

Abstract
The title molecular salt, C7H9N2 +.C8H7O3 −, was synthesized by reaction between benzamidine (benzenecarboximidamide) and 2-methoxybenzoic acid. In the cation, the amidinium group has two similar C—N bonds [1.3070 (17) and 1.3145 (16) Å] and is almost coplanar with the benzene ring, making a dihedral angle of 5.34 (12)°. In the anion, the methoxy substituent forces the carboxylate group to be twisted by 69.45 (6)° with respect to the plane of the aromatic fragment. In the crystal, the components are connected by two N+—H⋯O− (±)CAHB (charge-assisted hydrogen bonds), forming centrosymmetric ionic dimers with graph-set motif R 2 2(8). These ionic dimers are then joined in ribbons running along the b-axis direction by another R 4 2(8) motif involving the remaining N+—H⋯O− hydrogen bonds. Remarkably, at variance with the well known carboxylic dimer R 2 2(8) motif, the carboxylate–amidinium pair is not planar, the dihedral angle between the planes defined by the CN2 + and CO2 − atoms being 18.57 (12)°.
Related literature
For the biological and pharmacological relevance of benzamidine, see: Powers & Harper (1999 ▶). For structural analysis of proton-transfer adducts containing molecules of biological interest, see: Portalone (2010 ▶, 2013 ▶). For the supramolecular association in proton-transfer adducts containing benzamidinium cations, see: Portalone (2010 ▶, 2012 ▶, 2013 ▶); Irrera & Portalone (2012 ▶, 2013 ▶); Irrera et al. (2012 ▶). For 2-methoxybenzoic acid derivatives, see: Portalone (2011 ▶). For hydrogen-bond motifs, see: Bernstein et al. (1995 ▶).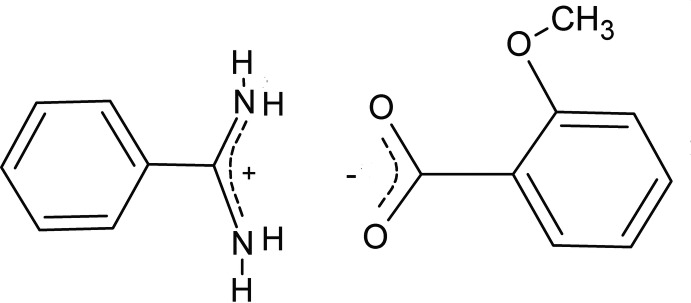
Experimental
Crystal data
C7H9N2 +·C8H7O3 −
M r = 272.30
Triclinic,
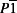
a = 7.5154 (3) Å
b = 9.1393 (4) Å
c = 11.6498 (5) Å
α = 69.612 (3)°
β = 80.500 (5)°
γ = 72.482 (4)°
V = 713.63 (6) Å3
Z = 2
Mo Kα radiation
μ = 0.09 mm−1
T = 298 K
0.12 × 0.09 × 0.05 mm
Data collection
Agilent Xcalibur Sapphire3 diffractometer
Absorption correction: multi-scan (CrysAlis PRO; Agilent, 2011 ▶) T min = 0.989, T max = 0.996
20559 measured reflections
4332 independent reflections
3188 reflections with I > 2σ(I)
R int = 0.025
Refinement
R[F 2 > 2σ(F 2)] = 0.062
wR(F 2) = 0.161
S = 1.05
4332 reflections
199 parameters
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.38 e Å−3
Δρmin = −0.18 e Å−3
Data collection: CrysAlis PRO (Agilent, 2011 ▶); cell refinement: CrysAlis PRO; data reduction: CrysAlis PRO; program(s) used to solve structure: SIR97 (Altomare et al., 1999 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: ORTEP-3 for Windows (Farrugia, 2012 ▶); software used to prepare material for publication: WinGX (Farrugia, 2012 ▶).
Supplementary Material
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S1600536813016395/nk2209sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536813016395/nk2209Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1A⋯O1 | 0.913 (19) | 1.87 (2) | 2.7777 (16) | 172.4 (17) |
| N1—H1B⋯O1i | 0.87 (2) | 1.97 (2) | 2.7926 (15) | 155.7 (17) |
| N2—H2A⋯O2 | 0.959 (19) | 1.93 (2) | 2.8863 (17) | 175.4 (16) |
| N2—H2B⋯O2ii | 0.86 (2) | 2.00 (2) | 2.8230 (16) | 160.3 (18) |
Symmetry codes: (i) 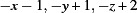 ; (ii)
; (ii) 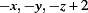 .
.
supplementary crystallographic information
Comment
The present study is a continuation of the work carried out in this Laboratory on proton-transfer adducts containing molecules of biological interest (Portalone, 2010, 2013), and deals with the single-crystal structure of the molecular salt, benzamidinium 2-methoxybenzoate, (I), obtained by a reaction between benzamidine (benzenecarboximidamide) and 2-methoxybenzoic acid in water solution. Benzamidine derivatives, which have shown strong biological and pharmacological activity (Powers & Harper, 1999), are being used in this Laboratory as bricks for supramolecular construction (Portalone, 2010). Indeed, these molecules are strong Lewis base and their cations can be easily anchored onto numerous inorganic and organic anions and polyanions, largely because of the presence of four potential donor sites for hydrogen-bonding.
The asymmetric unit of (I) comprises one planar benzamidinium cation and one 2-methoxybenzoate anion (Fig. 1).
In the cation the amidinium group forms a dihedral angle of 5.34 (12)° with the mean plane of the phenyl ring, at variance with the values observed in protonated benzamidinium ions (23.2 - 31.1°, Portalone, 2010; Portalone, 2013; Irrera et al., 2012; Irrera & Portalone, 2012, 2013). The pattern of bond lengths and bond angles of the benzamidinium cation agrees with that reported in previous structural investigations. In particular the amidinium group, true to one's expectations, features similar C—N bonds [1.3070 (17) and 1.3145 (16) Å], evidencing the delocalization of the π electrons and double-bond character.
In the 2-methoxybenzoate anion the benzene ring is essentially planar and the methoxy substituent forces the carboxylate group to be twisted with respect to the plane of the aromatic fragment by 69.45 (6)°. In the anion bond lengths and bond angles of the benzene ring are in accord with corresponding values obtained for both the orthorhombic and tetragonal forms of 2,6-dimethoxybenzoic acid (Portalone, 2011 and reference therein) and 4-methoxybenzamidinium 2,6-dimethoxybenzoate (Portalone, 2012). The C—O distances of the carboxylate group, 1.2441 (16) and 1.2488 (15) Å, indicate the delocalization of the negative charge.
The molecular components of the molecular salt are connected by two N+—H···O- (±)CAHB hydrogen bonds to form ionic dimers with graph-set motif R22(8) (Bernstein et al., 1995). Furthermore, centrosymmetric ionic dimers are joined in ribbons running along the b axis by another R24(8) motif involving the remaining N+—H···O- hydrogen bonds (Fig. 2). Remarkably, at variance with the well known carboxylic dimer R22 (8) motif, the carboxylate-amidinium pair is not planar, as the dihedral angle for the planes defined by the CN2+ and CO2- atoms is equal to 18.57 (12)°.
Experimental
Equimolar amounts (0.1 mmol) of benzamidine (Fluka at 96% purity) and 2-methoxybenzoic acid (Aldrich at 99% purity) were dissolved without further purification in 6 ml of hot water and heated under reflux for 6 h. After cooling the solution to an ambient temperature, colourless crystals suitable for single-crystal X-ray diffraction were grown by slow evaporation of the solvent after two weeks.
Refinement
All H atoms were identified in difference Fourier maps, but for refinement all C-bound H atoms were placed in calculated positions, with C—H = 0.97 Å (phenyl) and 1.01 Å (methyl), and refined as riding on their carrier atoms. The Uiso values were kept equal to 1.2Ueq(C, phenyl). and to 1.5Ueq(C, methyl). The hydrogen atoms of the methyl group were allowed to rotate with a fixed angle around the C–C bond to best fit the experimental electron density [HFIX 138 in the SHELX program suite (Sheldrick, 2008)]. Positional and thermal parameters of H atoms of the amidinium group were freely refined, giving N—H distances in the range 0.86 (2) - 0.96 (2) Å.
Figures
Fig. 1.
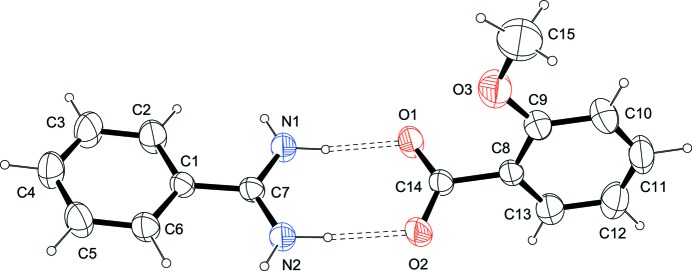
The asymmetric unit of (I), showing the atom-labelling scheme. Displacements ellipsoids are at the 50% probability level. The asymmetric unit was selected so that the two ions are linked by N—H+···O- hydrogen bonds. H atoms are shown as small spheres of arbitrary radii.
Fig. 2.

Crystal packing diagram for (I), viewed approximately down a. All atoms are shown as small spheres of arbitrary radii. For the sake of clarity, H atoms not involved in hydrogen bonding have been omitted. Hydrogen bonding is indicated by dashed lines.
Crystal data
| C7H9N2+·C8H7O3− | Z = 2 |
| Mr = 272.30 | F(000) = 288 |
| Triclinic, P1 | Dx = 1.267 Mg m−3 |
| Hall symbol: -P 1 | Mo Kα radiation, λ = 0.71069 Å |
| a = 7.5154 (3) Å | Cell parameters from 9004 reflections |
| b = 9.1393 (4) Å | θ = 3.2–32.5° |
| c = 11.6498 (5) Å | µ = 0.09 mm−1 |
| α = 69.612 (3)° | T = 298 K |
| β = 80.500 (5)° | Tablets, colourless |
| γ = 72.482 (4)° | 0.12 × 0.09 × 0.05 mm |
| V = 713.63 (6) Å3 |
Data collection
| Agilent Xcalibur Sapphire3 diffractometer | 4332 independent reflections |
| Radiation source: Enhance (Mo) X-ray Source | 3188 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.025 |
| Detector resolution: 16.0696 pixels mm-1 | θmax = 30.5°, θmin = 3.2° |
| ω and φ scans | h = −10→10 |
| Absorption correction: multi-scan (CrysAlis PRO; Agilent, 2011) | k = −13→13 |
| Tmin = 0.989, Tmax = 0.996 | l = −16→16 |
| 20559 measured reflections |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.062 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.161 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.05 | w = 1/[σ2(Fo2) + (0.0812P)2 + 0.1295P] where P = (Fo2 + 2Fc2)/3 |
| 4332 reflections | (Δ/σ)max < 0.001 |
| 199 parameters | Δρmax = 0.38 e Å−3 |
| 0 restraints | Δρmin = −0.18 e Å−3 |
Special details
| Geometry. All s.u.'s (except the s.u. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell s.u.'s are taken into account individually in the estimation of s.u.'s in distances, angles and torsion angles; correlations between s.u.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell s.u.'s is used for estimating s.u.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > 2σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| N1 | −0.44074 (18) | 0.33628 (15) | 0.94036 (13) | 0.0426 (3) | |
| H1A | −0.369 (3) | 0.355 (2) | 0.9866 (17) | 0.052 (5)* | |
| H1B | −0.550 (3) | 0.403 (2) | 0.9196 (17) | 0.053 (5)* | |
| N2 | −0.21153 (17) | 0.11282 (17) | 0.93377 (13) | 0.0464 (3) | |
| H2A | −0.139 (3) | 0.138 (2) | 0.9810 (17) | 0.055 (5)* | |
| H2B | −0.167 (3) | 0.022 (3) | 0.9197 (18) | 0.059 (5)* | |
| C1 | −0.50196 (17) | 0.16439 (15) | 0.84181 (11) | 0.0319 (3) | |
| C2 | −0.6776 (2) | 0.2674 (2) | 0.81080 (18) | 0.0531 (4) | |
| H2 | −0.7178 | 0.3693 | 0.8281 | 0.064* | |
| C3 | −0.7954 (2) | 0.2254 (2) | 0.75542 (18) | 0.0602 (5) | |
| H3 | −0.9172 | 0.2986 | 0.7333 | 0.072* | |
| C4 | −0.7414 (2) | 0.0816 (2) | 0.73164 (15) | 0.0508 (4) | |
| H4 | −0.8254 | 0.0522 | 0.6939 | 0.061* | |
| C5 | −0.5678 (2) | −0.0216 (2) | 0.76121 (16) | 0.0504 (4) | |
| H5 | −0.5291 | −0.1233 | 0.7435 | 0.060* | |
| C6 | −0.4480 (2) | 0.01930 (18) | 0.81621 (14) | 0.0418 (3) | |
| H6 | −0.3257 | −0.0539 | 0.8369 | 0.050* | |
| C7 | −0.38026 (17) | 0.20627 (15) | 0.90698 (12) | 0.0323 (3) | |
| O1 | −0.24816 (15) | 0.39582 (13) | 1.09618 (11) | 0.0513 (3) | |
| O2 | −0.00870 (15) | 0.18920 (14) | 1.08560 (11) | 0.0536 (3) | |
| O3 | −0.24579 (19) | 0.2238 (2) | 1.37136 (12) | 0.0694 (4) | |
| C8 | −0.00944 (18) | 0.31556 (15) | 1.23202 (12) | 0.0335 (3) | |
| C9 | −0.0897 (2) | 0.2793 (2) | 1.35187 (14) | 0.0434 (3) | |
| C10 | −0.0097 (3) | 0.2988 (2) | 1.44339 (15) | 0.0572 (4) | |
| H10 | −0.0654 | 0.2737 | 1.5268 | 0.069* | |
| C11 | 0.1493 (3) | 0.3540 (2) | 1.41503 (17) | 0.0578 (5) | |
| H11 | 0.2044 | 0.3678 | 1.4790 | 0.069* | |
| C12 | 0.2300 (3) | 0.3895 (2) | 1.29805 (18) | 0.0567 (4) | |
| H12 | 0.3417 | 0.4281 | 1.2788 | 0.068* | |
| C13 | 0.1494 (2) | 0.3694 (2) | 1.20632 (14) | 0.0466 (4) | |
| H13 | 0.2066 | 0.3940 | 1.1233 | 0.056* | |
| C14 | −0.09582 (18) | 0.29772 (15) | 1.13074 (12) | 0.0335 (3) | |
| C15 | −0.3237 (4) | 0.1697 (4) | 1.4934 (2) | 0.0938 (8) | |
| H15A | −0.2241 (19) | 0.083 (2) | 1.5466 (11) | 0.141* | |
| H15B | −0.430 (3) | 0.123 (2) | 1.4938 (3) | 0.141* | |
| H15C | −0.372 (3) | 0.2639 (17) | 1.5268 (9) | 0.141* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| N1 | 0.0387 (6) | 0.0361 (6) | 0.0566 (8) | 0.0057 (5) | −0.0185 (5) | −0.0255 (6) |
| N2 | 0.0370 (6) | 0.0443 (7) | 0.0641 (8) | 0.0087 (5) | −0.0192 (6) | −0.0341 (7) |
| C1 | 0.0328 (6) | 0.0316 (6) | 0.0304 (6) | −0.0023 (5) | −0.0065 (5) | −0.0122 (5) |
| C2 | 0.0485 (8) | 0.0434 (8) | 0.0714 (11) | 0.0097 (7) | −0.0279 (8) | −0.0309 (8) |
| C3 | 0.0469 (9) | 0.0595 (10) | 0.0779 (12) | 0.0077 (8) | −0.0332 (8) | −0.0317 (9) |
| C4 | 0.0538 (9) | 0.0555 (9) | 0.0506 (9) | −0.0146 (7) | −0.0188 (7) | −0.0189 (7) |
| C5 | 0.0590 (9) | 0.0429 (8) | 0.0573 (9) | −0.0076 (7) | −0.0158 (7) | −0.0253 (7) |
| C6 | 0.0410 (7) | 0.0373 (7) | 0.0483 (8) | 0.0006 (6) | −0.0120 (6) | −0.0202 (6) |
| C7 | 0.0325 (6) | 0.0305 (6) | 0.0331 (6) | −0.0015 (5) | −0.0057 (5) | −0.0133 (5) |
| O1 | 0.0498 (6) | 0.0453 (6) | 0.0616 (7) | 0.0158 (5) | −0.0293 (5) | −0.0328 (5) |
| O2 | 0.0462 (6) | 0.0537 (7) | 0.0690 (7) | 0.0137 (5) | −0.0211 (5) | −0.0442 (6) |
| O3 | 0.0628 (8) | 0.1107 (12) | 0.0494 (7) | −0.0439 (8) | 0.0064 (6) | −0.0300 (7) |
| C8 | 0.0341 (6) | 0.0297 (6) | 0.0363 (6) | 0.0028 (5) | −0.0105 (5) | −0.0156 (5) |
| C9 | 0.0415 (7) | 0.0506 (8) | 0.0409 (7) | −0.0066 (6) | −0.0066 (6) | −0.0208 (6) |
| C10 | 0.0624 (10) | 0.0752 (12) | 0.0384 (8) | −0.0115 (9) | −0.0104 (7) | −0.0258 (8) |
| C11 | 0.0645 (11) | 0.0638 (11) | 0.0548 (10) | −0.0087 (9) | −0.0253 (8) | −0.0279 (8) |
| C12 | 0.0536 (9) | 0.0614 (10) | 0.0652 (11) | −0.0192 (8) | −0.0166 (8) | −0.0232 (9) |
| C13 | 0.0472 (8) | 0.0541 (9) | 0.0420 (8) | −0.0150 (7) | −0.0045 (6) | −0.0175 (7) |
| C14 | 0.0344 (6) | 0.0301 (6) | 0.0366 (6) | −0.0001 (5) | −0.0088 (5) | −0.0156 (5) |
| C15 | 0.0826 (16) | 0.144 (2) | 0.0608 (13) | −0.0546 (17) | 0.0201 (12) | −0.0299 (14) |
Geometric parameters (Å, º)
| N1—C7 | 1.3070 (17) | O1—C14 | 1.2488 (15) |
| N1—H1A | 0.913 (19) | O2—C14 | 1.2441 (16) |
| N1—H1B | 0.87 (2) | O3—C9 | 1.369 (2) |
| N2—C7 | 1.3145 (16) | O3—C15 | 1.421 (3) |
| N2—H2A | 0.959 (19) | C8—C13 | 1.375 (2) |
| N2—H2B | 0.86 (2) | C8—C9 | 1.394 (2) |
| C1—C6 | 1.3869 (19) | C8—C14 | 1.5135 (17) |
| C1—C2 | 1.3898 (19) | C9—C10 | 1.393 (2) |
| C1—C7 | 1.4838 (18) | C10—C11 | 1.381 (3) |
| C2—C3 | 1.383 (2) | C10—H10 | 0.9700 |
| C2—H2 | 0.9700 | C11—C12 | 1.367 (3) |
| C3—C4 | 1.366 (2) | C11—H11 | 0.9700 |
| C3—H3 | 0.9700 | C12—C13 | 1.399 (2) |
| C4—C5 | 1.375 (2) | C12—H12 | 0.9700 |
| C4—H4 | 0.9700 | C13—H13 | 0.9700 |
| C5—C6 | 1.386 (2) | C15—H15A | 1.0120 |
| C5—H5 | 0.9700 | C15—H15B | 1.0120 |
| C6—H6 | 0.9700 | C15—H15C | 1.0120 |
| C7—N1—H1A | 119.2 (12) | C13—C8—C9 | 119.07 (13) |
| C7—N1—H1B | 120.3 (12) | C13—C8—C14 | 120.07 (12) |
| H1A—N1—H1B | 120.5 (17) | C9—C8—C14 | 120.86 (12) |
| C7—N2—H2A | 119.6 (11) | O3—C9—C10 | 124.03 (15) |
| C7—N2—H2B | 123.2 (13) | O3—C9—C8 | 115.99 (13) |
| H2A—N2—H2B | 116.7 (17) | C10—C9—C8 | 119.97 (15) |
| C6—C1—C2 | 118.65 (13) | C11—C10—C9 | 119.83 (16) |
| C6—C1—C7 | 121.19 (11) | C11—C10—H10 | 120.1 |
| C2—C1—C7 | 120.08 (12) | C9—C10—H10 | 120.1 |
| C3—C2—C1 | 120.39 (14) | C12—C11—C10 | 120.83 (15) |
| C3—C2—H2 | 119.8 | C12—C11—H11 | 119.6 |
| C1—C2—H2 | 119.8 | C10—C11—H11 | 119.6 |
| C4—C3—C2 | 120.44 (15) | C11—C12—C13 | 119.22 (16) |
| C4—C3—H3 | 119.8 | C11—C12—H12 | 120.4 |
| C2—C3—H3 | 119.8 | C13—C12—H12 | 120.4 |
| C3—C4—C5 | 119.99 (14) | C8—C13—C12 | 121.07 (15) |
| C3—C4—H4 | 120.0 | C8—C13—H13 | 119.5 |
| C5—C4—H4 | 120.0 | C12—C13—H13 | 119.5 |
| C4—C5—C6 | 120.16 (15) | O2—C14—O1 | 124.40 (12) |
| C4—C5—H5 | 119.9 | O2—C14—C8 | 118.04 (11) |
| C6—C5—H5 | 119.9 | O1—C14—C8 | 117.53 (11) |
| C5—C6—C1 | 120.36 (13) | O3—C15—H15A | 109.5 |
| C5—C6—H6 | 119.8 | O3—C15—H15B | 109.5 |
| C1—C6—H6 | 119.8 | H15A—C15—H15B | 109.5 |
| N1—C7—N2 | 118.81 (13) | O3—C15—H15C | 109.5 |
| N1—C7—C1 | 120.25 (11) | H15A—C15—H15C | 109.5 |
| N2—C7—C1 | 120.93 (12) | H15B—C15—H15C | 109.5 |
| C9—O3—C15 | 118.79 (15) | ||
| C6—C1—C2—C3 | −0.1 (3) | C14—C8—C9—O3 | 1.6 (2) |
| C7—C1—C2—C3 | −176.85 (16) | C13—C8—C9—C10 | 0.4 (2) |
| C1—C2—C3—C4 | 0.7 (3) | C14—C8—C9—C10 | −179.03 (14) |
| C2—C3—C4—C5 | −0.9 (3) | O3—C9—C10—C11 | 179.31 (17) |
| C3—C4—C5—C6 | 0.6 (3) | C8—C9—C10—C11 | −0.1 (3) |
| C4—C5—C6—C1 | 0.0 (3) | C9—C10—C11—C12 | −0.2 (3) |
| C2—C1—C6—C5 | −0.2 (2) | C10—C11—C12—C13 | 0.1 (3) |
| C7—C1—C6—C5 | 176.50 (14) | C9—C8—C13—C12 | −0.6 (2) |
| C6—C1—C7—N1 | −173.63 (14) | C14—C8—C13—C12 | 178.91 (14) |
| C2—C1—C7—N1 | 3.1 (2) | C11—C12—C13—C8 | 0.3 (3) |
| C6—C1—C7—N2 | 5.4 (2) | C13—C8—C14—O2 | 68.85 (18) |
| C2—C1—C7—N2 | −177.93 (15) | C9—C8—C14—O2 | −111.69 (16) |
| C15—O3—C9—C10 | −5.5 (3) | C13—C8—C14—O1 | −109.51 (16) |
| C15—O3—C9—C8 | 173.93 (19) | C9—C8—C14—O1 | 69.95 (18) |
| C13—C8—C9—O3 | −178.98 (14) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1A···O1 | 0.913 (19) | 1.87 (2) | 2.7777 (16) | 172.4 (17) |
| N1—H1B···O1i | 0.87 (2) | 1.97 (2) | 2.7926 (15) | 155.7 (17) |
| N2—H2A···O2 | 0.959 (19) | 1.93 (2) | 2.8863 (17) | 175.4 (16) |
| N2—H2B···O2ii | 0.86 (2) | 2.00 (2) | 2.8230 (16) | 160.3 (18) |
Symmetry codes: (i) −x−1, −y+1, −z+2; (ii) −x, −y, −z+2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: NK2209).
References
- Agilent (2011). CrysAlis PRO Agilent Technologies Ltd, Abingdon, England.
- Altomare, A., Burla, M. C., Camalli, M., Cascarano, G. L., Giacovazzo, C., Guagliardi, A., Moliterni, A. G. G., Polidori, G. & Spagna, R. (1999). J. Appl. Cryst. 32, 115–119.
- Bernstein, J., Davis, R. E., Shimoni, L. & Chang, N.-L. (1995). Angew. Chem. Int. Ed. Engl. 34, 1555–1573.
- Farrugia, L. J. (2012). J. Appl. Cryst. 45, 849–854.
- Irrera, S., Ortaggi, G. & Portalone, G. (2012). Acta Cryst. C68, o447–o451. [DOI] [PubMed]
- Irrera, S. & Portalone, G. (2012). Acta Cryst. E68, o3083. [DOI] [PMC free article] [PubMed]
- Irrera, S. & Portalone, G. (2013). Acta Cryst. E69, o56. [DOI] [PMC free article] [PubMed]
- Portalone, G. (2010). Acta Cryst. C66, o295–o301. [DOI] [PubMed]
- Portalone, G. (2011). Acta Cryst. E67, o3394–o3395. [DOI] [PMC free article] [PubMed]
- Portalone, G. (2012). Acta Cryst. E68, o268–o269. [DOI] [PMC free article] [PubMed]
- Portalone, G. (2013). Acta Cryst. E69, o14–o15. [DOI] [PMC free article] [PubMed]
- Powers, J. C. & Harper, J. W. (1999). Proteinase inhibitors, edited by A. J. Barrett & G. Salvesen, pp. 55–152. Amsterdam: Elsevier.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S1600536813016395/nk2209sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536813016395/nk2209Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report