-brain barrier integrity might be involved in the predilection of the optic nerve in NMO (Hofman et al., 2001; Liu et al., 2008). It has been proposed that non-specific inflammation may open the blood-brain barrier for transient passage of serum AQP4-IgG and plasma blasts secreting AQP4 autoantibodies. Interestingly, various gastrointestinal and other infections are reported in 25–30% of NMO exacerbations, which may provide the initial stimulus for AQP4-IgG entry into the central nervous system or for lymphocyte recruitment (Koga et al., 2011). Recent data suggests that NMO attacks might be triggered by Helicobacter and/or Clostridium infections (Li et al., 2009; Varrin-Doyer et al., 2012).
Figure 4.
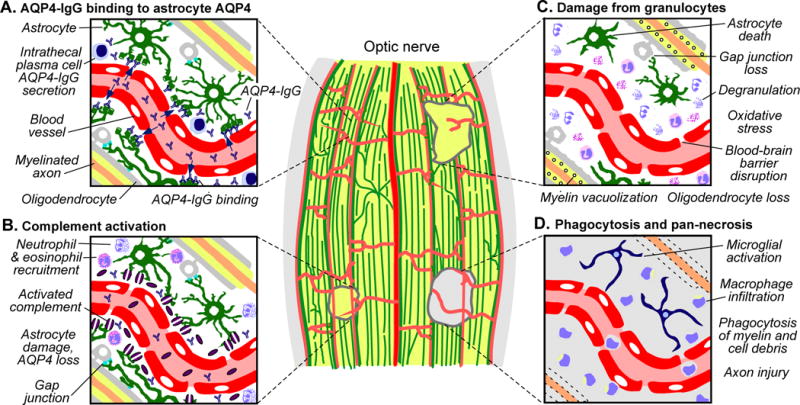
Mechanisms of optic nerve damage in NMO. Lesions of different ages are depicted in a longitudinal section through myelinated retrolaminar optic nerve. A. Pre-lesion, showing AQP4-IgG accessing and binding to AQP4 on perivascular astrocytes. B. Acute early lesion, showing AQP4-IgG deposition and complement activation (purple discs, membrane attack complexes) and astrocyte damage. Cytokine release and recruitment of granulocytes into perivascular space is also shown. C. Subacute lesion, showing neutrophil and eosinophil degranulation that exacerbate astrocytotoxicity. Secondary oligodendrocyte loss results from disruption of glial tight junction networks and oxidative stress. Myelination is relatively preserved, albeit with prominent vacuolization. D. Older lesion, showing fragmentation and loss of myelin and appearance of activated macrophages and resident microglia. Axonal injury may arise from both failed remeylination and inflammatory damage.
Optic nerve-specific pathology in NMO, however, is unlikely to be due solely to enhanced AQP4-IgG access. Restricted diffusion of AQP4-IgG and soluble pro-inflammatory factors (such as complement proteins) in the optic nerve may increase their concentration and contact time. Greatly slowed solute diffusion across white matter tracts in spinal cord has been found by elliptical photobleaching (Papadopoulos et al., 2005). The long, thin cylindrical dimension of the optic nerve is unique among myelinated tracks, and provides a potential mechanism for enhancing lesion formation due to restricted diffusion and clearance of debris (Ludwin, 1990; Hickman et al., 2004).
Optic nerve susceptibility in NMO might also arise from the high AQP4 expression in optic nerve compared to brain (Saini et al., 2010) and the abundance of large OAPs in perivascular astrocytic end-feet of optic nerve (Bäuerle and Wolburg, 1993; Amiry-Moghaddam et al., 2004; Nicchia et al., 2008), which enhance AQP4-IgG binding and CDC, as mentioned above. The presence of plasma blasts in the CSF secreting AQP4-IgG locally (Bennett et al., 2009) and/or regional variations in regulators of complement (CD46 and CD59) may also be involved in the unique susceptibility of the optic nerve in NMO. Finally, retinal Müller cell AQP4 could represent a second pathogenic ocular target of AQP-IgG as suggested by observations of focal retinal vascular attenuation, inner nuclear layer thickening, and microcystic edema in NMO patients (Green and Cree, 2009; Gelfand et al., 2012; Sotirchos et al., 2012).
4.6 Animal models of NMO
An animal model of NMO should recapitulate the key pathological features of human NMO lesions, and, ideally, manifest an AQP4-IgG autoimmune response with spontaneous development of NMO pathology in optic nerve and spinal cord. However, despite considerable effort there are no good models of AQP4-IgG-positive NMO. Challenges in the development of NMO animal models include differences in murine and human complement activity (Bergman et al., 2000), and in the ratio of astrocytes to neurons in central nervous system tissues (Jacobson, 1978).
The CSF plasma cell repertoire in NMO displays features of an antigen-driven, T cell-mediated germinal center humoral immune response (Bennett et al., 2009). As such, some investigators are exploring possible NMO animal models based on direct immunization with AQP4 peptide fragments or adoptive transfer of AQP4-sensitized T cells, possibly in combination with CNS exposure to AQP4-IgG. Combining adoptive transfer of AQP4-specific rat T cells and peripheral infusion of AQP4-IgG-containing patient serum induced lesions in rat spinal cord and brain, but not in optic nerve (Pohl et al., 2011).
AQP4-specific T cells exhibit a differentiation bias toward the Th17 sub-type, driven by the cytokines interkleuken-6, -21, and -23 and TGF-β (Varrin-Doyer et al., 2012). One mouse model involving both T and B cell activation against myelin oligodendrocyte glycoprotein manifests a Th17 bias and selective optic nerve and spinal cord pathology reminiscent of NMO (Bettelli et al., 2006). Interestingly, a mouse model of EAE induced by Th17 (but not Th1) cells demonstrated neutrophilic infiltration in optic nerves and spinal cord, recapitulating several aspects of NMO (Herges et al., 2012). Although EAE is associated with inflammatory demyelination in optic nerve, brain and spinal cord, the underlying mechanism, a T-cell response against myelin-derived peptides, is quite different from the antibody-dependent astrocytopathy in NMO.
In support of a pathogenic role of AQP4-IgG include several studies showing that administered AQP4-IgG can alter the pathology of pre-existing EAE in rats to that of NMO (Bennett et al., 2009; Bradl et al., 2009). However, the background hyper-inflammatory environment and presence of myelin-reactive T cell preclude an unambiguous conclusion about the sufficiency of AQP4 antibodies for lesion formation. Compelling evidence of a pathogenic role of AQP4-IgG in seropositive NMO came from passive transfer of AQP4-IgG and human complement via intracerebral injection in mice. This model reproduced the major features of human NMO lesions, including loss of AQP4 and GFAP, granulocyte infiltration, perivascular deposition of activated complement, and demyelination (Saadoun et al., 2010). Pathology was not seen in key control studies, including injection of complement with IgG from non-NMO humans or with AQP4-IgG depleted NMO serum, injection of AQP4-IgG in the absence of complement, or injection of AQP4-IgG and complement in AQP4-knockout mice. Human complement was used in this model because of the very low endogenous activity of mouse complement.
The intracerebral injection model has been used to study the role of specific immune cells (Saadoun et al., 2011; Saadoun et al., 2012b; Ratelade et al., 2012; Zhang and Verkman, 2013) and potential therapeutics (Tradtrantip et al., 2012a, 2012b, 2013). Attempts to create NMO disease by intravenous administration of AQP4-IgG to naïve rodents have thus far been unsuccessful (Ratelade et al., 2011b). An optic nerve-specific animal model, perhaps involving optic nerve exposure to AQP4-IgG, would be useful in studying NMO ON.
Ex vivo organ culture models of NMO ON and spinal cord pathology have been developed (Zhang et al., 2011) and used to test the role of inflammatory cells and soluble factors and potential therapeutics. Organ culture models allow exposure of tissues to defined conditions and factors. The ex vivo model of NMO ON involves culture of mouse optic nerve culture for 1 day, in which NMO pathology, with loss of AQP4 and GFAP, was produced by incubation of optic nerve cultures with AQP4-IgG and complement (Figure 5). For ex vivo culture of spinal cord, 300 μm-thick vibratome-cut transverse slices of mouse spinal cord were cultured on transwell porous supports, which preserved basic spinal cord architecture and cellular structure, including astrocytes, microglia, neurons and myelin. After 7 days in culture, spinal cord slices were exposed to AQP4-IgG and complement for 2–3 days and analyzed by immunofluorescence. Exposure to AQP4-IgG and complement produced marked loss of GFAP, AQP4 and myelin, and deposition of activated complement and microglial cell activation. In key controls, lesions were not seen in optic nerve and spinal cord slice cultures cultured with either AQP4-IgG or complement alone, or in tissues from AQP4 null mice exposed to AQP4-IgG and complement together.
Figure 5.
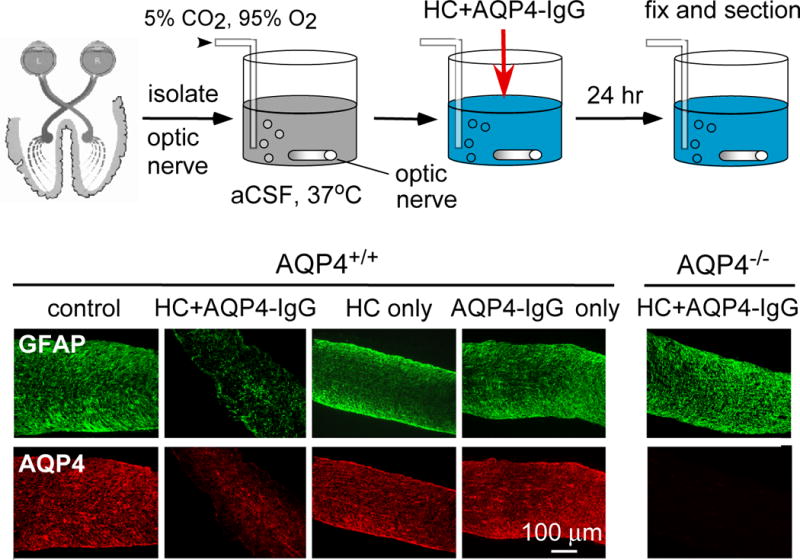
Ex vivo optic nerve culture model of NMO. Top, schematic showing mouse optic nerves cultured for 24 h in CO2/O2-bubbled artificial cerebrospinal fluid, with human complement (HC) and/or AQP4-IgG. Bottom, immunofluorescence for GFAP (green) and AQP4 (red) and myelin basic protein (MBP) (red) in wildtype (AQP4+/+) and AQP4 knockout (AQP4−/−) mice. ‘Control’ indicates no added AQP4-IgG or HC.
Issues of oxygen diffusion present difficulty in maintaining viable optic nerve organotypic cultures for the period (3–5 days) necessary to study the predicted sequence of events in NMO ON. Protocols involving optic nerve explants cultured with their retinas attached may prolong the viability of rodent optic nerves ex vivo (Azim et al., 2011; Fu and Sretavan, 2012). Notwithstanding technical challenges in maintaining optic nerve cultures, further optimization of this model is warranted to address questions that are not easily investigated using in vivo models.
5. Emerging therapeutics
Current abortive and preventative therapies for NMO ON, such as immunosuppression and immunomodulation are non-selective and hence can have off-target side effects such as infections and malignancy. An ideal NMO therapy would target specific NMO pathogenesis mechanism(s) with high precision. Recent advances in understanding NMO pathogenesis mechanisms suggest potential therapeutic targets. As diagrammed in Figure 6, NMO therapies can target AQP4-IgG producing immune cells, AQP4-IgG effector functions, access of AQP4-IgG across the BBB, components involved in CDC or ADCC, binding of AQP4-IgG to AQP4 on astrocytes, AQP4 cell surface expression or supramolecular assembly, and/or specific immune cells and soluble factors.
Figure 6.
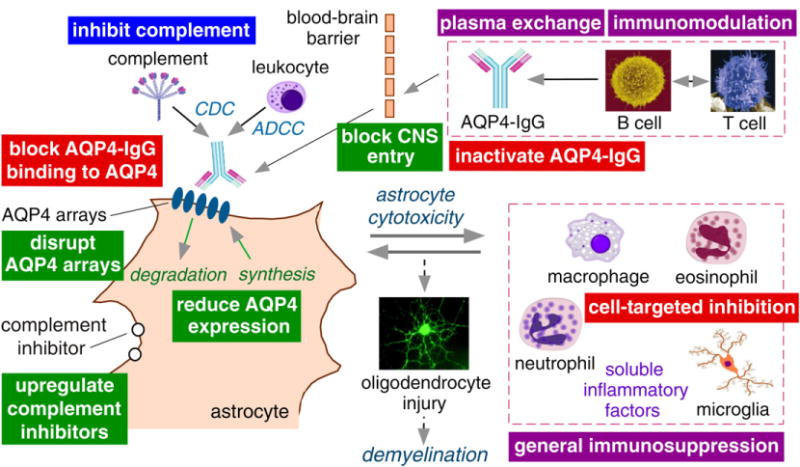
Approaches to treat NMO optic neuritis. Schematic of NMO pathogenesis showing AQP4-IgG production by lymphocytes, penetration through the BBB, binding to AQP4 on astrocytes, CDC (complement-dependent cytotoxicity) and ADCC (antibody-dependent cell-mediated cytotoxicity), and the inflammatory reaction. Purple boxes, current NMO therapies; blue box, therapy in clinical trials; red boxes, therapies in pre-clinical development; green boxes, alternative possible therapeutic targets.
Some of these potential targets are being exploited to develop a new generation of NMO therapeutics. An attractive target is the binding of AQP4-IgG to AQP4 on astrocytes, which is the major initiating event in NMO. Several blocking strategies have been introduced to prevent AQP4-IgG binding to AQP4. One blocking strategy utilizes a non-pathogenic, high-affinity monoclonal antibody (called ‘aquaporumab’) that binds tightly to AQP4, but is mutated to eliminate its CDC and ADCC effector functions (Tradtrantip et al., 2012b). Aquaporumab prevented the binding of AQP4-IgG in NMO patient sera to AQP4, resulting in reduced pathology in cell culture, organ culture and mouse models of NMO.
A variation of this strategy is enzymatic deglycosylation of patient AQP4-IgG by the IgG-selective enzyme endoglycosidase S (Tradtrantip et al., 2013). Treatment of NMO patient serum with endoglycosidase S converts pathogenic AQP4-IgG to therapeutic blocking antibodies, as AQP4-IgG deglycosylation does not interfere with its binding to AQP4. Blocking antibodies or small molecules might be delivered by systemic, intraocular or intrathecal routes; enzymatic deglycosylation might be accomplished by therapeutic apheresis in which blood is passed over surface-immobilized endoglycosidase S. An alternative blocking strategy under development utilizes small-molecule drugs that interfere with AQP4-IgG binding to AQP4, which have been identified by high-throughput screening (Tradtrantip et al., 2012a).
The involvement of neutrophils and eosinophils in NMO pathogenesis has suggested that granulocyte-targeted therapies may be beneficial. Neutropenia greatly reduced pathology in a mouse model of NMO, while neutrophilia increased NMO pathology (Saadoun et al., 2012a). In one case report, inadvertent administration of granulocyte colony stimulating factor to an NMO patient produced neutrophilia and greatly exacerbated NMO clinical disease (Jacob et al., 2012). Neutrophil elastase inhibitors, including Sivelastat, greatly reduced pathology in mouse and spinal cord slice culture models of NMO (Zhang et al., 2011; Saadoun et al., 2012a). Sivelastat is approved in Japan for therapy of lung inflammation associated with respiratory distress syndrome. Eosinophils have also been shown to exacerbate AQP4-IgG-dependent NMO pathology, and certain second-generation antihistamines with eosinophil-stabilizing actions reduced NMO pathology in spinal cord slice and mouse models (Zhang and Verkman, 2013). The repurposing of existing granulocyte-targeted drugs may thus have utility for NMO therapy.
Various other approved drugs are under consideration for repurposing in NMO, including the complement inhibitor eculizumab (Laino, 2012) and the interleuken-6 receptor inhibitor tocilizumab (Araki et al., 2012). Other NMO targets remain theoretical possibilities, including tolerizing therapies targeting immune cells, inhibitors of AQP4 expression or supramolecular assembly, and modulators of astrocyte complement inhibitor proteins.
6. Future directions
Though substantial recent progress has been made in the pathogenesis and treatment of ON in NMO, many fundamental questions remain unanswered. How is NMO initiated and is the antigenic target restricted to AQP4? Why does the disease manifest ON and spinal cord specificity? What are the roles of granulocytes, macrophages, lymphocytes and inflammatory cytokines, and how do they contribute to optic nerve astrocytopathy and retinal ganglion cell injury? Is Müller cell AQP4 a significant target of AQP-IgG? Animal models of AQP4-IgG pathology in the anterior visual system will be important in addressing these questions. Improvement in NMO prognosis is anticipated, as new compounds and monoclonal antibodies are entering the therapeutics pipeline, and approved drugs are repurposed for NMO therapy. However, the relative rarity of NMO and its highly variable course remain challenges in the evaluation of new therapeutics and the optimization of existing therapies.
Acknowledgments
This work was supported by grants from the Guthy-Jackson Charitable Foundation (A.S.V. and J.L.B.), grants EY13574, EB00415, DK35124, HL73856, DK86125 and DK72517 from the National Institutes of Health (to A.S.V.), and Grant RG4320 from the National Multiple Sclerosis Society (to J.L.B.). Dr. Levin was supported by the Heed Ophthalmic Foundation as a Heed Fellow during the generation of this manuscript.
Abbreviations
- ADCC
antibody-dependent cell-mediated cytotoxicity
- AQP4
aquaporin-4
- BBB
blood-brain barrier
- CDC
complement-dependent cytotoxicity
- CDCC
complement-dependent cell-mediated cytotoxicity
- CSF
cerebral spinal fluid
- Cx
connexin
- GFAP
glial fibrillary acidic protein
- IVMP
intravenous methylprednisone
- MRI
magnetic resonance imaging
- MS
multiple sclerosis
- NK
natural killer
- NMO
neuromyelitis optica
- NMOSD
neuromyelitis optica spectrum disorder
- OAP
orthogonal arrays of particles
- OCT
optical coherence tomography
- ON
optic neuritis
- PE
plasma exchange
- RNFL
retinal nerve fiber layer
- TM
transverse myelitis
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amiry-Moghaddam M, Xue R, Haug FM, Neely JD, Bhardwaj A, Agre P, Adams ME, Froehner SC, Mori S, Ottersen OP. Alpha-syntrophin deletion removes theperivascular but not endothelial pool of aquaporin-4 at the blood-brain barrier and delays the development of brain edema in an experimental model of acute hyponatremia. FASEB J. 2004;18:542–544. doi: 10.1096/fj.03-0869fje. [DOI] [PubMed] [Google Scholar]
- Ara J, See J, Mamontov P, Hahn A, Bannerman P, Pleasure D, Grinspan JB. Bone morphogenetic proteins 4, 6, and 7 are up-regulated in mouse spinal cord during experimental autoimmune encephalomyelitis. J Neurosci Res. 2008;86:125–135. doi: 10.1002/jnr.21462. [DOI] [PubMed] [Google Scholar]
- Araki M, Aranami T, Matsuoka T, Nakamura M, Miyake S, Yamamura T. Clinical improvement in a patient with neuromyelitis optica following therapy with the anti-IL-6 receptor monoclonal antibody tocilizumab. Mod Rheumatol. 2012 doi: 10.1007/s10165-012-0715-9. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asgari N, Lillevang ST, Skejoe HP, Falah M, Stenager E, Kyvik KO. A population-based study of neuromyelitis optica in Caucasians. Neurology. 2011;76:1589–1595. doi: 10.1212/WNL.0b013e3182190f74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asgari N, Nielsen C, Stenager E, Kyvik KO, Lillevang ST. HLA, PTPN22, and PD-1 associations as markers of autoimmunity in neuromyelitis optica. Mult Scler. 2012;18:23–30. doi: 10.1177/1352458511417480. [DOI] [PubMed] [Google Scholar]
- Auguste KI, Jin S, Uchida K, Yan D, Manley GT, Papadopoulos MC, Verkman AS. Greatly impaired migration of implanted aquaporin-4-deficient astroglial cells in mouse brain toward a site of injury. FASEB J. 2007;21:108–116. doi: 10.1096/fj.06-6848com. [DOI] [PubMed] [Google Scholar]
- Azim K, Butt AM. GSK3p negatively regulates oligodendrocyte differentiation and myelination in vivo. Glia. 2011;50:540–553. doi: 10.1002/glia.21122. [DOI] [PubMed] [Google Scholar]
- Barboni P, Carelli V, Svini G, Carbonelli M, La Morgia C, Sadun AA. Microcystic macular degeneration from optic neuropathy: not inflammatory, not trans-synaptic degeneration. 2013. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Bäuerle C, Wolburg H. Astrocytes in the nonmyelinated lamina cribosa of the rat are less polarized than in the optic nerve proper: a freeze-fracture study. Glia. 1993;9:238–241. doi: 10.1002/glia.440090309. [DOI] [PubMed] [Google Scholar]
- Bennett JL, Lam C, Kalluri SR, Saikali P, Bautista K, Dupree C, Glogowska M, Case D, Antel JP, Owens GP, Gilden D, Nessler S, Stadelmann C, Hemmer B. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann Neurol. 2009;66:617–629. doi: 10.1002/ana.21802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck RW, Cleary PA, Anderson MM, Jr, Keltner JL, Shults WT, Kaufman DI, Buckley EG, Corbett JJ, Kupersmith MJ, Miller NR, et al. A randomized controlled trialof corticosteroids in the treatment of acute optic neuritis. The Optic Neuritis Study Group. N Engl J Med. 1992;326:581–588. doi: 10.1056/NEJM199202273260901. [DOI] [PubMed] [Google Scholar]
- Bergamaschi R, Jarius S, Robotti M, Pichiecchio A, Wildemann B, Meola G. Two cases of benign neuromyelitis optica in patients with celiac disease. J Neurol. 2009;256:2097–2099. doi: 10.1007/s00415-009-5288-y. [DOI] [PubMed] [Google Scholar]
- Berman I, Basse PH, Barmada MA, Griffin JA, Cheung NK. Comparison of in vitro antibody-targeted cytotoxicity using mouse, rat, and human effectors. Cancer Immunol Immunother. 2000;59:266–269. doi: 10.1007/s002620000120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E, Baeten D, Jäger A, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T and B cells cooperate to induce a Devic-like disease in mice. J Clin Invest. 2006;116:2393–2402. doi: 10.1172/JCI28334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder DK, Yao X, Sick TJ, Verkman AS, Manley GT. Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin-4 water channels. Glia. 2006;53:631–636. doi: 10.1002/glia.20318. [DOI] [PubMed] [Google Scholar]
- Bradl M, Misu T, Takahashi T, Watanabe M, Mader S, Reindl M, Adzemovic M, Bauer J, Berger T, Fujihara K, Itoyama Y, Lassmann H. Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann Neurol. 2009;66:630–643. doi: 10.1002/ana.21837. [DOI] [PubMed] [Google Scholar]
- Brenner M, Johnson AB, Boespflug-Tanguy O, Rodriguez D, Goldmann JE, Messing A. Mutations in GFAB, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat Genet. 2001;27:117–120. doi: 10.1038/83679. [DOI] [PubMed] [Google Scholar]
- Cabre P, Signate Al, Lindo S, Merle H, Caparros-Lefebvre D, Béra O, Smadja D. Role of return migration in the emergence of multiple sclerosis in the French West Indies. Brain. 2005;128:2899–2910. doi: 10.1093/brain/awh624. [DOI] [PubMed] [Google Scholar]
- Cabrera-Gómez JA, Kurtzke JF, González-Quevedo A, Lara-Rodríguez R. An epidemiological study of neuromyelitis optica in Cuba. J Neurol. 2009;256:1891–1898. doi: 10.1007/s00415-009-0009-0. [DOI] [PubMed] [Google Scholar]
- Collongues N, Marignier R, Zéphir H, Papeix C, Fontaine B, Blanc F, Rodriguez D, Fleury M, Vukesic S, Pelletier J, Audoin B, Thouvenot E, Camu W, Barroso B, Ruet A, Brochet B, Confavreux C, de Seze J. Long-term follow-up of neuromyelitis optic with a pediatric onset. Neurology. 2010;75:1084–1088. doi: 10.1212/WNL.0b013e3181f39a66. [DOI] [PubMed] [Google Scholar]
- Costa C, Arrambide G, Tintore M, Castilló J, Sastre-Garriga J, Tur C, Río J, Saiz A, Vidal-Jordana A, Auger C, Nos C, Rovira A, Comabella M, Horga A, Montalban S. Value of NMO-IgG determination at the time of presentation as CIS. Neurology. 2012;78:1608–1611. doi: 10.1212/WNL.0b013e3182563b32. [DOI] [PubMed] [Google Scholar]
- Costa RM, Santos AC, Costa LS. An unusual chiasmal visual field defect in a patient with neuromyelitis optica: case report. Arq Bras Oftalmol. 2007;70:153–155. doi: 10.1590/s0004-27492007000100029. [DOI] [PubMed] [Google Scholar]
- Cossburn M, Tackley G, Baker K, Ingram G, Burtonwood M, Malik G, Pickersgill T, te Water Naudé J, Robertson N. The prevalence of neuromyelitis optica in South East Wales. Eur J Neurol. 2012;18:655–659. doi: 10.1111/j.1468-1331.2011.03529.x. [DOI] [PubMed] [Google Scholar]
- Cotrina ML, Nedergaard M. Brain connexins in demyelinating disease: Therapeutic potential of glial targets. Brain Res. 2012;1487:61–68. doi: 10.1016/j.brainres.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzi C, Matiello M, Lucchinetti CF, Weinshenker BG, Pittock SJ, Mandrekar J, Thapa P, McKeon A. Azathioprine: tolerability, efficacy, and predictors of benefit in neuromyelitis optica. Neurology. 2011;77:659–666. doi: 10.1212/WNL.0b013e31822a2780. [DOI] [PubMed] [Google Scholar]
- Crane JM, Verkman AS. Determinants of aquaporin-4 assembly in orthogonal arrays revealed by live cell single molecule fluorescence imaging. J Cell Sci. 2009;122:813–821. doi: 10.1242/jcs.042341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane JM, Lam C, Rossi A, Gupta T, Bennett JL, Verkman AS. Binding affinity and specificity of neuromyelitis optica autoantibodies to aquaporin-4 M1/M23 isoforms and orthogonal arrays. J Biol Chem. 2011;286:16516–16524. doi: 10.1074/jbc.M111.227298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Q, Yin Y, Benowitz LI. The role of macrophages in optic nerve regeneration. Neuroscience. 2009;158:1039–1048. doi: 10.1016/j.neuroscience.2008.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daily J, Pula J, Keung B, Wang H, Kattah K. Longitudinally extensive optic neuritis distinguishes neuromyelitis optica from multiple sclerosis. American Academy of Neurology. 2013 Abstracts IN4-1.003. [Google Scholar]
- Devic E. Myelite subaigue compliquee de neurite optique. Bull Med. 1894;8:1033–1034. [Google Scholar]
- Domercq M, Etxebarria E, Pérez-Samartín A, Matute C. Excitotoxic oligodendrocyte death and axonal damage induced by glutamate transporter inhibition. Glia. 2005;52:36–46. doi: 10.1002/glia.20221. [DOI] [PubMed] [Google Scholar]
- Fancy SP, Harrington EP, Yuen TJ, Silbereis JC, Zhao C, Baranzini SE, Bruce CC, Otero JJ, Huang EJ, Nusse R, Franklin RJ, Rowitch DH. Axin2 as regulatory and therapeutic target in newborn brain injury and remyelination. Nat Neurosci. 2011;14:1009–1016. doi: 10.1038/nn.2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairless R, Williams SK, Hoffmann DB, Stojic A, Hochmesiter S, Shmitz F, Storch MK, Diem R. Preclinical retinal neurodegeneration in a model of multiple sclerosis. J Neurosci. 2012;32:5585–5597. doi: 10.1523/JNEUROSCI.5705-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes DB, Ramos Rde I, Falcochio C, Apóstolos-Pereira S, Callegaro D, Monteiro ML. Comparison of visual acuity and automated perimetry findings in patients with neuromyelitis optica or multiple sclerosis after single or multiple attacks of optic neuritis. J Neuroophthalmol. 2012a;32:102–106. doi: 10.1097/WNO.0b013e31823a9ebc. [DOI] [PubMed] [Google Scholar]
- Fernandes DB, Raza AS, Nogueira RG, Wang D, Callegaro D, Hood DC, Monteiro ML. Evaluation of inner retinal layers in patients with multiple sclerosis or neuromyelitis optica using optical coherence tomography. Ophthalmology. 2012b doi: 10.1016/j.ophtha.2012.07.066. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frigeri A, Gropper M, Turck CW, Verkman AS. Immunolocalization of the mercurial-insensitive water channel and glycerol intrinsic protein in epithelial cell plasma membranes. Proc Natl Acad Sci U S A. 1995;92:4328–4331. doi: 10.1073/pnas.92.10.4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu CT, Sretavan D. Involvement of EphB/Ephrin-B signaling in axonal survival in mouse experimental glaucoma. Invest Ophthalmol Vis Sci. 2012;53:76–84. doi: 10.1167/iovs.11-8546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galetta SL, Cornblath WT. Should most patients with optic neuritis be tested for neuromyelitis optica antibodies and should this affect their treatment? J. Neuroophthalmol. 2010;30:376–379. doi: 10.1097/WNO.0b013e3181f68c19. [DOI] [PubMed] [Google Scholar]
- Gault F. De la neuromyelite optique aigue [thesis] University of Lyon; Lyon, France: 1894. [Google Scholar]
- Gelfand JM, Cree BA, Calabresi P, Frohman E, Balcer L, Nolan R, Green AJ. Microcytic macular oedema in neuromyelitis optica. ECTRIMS. 2012 Meeting Abstracts P599. [Google Scholar]
- Gelfand JM, Nolan R, Schwartz DM, Graves J, Green AJ. Microcystic macular oedema in multiple sclerosis is associated with disease severity. Brain. 2012;135:1786–1793. doi: 10.1093/brain/aws098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green AJ, Cree BA. Distinctive retinal nerve fibre layer and vascular changes in neuromyelitis optica following optic neuritis. J Neurol Neurosurg Psychiatry. 2009;80:1002–1005. doi: 10.1136/jnnp.2008.166207. [DOI] [PubMed] [Google Scholar]
- Hamann S, Zeuthen T, La Cour M, Nagelhus EA, Ottersen OP, Agre P, Nielsen S. Aquaporin in complex tissues: distribution of aquaporins 1–5 in human and rat eye. Am J Physiol. 1998;274:C1332–C1345. doi: 10.1152/ajpcell.1998.274.5.C1332. [DOI] [PubMed] [Google Scholar]
- Hasegawa H, Ma T, Skach W, Matthay M, Verkman AS. Molecular cloning of a mercurial-insensitive water channel expressed in selected water transporting tissues. J Biol Chem. 1994;269:5497–5500. [PubMed] [Google Scholar]
- Herges K, De Jong BA, Kolkowitz I, Dunn C, Mandelbaum G, Ko RM, Maini A, Han MH, Killestein J, Polman C, Goodyear AL, Dunn J, Steinman L, Axtell RC. Protective effect of an elastase inhibitor in a neuromyelitis optica-like disease driven by a peptide of myelin oligodendroglial glycoprotein. Mult Scler. 2012;18:398–408. doi: 10.1177/1352458512440060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman SJ, Kapoor R, Jones SJ, Altmann DR, Plant GT, Miller DH. Corticosteroids do not prevent optic nerve atrophy following optic neuritis. J Neurol Neurosurg Psychiatry. 2003;74:1139–1141. doi: 10.1136/jnnp.74.8.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman SJ, Toosy AT, Jones SJ, Altmann DR, Miszkiel KA, MacManus DG, Barker GJ, Plant GT, Thompson AJ, Miller DH. Serial mangetization transfer imaging in acute optic neuritis. Brain. 2004;127:692–700. doi: 10.1093/brain/awh076. [DOI] [PubMed] [Google Scholar]
- Hinson SR, Roemer SF, Lucchinetti CF, Fryer JP, Kryzer TJ, Chamberlain JL, Howe CL, Pittock SJ, Lennon VA. Aquaporin-4-binding autoantibodies in patients with neuromyelitis optica impair glutamate transport by down-regulating EAAT2. J Exp Med. 2008;205:2473–2481. doi: 10.1084/jem.20081241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinson SR, Romero MF, Popescu BF, Lucchinetti CF, Fryer JP, Wolburg H, Fallier-Becker P, Noell S, Lennon VA. Molecular outcomes of neuromyelitis optica (NMO)-IgG binding to aquaporin-4 in astrocytes. Proc Natl Acad Sci USA. 2012;109:1245–1250. doi: 10.1073/pnas.1109980108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho JD, Yeh R, Sandstrom A, Chorny I, Harries WE, Robbins RA, Miercke LJ, Stroud RM. Crystal structure of human aquaporin 4 at 1.8 A and its mechanism of conductance. Proc Natl Acad Sci USA. 2009;106:7437–7442. doi: 10.1073/pnas.0902725106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofman P, Hoyng P, vanderWerf F, Vrensen GF, Schlingemann RO. Lack of blood-brain barrier properties in microvessels of the prelaminar optic nerve head. Invest Ophthalmol Vis Sci. 2001;42:895–901. [PubMed] [Google Scholar]
- Iorio R, Lucchinetti CF, Lennon VA, Costanzi C, Hinson S, Weinshenker BG, Pittock SJ. Syndrome of inappropriate antidiuresis may herald or accompany neuromyelitis optica. Neurology. 2011;77:1644–1646. doi: 10.1212/WNL.0b013e3182343377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob A, Matiello M, Weinshenker BG, Wingerchuk DM, Lucchinetti C, Shuster E, Carter J, Keegan BM, Kantarci OH, Pittock SJ. Treatment of neuromyelitis optica with mycophenolate mofetil: retrospective analysis of 24 patients. Arch Neurol. 2009;9:1128–1133. doi: 10.1001/archneurol.2009.175. [DOI] [PubMed] [Google Scholar]
- Jacob A, Weinshenker BG, Violich I, McLinkskey N, Krupp L, Wingerchuk DM, Boggild M, Constantinescu CS, Miller A, De Angelis T, Matiello M, Cree BA. Treatment of neuromyelitis optica with rituximab: retrospective analysis of 25 patients. Arch Neurol. 2008;65:1443–1448. doi: 10.1001/archneur.65.11.noc80069. [DOI] [PubMed] [Google Scholar]
- Jacob A, Saadoun S, Kitley J, Leite MI, Palace J, Schon F, Papadopoulos MC. Detrimental role of granulocyte-colony stimulating factor in neuromyelitis optica: clinical case and histological evidence. Mult Scler. 2012 doi: 10.1177/1352458512443994. In press. [DOI] [PubMed] [Google Scholar]
- Jacobson M. Developmental Neurobiology. 2. Plenum Press; New York: 1978. [Google Scholar]
- Jarius S, Aboul-Enein F, Waters P, Kuenz B, Hauser A, Berger T, Lang W, Reindl M, Vicent A, Kristoferitsch W. Antibody to aquaporin-4 in the long-term course of neuromyelitis optica. Brain. 2008;131:1372–1380. doi: 10.1093/brain/awn240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarius S, Frederikson J, Waters P, Paul F, Akman-Demir G, Marignier R, Franciotta D, Ruprecht K, Kuenz B, Rommer P, Kritoferitsch W, Wildemann B, Vincent A. Frequency and prognostic impact of antibodies to aquaporin-4 in patients with optic neuritis. 2010a. [DOI] [PubMed] [Google Scholar]
- Jarius S, Franciotta D, Paul F, Ruprecht K, Bergamaschi R, Rommer PS, Reuss R, Probst C, Kristoferitsch W, Wandinger KP, Wildemann B. Cerebrospinal fluid antibodies to aquaporin-4 in neuromyelitis optica and related disorders: frequency, origin, and diagnostic relevance. J Neuroinflammation. 2010b;7:52. doi: 10.1186/1742-2094-7-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarius S, Jacobi C, de Seze J, Zéphir H, Paul F, Franciotta D, Rommer P, Mader S, Kleiter Il, Reindl M, Akman-Demir G, Seifert-Held T, Kristoferitsch W, Melms A, Wandinger KP, Wildemann B. Frequency and syndrome specificity of antibodies to aquaporin-4 in neurological patients with rheumatic disorders. Mult Scler. 2011;17:1067–1073. doi: 10.1177/1352458511403958. [DOI] [PubMed] [Google Scholar]
- Jarius S, Wandinger KP, Borowski K, Stoecker W, Wildemann B. Antibodies to CV2/CRMP5 in neuromyelitis optica-like disease: Case report and review of the literature. Clin Neurol Neurosurg. 2012;114:331–335. doi: 10.1016/j.clineuro.2011.10.048. [DOI] [PubMed] [Google Scholar]
- John GR, Shankar SL, Shafit-Zagardo B, Massimi A, Lee SC, Raine CS, Brosnan CF. Multiple sclerosis: re-expression of a developmental pathway that restricts oligodendrocyte maturation. Nat Med. 2002;8:1115–1121. doi: 10.1038/nm781. [DOI] [PubMed] [Google Scholar]
- Khatri BO, Kramer J, Dikuc M, Palencia M, Verre W. Maintenance plasma exchange therapy for steroid-refractory neuromyelitis optica. J Clin Apher. 2012;27:183–192. doi: 10.1002/jca.21215. [DOI] [PubMed] [Google Scholar]
- Kim SH, Kim W, Li XF, Jung IJ, Kim HJ. Repeated treatment with rituximab based on the assessment of peripheral circulating memory B cells in patients with relapsing neuromyelitis optica over 2 years. Arch Neurol. 2011;11:1412–1420. doi: 10.1001/archneurol.2011.154. [DOI] [PubMed] [Google Scholar]
- Kira J. Multiple sclerosis in the Japanese population. Lancet Neurol. 2003;2:117–127. doi: 10.1016/s1474-4422(03)00308-9. [DOI] [PubMed] [Google Scholar]
- Kleiter I, Hellwig K, Berthele A, Kümpfel T, Linker RA, Hartung HP, Paul F, Aktas O, for the Neuromyelitis Optica Study Group Failure of natalizumab to prevent relapses in neuromyelitis optica. Arch Neurol. 2012;69:239–245. doi: 10.1001/archneurol.2011.216. [DOI] [PubMed] [Google Scholar]
- Koga M, Takahashi T, Kawai M, Fujihara K, Kanda T. A serological analysis of viral and bacterial infections associated with neuromyelitis optica. J Neurol Sci. 2011;300:19–22. doi: 10.1016/j.jns.2010.10.013. [DOI] [PubMed] [Google Scholar]
- Laino C. News from the American Neurological Association Annual Meeting: Eculizumab shows promise for preventing NMO attacks, keeping disease in check. Neurol Today. 2012;12:24–27. [Google Scholar]
- Leite MI, Coutinho E, Lana-Peixoto M, Apostolos S, Waters P, Sato D, Melamud L, Marta M, Graham A, Spillane J, Villa AM, Callegaro D, Santos E, da Silva AM, Jarius S, Howard R, Nakashima I, Giovannoni G, Buckley C, Hilton-Jones D, Vincent A, Palace J. Myasthenia gravis and neuromyelitis optica spectrum disorder: A multicenter study of 16 patients. Neurology. 2012;78:1601–1607. doi: 10.1212/WNL.0b013e31825644ff. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, Nakashima I, Weinshenker BG. A serum autoantibody marker for neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004;364:2106–2112. doi: 10.1016/S0140-6736(04)17551-X. [DOI] [PubMed] [Google Scholar]
- Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202:473–477. doi: 10.1084/jem.20050304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke W, Edelmann W, Bieri PL, Chiu FC, Cowan NJ, Kucherlapati R, Raine CS. GFAP is necessary for the integrity of CNS white matter architecture and long-term maintenance of myelination. Neuron. 1996;17:607–615. doi: 10.1016/s0896-6273(00)80194-4. [DOI] [PubMed] [Google Scholar]
- Li J, Verkman AS. Impaired hearing in mice lacking aquaporin-4 water channels. J Biol Chem. 2001;276:31233–31237. doi: 10.1074/jbc.M104368200. [DOI] [PubMed] [Google Scholar]
- Li J, Patil RV, Verkman AS. Mildly abnormal retinal function in transgenic mice without Müller cell aquaporin-4 water channels. Invest Ophthalmol Vis Sci. 2002;43:573–579. [PubMed] [Google Scholar]
- Li L, Zhang H, Varrin-Doyer M, Zamvil SS, Verkman AS. Proinflammatory role of aquaporin-4 in autoimmune neuroinflammation. FASEB J. 2011;25:1556–1566. doi: 10.1096/fj.10-177279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Minohara M, Piao H, Matsushita T, Masaki K, Matsuoka T, Isobe N, Su JJ, Ohyagi Y, Kira J. Association of anti-Helicobacter pylori neutrophil-activating protein antibody response with anti-aquaporin-4 autoimmunity in Japanese patients with multiple sclerosis and neuromyelitis optica. Mult Scler. 2009;15:1411–1421. doi: 10.1177/1352458509348961. [DOI] [PubMed] [Google Scholar]
- Li Y, Xie P, Lv F, Mu J, Li Q, Yang Q, Hu M, Tang H, Yi J. Brain magnetic resonance imaging abnormalities in neuromyelitis optica. Acta Neurol Scand. 2008;118:218–225. doi: 10.1111/j.1600-0404.2008.01012.x. [DOI] [PubMed] [Google Scholar]
- Liu LY, Zheng H, Xiao HL, She ZJ, Zhao SM, Chen ZL, Zhou GM. Comparison of blood-nerve barrier disruption and matrix metalloprotease-9 expression in injured central and peripheral nerves in mice. Neurosci Lett. 2008;434:155–159. doi: 10.1016/j.neulet.2007.12.052. [DOI] [PubMed] [Google Scholar]
- Lu DC, Zhang H, Zador Z, Verkman AS. Impaired olfaction in mice lacking aquaporin-4 water channels. FASEB J. 2008;22:3216–3223. doi: 10.1096/fj.07-104836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwin SK. Phagocytosis in the rat optic nerve following Wallerian degeneration. Acta Neuropathol. 1990;80:266–273. doi: 10.1007/BF00294644. [DOI] [PubMed] [Google Scholar]
- Lucchinetti CF, Mandler RN, McGavern D, Bruck W, Gleich G, Ransohoff RM, Trebst C, Weinshenker B, Wingerchuk D, Parisi JE, Lassmann H. A role for humoral mechanisms in the pathogenesis of Devic’s neuromyelitis optica. Brain. 2002;125:1450–1461. doi: 10.1093/brain/awf151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Yang B, Gillespie A, Carlson EJ, Epstein CJ, Verkman AS. Generation and phenotype of a transgenic knock-out mouse lacking the mercurial-insensitive water channel aquaporin-4. J Clin Invest. 1997;100:957–962. doi: 10.1172/JCI231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mader S, Gredler V, Schanda K, Rostasy K, Dujmovic I, Pfaller K, Lutterotti A, Jarius S, Di Pauli F, Kuenz B, Ehling R, Hegen H, Deisenhammer F, Aboul-Enein F, Storch MK, Koson P, Drulovic J, Kristoferitsch W, Berger T, Reindl M. Complement activating antibodies to myelin oligodendrocyte glycoprotein in neuromyelitis optica and related disorders. J Neuroinflammation. 2011;8:184. doi: 10.1186/1742-2094-8-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magraner MJ, Coret F, Casanova B. The effect of intravenous immunoglobulin on neuromyelitis optica. Neurologia. 2013;28:65–72. doi: 10.1016/j.nrl.2012.03.014. [DOI] [PubMed] [Google Scholar]
- Mandler RN, Davis LE, Jeffery DR, Kornfeld M. Devic’s neuromyelitis optica: a clinicopathological study of 8 patients. Ann Neurol. 1993;34:162–168. doi: 10.1002/ana.410340211. [DOI] [PubMed] [Google Scholar]
- Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, Chan P, Verkman AS. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000;6:159–163. doi: 10.1038/72256. [DOI] [PubMed] [Google Scholar]
- Matiello M, Kim HJ, Kim W, Brum DG, Barreira AA, Kingsbury DJ, Plant GT, Adoni T, Weinshenker BG. Familial neuromyelitis optica. Neurology. 2010;75:310–315. doi: 10.1212/WNL.0b013e3181ea9f15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga Y, Kezuka T, An X, Fujita K, Matsuyama N, Matsuda R, Usui Y, Yamakawa N, Kuroda M, Goto H. Visual functional and histopathological correlation in experimental autoimmune optic neuritis. Invest Ophthalmol Vis Sci. 2012;53:6964–6971. doi: 10.1167/iovs.12-10559. [DOI] [PubMed] [Google Scholar]
- Matule C. Characteristics of acute and chronic kainite excitotoxic damage to the optic nerve. Proc Natl Acad Sci USA. 1998;95:10229–10234. doi: 10.1073/pnas.95.17.10229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mealy MA, Wingerchuk DM, Greenberg BM, Levy M. Epidemiology of neuromyelitis optica in the United States. Arch Neurol. 2012;69:1176–1180. doi: 10.1001/archneurol.2012.314. [DOI] [PubMed] [Google Scholar]
- Melamud L, Fernandez JM, Rivarola V, Di Giusto G, Ford P, Villa A, Capurro C. Neuromyelitis Optica Immunoglobulin G present in sera from neuromyelitis optica patients affects aquaporin-4 expression and water permeability of the astrocyte plasma membrane. J Neurosci Res. 2012;90:1240–1248. doi: 10.1002/jnr.22822. [DOI] [PubMed] [Google Scholar]
- Menichella DM, Goodenough DA, Sirowski E, Scherer SS, Paul DL. Connexins are critical for normal myelination in the CNS. J Neurosci. 2003;23:5963–5973. doi: 10.1523/JNEUROSCI.23-13-05963.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menichella DM, Majdan M, Awatramani R, Goodenough DA, Sirkowski E, Scherer SS, Paul DL. Genetic and physiological evidence that oligodendrocyte gap junctions contribute to spatial buffering of potassium released during neuronal activity. J Neurosci. 2006;26:10984–10991. doi: 10.1523/JNEUROSCI.0304-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merle H, Olindo S, Bonnan M, Donnio A, Richer R, Smadja D, Cabre P. Natural history of the visual impairment of relapsing neuromyelitis optica. Ophthalmology. 2007;114:810–815. doi: 10.1016/j.ophtha.2006.06.060. [DOI] [PubMed] [Google Scholar]
- Merle H, Olindo S, Donnio A, Richer R, Smadja D, Cabre P. Retinal peripapillary nerve fiber layer thickness in neuromyelitis optica. Invest Ophthalmol Vis Sci. 2008;49:4412–4417. doi: 10.1167/iovs.08-1815. [DOI] [PubMed] [Google Scholar]
- Merle H, Olindo S, Jeannin S, Valentino R, Mehdaoui H, Cabot F, Donnio A, Hage R, Richer R, Smadja D, Cabre P. Treatment of optic neuritis by plasma exchange (add-on) in neuromyelitis optica. Arch Ophthalmol. 2012;130:858–862. doi: 10.1001/archophthalmol.2012.1126. [DOI] [PubMed] [Google Scholar]
- Micu I, Jiang Q, Coderre E, Ridsdale A, Zhang L, Woulfe J, Yin X, Trapp BD, McRory JE, Rehak R, Zamponi GW, Wang W, Stys PK. NMDA receptors mediate calcium accumulation in myelin during chemical ischaemia. Nature. 2006;439:988–992. doi: 10.1038/nature04474. [DOI] [PubMed] [Google Scholar]
- Mignot C, Boespflug-Tanguy O, Gelot A, Dautigny A, Pham-Dinh D, Rodriguez D. Alexander disease: putative mechanisms of an astrocytic encephalopathy. Cell Mol Life Sci. 2004;61:369–385. doi: 10.1007/s00018-003-3143-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min JH, Kim BJ, Lee KH. Development of extensive brain lesions following fingolimod (FTY720) treatment in a patient with neuromyelitis optica spectrum disorder. Mult Scler. 2012;18:113–115. doi: 10.1177/1352458511431973. [DOI] [PubMed] [Google Scholar]
- Misu T, Fijihara K, Kakita A, Konno H, Nakamura W, Watanabe S, Takahashi T, Nakashima I, Takahashi H, Itoyama Y. Loss of aquaporin 4 in lesions of neuromyelitis optica: distinction from multiple sclerosis. Brain. 2007;130:1224–1234. doi: 10.1093/brain/awm047. [DOI] [PubMed] [Google Scholar]
- Mizokami J, Kanamori A, Negi A, Nakamura M. A preliminary study of reduced expression of aquaporin-9 in the optic nerve of private and human eyes with glaucoma. Curr Eye Res. 2011;36:1064–1067. doi: 10.3109/02713683.2011.611610. [DOI] [PubMed] [Google Scholar]
- Monteiro ML, Fernandes DB, Apóstolos-Pereira SL, Callegaro D. Quantification of retinal neural loss in patients with neuromyelitis optica and multiple sclerosis with or without optic neuritis using Fourier-domain optical coherence tomography. Invest Ophthalmol Vis Sci. 2012;53:3959–3966. doi: 10.1167/iovs.11-9324. [DOI] [PubMed] [Google Scholar]
- Nagelhus EA, Veruki ML, Torp R, Haug FM, Laake JH, Nielsen S, Agre P, Ottersen OP. Aquaporin-4 water channel protein in the rat retina and optic nerve: polarized expression in Müller cells and fibrous astrocytes. J Neurosci. 1998;18:2506–2519. doi: 10.1523/JNEUROSCI.18-07-02506.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naismith RT, Tutlam NT, Xu J, Klawiter EC, Shepherd J, Trinkaus K, Song SK, Cross AH. Optical coherence tomography differs in neuromyelitis optica compared with multiple sclerosis. Neurology. 2009;72:1077–1082. doi: 10.1212/01.wnl.0000345042.53843.d5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicchia GP, Cogotzi L, Rossi A, Basco D, Brancaccio A, Svelto M, Frigeri A. Expression of multiple AQP4 pools in the plasma membrane and their association with the dystrophin complex. J Neurochem. 2008;105:2156–2165. doi: 10.1111/j.1471-4159.2008.05302.x. [DOI] [PubMed] [Google Scholar]
- Nicchia GP, Mastrototaro M, Rossi A, Pisani F, Tortorella C, Ruggieri M, Lia A, Trojano M, Frigeri A, Svelto M. Aquaporin-4 orthogonal arrays of particles are the target for neuromyelitis optica autoantibodies. Glia. 2009;57:1363–1373. doi: 10.1002/glia.20855. [DOI] [PubMed] [Google Scholar]
- Nakamura M, Nakazawa T, Doi H, Hariya T, Omodaka K, Misu T, Takahashi T, Fujihara K, Nishida K. Early high-dose intravenous methylprednisolone is effective in preserving retinal nerve fiber layer thickness in patients with neuromyelitis optica. Graefes Arch Clin Exp Ophthalmol. 2010;248:1777–1785. doi: 10.1007/s00417-010-1344-7. [DOI] [PubMed] [Google Scholar]
- Nielsen S, Nagelhus EA, Amiry-Moghaddam M, Bourque C, Agre P, Ottersen OP. Specialized membrane domains for water transport in glial cells: high-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J Neurosci. 1997;17:171–180. doi: 10.1523/JNEUROSCI.17-01-00171.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noseworthy JH, O’Brien PC, Petterson TM, Weis J, Stevens L, Peterson WK, Sneve D, Cross SA, Leavitt JA, Auger RG, Weinshenker BG, Dodick DW, Wingerchuk DM, Rodriguez M. A randomized trial of intravenous immunoglobulin in inflammatory demyelinating optic neuritis. Neurology. 2001;56:1514–1522. doi: 10.1212/wnl.56.11.1514. [DOI] [PubMed] [Google Scholar]
- Optic Neuritis Study Group. Visual function 15 years after optic neuritis: a final follow-up report from the Optic Neuritis Treatment Trial. Ophthalmology. 2008;115:1079–1082. doi: 10.1016/j.ophtha.2007.08.004. [DOI] [PubMed] [Google Scholar]
- O’Riordan JI, Gallagher HL, Thompson AJ, Howard RS, Kingsley DP, Thompson EJ, McDonald WI, Miller DH. Clinical, CSF and MRI findings in Devic’s neuromyelitis optica. J Neurol Neurosurg Psychiat. 1996;60:382–387. doi: 10.1136/jnnp.60.4.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyama T, Abe H, Ushiki T. The connective tissue and glial framework in the optic nerve head of the normal human eye: light and scanning electron microscopic studies. Arch Histol Cytol. 2006;69:341–356. doi: 10.1679/aohc.69.341. [DOI] [PubMed] [Google Scholar]
- Palace J, Leite MI, Nairne A, Vincent A. Interferon Beta treatment in neuromyelitis optica: increase in relapses and aquaporin 4 antibody titers. Arch Neurol. 2010;67:1016–1017. doi: 10.1001/archneurol.2010.188. [DOI] [PubMed] [Google Scholar]
- Papadopoulos MC, Krishna S, Manley GT, Verkman AS. Aquaporin-4 facilitates the reabsorption of excess fluid in vasogenic brain edema. FASEB J. 2004;18:1291–1293. doi: 10.1096/fj.04-1723fje. [DOI] [PubMed] [Google Scholar]
- Papadopoulos MC, Kim JK, Verkman AS. Extracellular space diffusion in central nervous system: anisotropic diffusion measured by elliptical surface photobleaching. Biophys J. 2005;89:3660–3668. doi: 10.1529/biophysj.105.068114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papais-Alvarenga RM, Carellos SC, Alvarenga MP, Holander C, Bichara RP, Thuler LC. Clinical course of optic neuritis in patients with relapsing neuromyelitis optica. Arch Ophthalmol. 2008;126:12–16. doi: 10.1001/archophthalmol.2007.26. [DOI] [PubMed] [Google Scholar]
- Parratt JD, Prineas JW. Neuromyelitis optica: a demyelinating disease characterized by acute destruction and regeneration of perivascular astrocytes. Mult Scler. 2010;16:1156–1172. doi: 10.1177/1352458510382324. [DOI] [PubMed] [Google Scholar]
- Petzold A, Pittock S, Lennon V, Maggiore C, Weinshenker BG, Plant GT. Neuromyelitis optica-IgG (aquaporin-4) autoantibodies in immune mediated optic neuritis. J Neurol Neurosurg Psychiatry. 2010;81:109–111. doi: 10.1136/jnnp.2008.146894. [DOI] [PubMed] [Google Scholar]
- Phuan PW, Ratelade J, Rossi A, Tradtrantip L, Verkman AS. Complement-dependent cytotoxicity in neuromyelitis optica requires aquaporin-4 protein assembly in orthogonal arrays. J Biol Chem. 2012;287:13829–13839. doi: 10.1074/jbc.M112.344325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittock SJ, Lennon VA, de Seze J, Vermersch P, Homburger HA, Wingerchuk DM, Lucchinetti CF, Zéphir H, Moder K, Weinshenker BG. Neuromyelitis optica and non organ-specific autoimmunity. Arch Neurol. 2008;65:78–83. doi: 10.1001/archneurol.2007.17. [DOI] [PubMed] [Google Scholar]
- Pohl M, Fischer MT, Mader S, Schanda K, Kitic M, Sharma R, Wimmer I, Misu T, Fujihara K, Reindl M, Lassmann H, Bradl M. Pathogenic T cell responses against aquaporin 4. Acta Neuropathol. 2011;122:21–34. doi: 10.1007/s00401-011-0824-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popescu BF, Parisi JE, Cabrera-Gómez JA, Newell K, Mandler RN, Pittock SJ, Weinshenker BG, Lucchinetti CF. Absence of cortical demyelination in neuromyelitis optica. Neurology. 2010;75:2103–2109. doi: 10.1212/WNL.0b013e318200d80c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popescu BF, Lennon VA, Parisi JE, Howe CL, Weigand SD, Cabrera-Gómez JA, Newell K, Mandler RN, Pittock SJ, Weinshenker BG, Lucchinetti CF. Neuromyelitis optica unique area postrema lesions: nausea, vomiting, and pathogenic implications. Neurology. 2011;76:1229–1237. doi: 10.1212/WNL.0b013e318214332c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quek AM, McKeon A, Lennon VA, Mandrekar JN, Iorio R, Jiao Y, Costanzi C, Weinshenker BG, Wingerchuk DM, Lucchinetti CF, Shuster EA, Pittock SJ. Effects of age and sex on aquaporin-4 autoimmunity. Arch Neurol. 2012;69:1039–1043. doi: 10.1001/archneurol.2012.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratchford JN, Quigg ME, Conger A, Frohman T, Frohman E, Balcer LJ, Calabresi PA, Kerr DA. Optical coherence tomography helps differentiate neuromyelitis optica and MS optic neuropathies. Neurology. 2009;73:302–308. doi: 10.1212/WNL.0b013e3181af78b8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratelade J, Bennett JL, Verkman AS. Evidence against cellular internalization in vivo of NMO-IgG, aquaporin-4, and excitatory amino acid transporter 2 in neuromyelitis optica. J Biol Chem. 2011a;286:45156–45164. doi: 10.1074/jbc.M111.297275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratelade J, Bennett JL, Verkman AS. Intravenous neuromyelitis optica autoantibody in mice targets aquaporin-4 in peripheral organs and area postrema. PLoS One. 2011b;6:e27412. doi: 10.1371/journal.pone.0027412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratelade J, Zhang H, Saadoun S, Bennett JL, Papadopoulos MC, Verkman AS. Neuromyelitis optica IgG and natural killer cells produce NMO lesions in mice without myelin loss. Acta Neuropathol. 2012;123:861–872. doi: 10.1007/s00401-012-0986-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz N, Vaknin A, Chokron S, Ben-Hur T, Levin N. Functional MRI as a tool for assessing chiasmal visual defect in a patient with neuromyelitis optica. J Neurol Neurosurg Psychiatry. 2010;81:1174–1175. doi: 10.1136/jnnp.2009.183749. [DOI] [PubMed] [Google Scholar]
- Reid MV, Murray KA, Marsh ED, Golden JA, Simmons RA, Grinspan JB. Delayed myelination in an intrauterine growth retardation model is mediated by oxidative stress upregulating bone morphogenetic protein 4. J Neuropathol Exp Neurol. 2012;71:640–653. doi: 10.1097/NEN.0b013e31825cfa81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera JF, Kurtzke JF, Alatriste Booth VJ, Corona T. Characteristics of Devic’s disease (neuromyelitis optica) in Mexico. J Neurol. 2008;255:710–715. doi: 10.1007/s00415-008-0781-2. [DOI] [PubMed] [Google Scholar]
- Roed HG, Langkilde A, Sellebjerg F, Lauritzen M, Bang P, Morup A, Frederiksen JL. A double-blind, randomized trial of IV immunoglobulin treatment in acute optic neuritis. Neurology. 2005;64:804–810. doi: 10.1212/01.WNL.0000152873.82631.B3. [DOI] [PubMed] [Google Scholar]
- Roh M, Zhang Y, Murakami Y, Thanos A, Lee SC, Vavvas DG, Benowitz LI, Miller JW. Etanercept, a widely used inhibitor of tumor necrosis factor-α (TNF-α), prevents retinal ganglion cell loss in a rat model of glaucoma. PLoS One. 2012;7:e40065. doi: 10.1371/journal.pone.0040065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero RS, Gutierrez I, Wang E, Reder AT, Bhatti MT, Bernard JT, Javed A. Homonymous hemimacular thinning: a unique presentation of optic tract injury in neuromyelitis optica. J Neuroophthalmol. 2012;32:150–153. doi: 10.1097/WNO.0b013e3182504688. [DOI] [PubMed] [Google Scholar]
- Rossi A, Moritz TJ, Ratelade J, Verkman AS. Super-resolution imaging of aquaporin-4 orthogonal arrays of particles in cell plasma membranes. J Cell Sci. 2012a;125:4405–4412. doi: 10.1242/jcs.109603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi A, Ratelade J, Papadopoulos MC, Bennett JL, Verkman AS. Neuromyelitis optica (NMO) IgG does not alter aquaporin-4 water permeability, plasma membrane M1/M23 isoform content or supramolecular assembly. Glia. 2012b;60:2027–2039. doi: 10.1002/glia.22417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostasy K, Mader S, Schanda K, Huppke P, Gärtner J, Kraus V, Karenfort M, Tibussek D, Blaschek A, Bajer-Kornek B, Leitz S, Schimmel M, Di Pauli F, Berger T, Reindl M. Anti-myelin oligodendrocyte glycoprotein antibodies in pediatric patients with optic neuritis. Arch Neurol. 2012;69:752–756. doi: 10.1001/archneurol.2011.2956. [DOI] [PubMed] [Google Scholar]
- Ruprecht K, Klinker E, Dintelmann T, Rieckmann P, Gold R. Plasma exchange for severe optic neuritis: treatment of 10 patients. Neurology. 2004;63:1081–1083. doi: 10.1212/01.wnl.0000138437.99046.6b. [DOI] [PubMed] [Google Scholar]
- Saadoun S, Papadopoulos MC, Watanabe H, Yan D, Manley GT, Verkman AS. Involvement of aquaporin-4 in astroglial cell migration and glial scar formation. J Cell Sci. 2005;118:5691–5698. doi: 10.1242/jcs.02680. [DOI] [PubMed] [Google Scholar]
- Saadoun S, Waters P, Bell BA, Vincent A, Verkman AS, Papadopoulos MC. Intracerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain. 2010;133:349–361. doi: 10.1093/brain/awp309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadoun S, Waters P, Macdonald C, Bridges LR, Bell BA, Vincent A, Verkman AS, Papadopoulos MC. T cell deficiency does not reduce lesions in mice produced by intracerebral injection of NMO-IgG and complement. J Neuroimmunol. 2011;235:27–32. doi: 10.1016/j.jneuroim.2011.03.007. [DOI] [PubMed] [Google Scholar]
- Saadoun S, Bridges LR, Verkman AS, Papadopoulos MC. Paucity of natural killer and cytotoxic T cells in human neuromyelitis optica lesions. Neuroreport. 2012a;23:1044–1047. doi: 10.1097/WNR.0b013e32835ab480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadoun S, Waters P, MacDonald C, Bell BA, Vincent A, Verkman AS, Papadopoulos MC. Neutrophil protease inhibition reduces neuromyelitis optica-immunoglobulin G-induced damage in mouse brain. Ann Neurol. 2012b;71:323–333. doi: 10.1002/ana.22686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saidha S, Sotirchos ES, Ibrahim MA, Crainiceanu CM, Gelfand JM, Sepah YJ, Ratchford JN, Oh J, Seigo MA, Newsome SD, Balcer LJ, Frohman EM, Green AJ, Nguyen QD, Calabresi PA. Microcystic macular oedema, thickness of the inner nuclear layer of the retina, and disease characteristics in multiple sclerosis: a retrospective study. Lancet Neurol. 2012;11:963–972. doi: 10.1016/S1474-4422(12)70213-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini H, Fernandez G, Kerr D, Levy M. Differential expression of aquaporin-4 isoforms localizes with neuromyelitis optica disease activity. J Neuroimmunol. 2010;221:68–72. doi: 10.1016/j.jneuroim.2010.02.007. [DOI] [PubMed] [Google Scholar]
- Sawaishi Y. Review of Alexander disease: beyond the classical concept of leukodystrophy. Brain Dev. 2009;31:493–498. doi: 10.1016/j.braindev.2009.03.006. [DOI] [PubMed] [Google Scholar]
- Sharma R, Fischer MT, Bauer J, Felts PA, Smith KJ, Misu T, Fujihara K, Bradl M, Lassmann H. Inflammation induced by innate immunity in the central nervous system leads to primary astrocyte dysfunction followed by demyelination. Acta Neuropathol. 2010;120:223–236. doi: 10.1007/s00401-010-0704-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotirchos ES, Saidha S, Byraiah G, Mealy MA, Ibrahim MA, Sepah YJ, Newsome SD, Ratchford JN, Frohman EM, Balcer LJ, Nguyen QD, Levy M, Calabresi PA. In-vivo identification of a novel retinal pathology in neuromyelitis optica. ECTRIMS. 2012 doi: 10.1212/WNL.0b013e31828c2f7a. 2012 Meeting Abstracts P657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Lye-Barthel M, Masland RH, Jakobs TC. The morphology and spatial arrangement of astrocytes in the optic nerve head of the mouse. J Compar Neurol. 2009;516:1–19. doi: 10.1002/cne.22058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syc SB, Saidha S, Newsome SD, Ratchford JN, Levy M, Ford E, Crainiceanu CM, Durbin MK, Oakley JD, Meyer SA, Frohman EM, Calabresi PA. Optical coherence tomography segmentation reveals ganglion cell layer pathology after optic neuritis. Brain. 2012;135:521–533. doi: 10.1093/brain/awr264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima M, Crane JM, Verkman AS. Aquaporin-4 (AQP4) associations and array dynamics probed by photobleaching and single-molecule analysis of green fluorescent protein-AQP4 chimeras. J Biol Chem. 2010;285:8163–8170. doi: 10.1074/jbc.M109.093948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tradtrantip L, Zhang H, Anderson MO, Saadoun S, Phuan PW, Papadopoulos MC, Bennett JL, Verkman AS. Small-molecule inhibitors of NMO-IgG binding to aquaporin-4 reduce astrocyte cytotoxicity in neuromyelitis optica. FASEB J. 2012a;26:2197–2208. doi: 10.1096/fj.11-201608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tradtrantip L, Zhang H, Saadoun S, Phuan PW, Lam C, Papadopoulos MC, Bennett JL, Verkman AS. Anti-aquaporin-4 monoclonal antibody blocker therapy for neuromyelitis optica. Ann Neurol. 2012b;71:314–322. doi: 10.1002/ana.22657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tradtrantip L, Ratelade J, Zhang H, Verkman AS. Enzymatic deglycosylation converts pathogenic neuromyelitis optica anti-aquaporin-4 immunoglobulin G into therapeutic antibody. Ann Neurol. 2013;73:77–85. doi: 10.1002/ana.23741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triviño A, Ramírez JM, Salazar JJ, Ramírez AI, García-Sanchez J. Immunohistochemical study of human optic nerve head astroglia. Vision Res. 1996;36:2015–2028. doi: 10.1016/0042-6989(95)00317-7. [DOI] [PubMed] [Google Scholar]
- Varrin-Doyer M, Spencer CM, Schulze-Topphoff U, Nelson PA, Stroud RM, Cree BA, Zamvil SS. Aquaporin-4-specific T cells in neuromyelitis optica exhibit a Th17 bias and recognize Clostridium ABC transporter. Ann Neurol. 2012;72:53–64. doi: 10.1002/ana.23651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkman AS. Aquaporins at a glance. J Cell Sci. 2011;124:2107–2112. doi: 10.1242/jcs.079467. [DOI] [PubMed] [Google Scholar]
- Wandinger KP, Stangel M, Witte T, Venables P, Charles P, Jarius S, Wildemann B, Probst C, Iking-Konert C, Schneider M. Autoantibodies against aquaporin-4 in patients with neuropsychiatric systemic lupus erythematous and primary Sjogren’s syndrome. Arthritis Rheum. 2010;62:1198–1200. doi: 10.1002/art.27337. [DOI] [PubMed] [Google Scholar]
- Wang H, Dai Y, Qiu W, Zhong X, Wu A, Wang Y, Lu Z, Bao J, Hu X. HLA-DPB10501 is associated with susceptibility to anti-aquaporin-4 antibodies positive neuromyelitis optica in southern Han Chinese. J Neuroimmunol. 2011;233:181–184. doi: 10.1016/j.jneuroim.2010.11.004. [DOI] [PubMed] [Google Scholar]
- Watanabe S, Nakashima I, Misu T, Miyazawa I, Shiga Y, Fujihara K, Itoyama Y. Therapeutic efficacy of plasma exchange in NMO-IgG-positive patients with neuromyelitis optica. Mult Scler. 2007;13:968–974. doi: 10.1177/1352458506071174. [DOI] [PubMed] [Google Scholar]
- Waters PJ, McKeon A, Leite MI, Rajasekharan S, Lennon VA, Villalobos A, Palace J, Mandrekar JN, Vincent A, Bar-Or A, Pittock SJ. Serologic diagnosis of NMO: a multicenter comparison of aquaporin-4-IgG assays. Neurology. 2012;78:665–671. doi: 10.1212/WNL.0b013e318248dec1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinshenker BG, Wingerchuk DM, Vukusic S, Linbo L, Pittock SJ, Lucchinetti CF, Lennon VA. Neuromyelitis optica IgG predicts relapse after longitudinally extensive transverse myelitis. Ann Neurol. 2006;59:566–569. doi: 10.1002/ana.20770. [DOI] [PubMed] [Google Scholar]
- Wilke S, Thomas R, Allcock N, Fern R. Mechanism of acute injury to oligodendroglia in early myelinating white matter: the importance of astrocyte injury and glutamate release. J Neuropathol Exp Neurol. 2004;63:872–881. doi: 10.1093/jnen/63.8.872. [DOI] [PubMed] [Google Scholar]
- Wingerchuk DM, Hogancamp WE, O’Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic’s syndrome) Neurology. 1999;53:1107–1114. doi: 10.1212/wnl.53.5.1107. [DOI] [PubMed] [Google Scholar]
- Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology. 2006;66:1485–1489. doi: 10.1212/01.wnl.0000216139.44259.74. [DOI] [PubMed] [Google Scholar]
- Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol. 2007;6:805–815. doi: 10.1016/S1474-4422(07)70216-8. [DOI] [PubMed] [Google Scholar]
- Wohl SG, Schmeer CW, Witte OW, Isenmann S. Proliferative response of microglia and macrophages in the adult mouse eye after optic nerve lesion. Invest Ophthalmol Vis Sci. 2010;51:2686–2696. doi: 10.1167/iovs.09-4537. [DOI] [PubMed] [Google Scholar]
- Yang B, Brown D, Verkman AS. The mercurial-insensitive water channel (AQP-4) forms orthogonal arrays in stably transfected CHO cells. J Biol Chem. 1996;271:4577–4580. [PubMed] [Google Scholar]
- Yang B, Ma T, Verkman AS. cDNA cloning, gene organization and chromosomal localization of a human mercurial-insensitive water channel: evidence for distinct transcriptional units. J Biol Chem. 1995;270:22907–22913. doi: 10.1074/jbc.270.39.22907. [DOI] [PubMed] [Google Scholar]
- Zéphir H, Fajardy I, Outteryck O, Blanc F, Roger N, Fleury M, Rudolf G, Marignier R, Vukusic S, Confavreux C, Vermersch P, de Seze J. Is neuromyelitis optica associated with Human Leukocyte antigen? Mult Scler. 2009;15:571–579. doi: 10.1177/1352458508102085. [DOI] [PubMed] [Google Scholar]
- Zhang H, Bennett JL, Verkman AS. Ex vivo spinal cord slice model of neuromyelitis optica reveals novel immunopathogenic mechanisms. Ann Neurol. 2011;201170:943–954. doi: 10.1002/ana.22551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Verkman AS. Eosinophil pathogenicity mechanisms and therapeutics in neuromyelitis optica. J Clin Invest. 2013 doi: 10.1172/JCI67554. 2013 In press. [DOI] [PMC free article] [PubMed] [Google Scholar]