

Abstract
Prolonged highly active anti-retroviral therapy (HAART) with multiple nucleoside reverse transcriptase inhibitors (NRTI) for the treatment of human immunodeficiency virus type 1 (HIV-1) infected patients can induce the development of an HIV-1 RT harboring a dipeptide insertion at the RT fingers domain with a background thymidine analog mutation (TAM). This mutation renders viral resistance to multiple NRTIs. We investigated the effect of the dipeptide fingers domain insertion mutation on strand transfer activity using two clinical RT variants isolated during pre- and post-treatment of an infected patient, termed pre-drug RT without the dipeptide insertion and post-drug RT with the Ser-Gly insertion mutation, respectively. First, the post-drug RT displayed elevated strand transfer activity, compared to the pre-drug RT, with two different RNA templates. Second, the post-drug RT exhibited less RNA template degradation than the pre-drug RT, but higher polymerization-dependent RNase H activity. Third, the post-drug RT had a faster association rate for template binding (kon) and lower equilibrium binding constant KD to template, leading to the tighter template binding affinity than the pre-drug RT. The koff rates for pre-drug RT and post-drug RTs were similar. Finally, the removal of the dipeptide insertion from the post-drug RT abolished the elevated strand transfer activity and RNase H activity in addition to the loss of AZT resistance. These biochemical data suggests that the dipeptide insertion mutation elevates strand transfer activity by increasing the interaction of the RT with RNA donor template, promoting cleavage that generates more invasion site for the acceptor template during DNA synthesis.
Keywords: HIV-1, reverse transcriptase, multiple-drug resistance, finger domain insertion, strand transfer
There are currently more than 17 anti-HIV-1 drugs clinically approved for the treatment of HIV-1 infected patients 1. These anti-HIV drugs belong to six different classes, which include the most widely used anti-HIV-1 inhibitors targeting HIV-1 reverse transcriptase (RT), nucleoside and non-nucleoside RT inhibitors, NRTIs and NNRTIs, respectively 1. Even with the advent of combination drug therapy, highly active anti-retroviral therapy (HAART), the emergence of HIV-1 drug resistant mutations remains an issue due to the high capability of HIV-1 to mutate and escape drug efficacy1; 2; 3.
The low-fidelity, error-prone nature of HIV-1 replication provides the virus with numerous genomic mutations, which are mixed by frequent recombination events between the two single stranded HIV-1 RNA genomes during reverse transcription 2; 3; 4; 5; 6; 7. This leads to the enhancement of the ability of HIV-1 to accumulate viral mutations rendering resistance to the applied anti-HIV-1 agents 5; 8; 9. Therefore, recombination may allow the virus to efficiently combine individual pre-existing drug resistant mutations encoded in separate viral genomes for the generation of an HIV-1 variant that is multidrug resistant5; 9; 10; 11. Adding to these problems, HAART selects a particular RT population harboring multiple genomic mutations, which yields simultaneous cross-resistance to similar types of HIV-1 drugs used, leaving fewer choices for further anti-viral treatment 12; 13.
A particular group of clinical HIV-1 RT variants, which causes viral resistance to multiple NRTIs, contains RT mutations with peptide insertions at the β3–β4 fingers domain of HIV-1 RT along with background thymidine analog mutation (TAM)8; 14; 15; 21. The T69S substitution mutation generally appears in combination with an inserted dipeptide, typically Ser and Gly, between position 69 and 70, respectively16; 17; 18; 19; 20; 21. Several biochemical studies have shown that dipeptide insertion mutation along with background thymidine analog mutation (TAM) are capable of unblocking primers21; 22; 23; 24; 25. Furthermore, it has been shown that retroviral recombination under anti-viral drug selective pressure can generate viral progeny with enhanced or dual resistant mutations10; 11; 26. The impact of the RT dipeptide insertion on other biochemical activities, such as strand transfer activity of HIV-1 RT remains to be explored.
In this study, we characterized the biochemical recombination capability of two clinical RT variants previously isolated from a single HIV-1 patient before and after HAART, termed pre-drug and post-drug RTs: the post-drug RT is a dipeptide insertion RT containing a Ser-Gly insertion mutation at positions 69 and 70 of the fingers domain along with a T215Y TAM. Our study reveals that the post drug-RT has elevated strand transfer activity and altered interaction with templates, compared to the pre-drug RT. More importantly, the removal of the dipeptide insertion mutation from post-drug RT simultaneously inactivates the elevated strand transfer activity and azidothymidine (AZT) resistance, supporting a mechanistic link between strand transfer and multiple drug resistance of the dipeptide insertion RT containing T215Y.
Results
Characteristics of an NRTI-resistant HIV-1 RT isolate with a dipeptide insertion in the fingers domain
We obtained two HIV-1 RT clinical clones (through a collaboration with Dr. Jaap Goudsmit), which were previously isolated from a single patient before and after prolonged treatments with multiple NRTIs, termed pre-drug RT and post-drug RT in this study, respectively. The RT genes were sequenced completely. A portion of the polymerase domain sequence alignment is shown in Fig. 1A. The pre-drug RT does not harbor any previously known RT mutations rendering viral resistance to currently available NRTIs and NNRTIs, and has 97% sequence similarity to the RT of the common laboratory NL4-3 strain. In contrast, the post-drug RT clone contains a series of RT mutations throughout the fingers, palm and thumb domains of HIV-1 RT, including a Ser-Gly (SG) insertion between residues 69 and 70 of the fingers domain and a T215Y TAM mutation. Amino acid substitution mutation differences in the post-drug RT compared to the NL4-3 WT and pre-drug RT showed mutations at I329V, T377R, K431T, and I435V in the connection domain and R461K, P468S, L469I, and K512Q in the RNase H domain. The genome sequence variations among NL4-3 RT, pre-drug RT and post-drug RT are summarized in Supplementary figure 1 characterizing all three RT domains. While several additional mutations were identified in the post-drug RT, compared to the pre-drug RT, none of these mutations are known to confer RT drug resistance. Specific amino acid differences in the polymerase domain are shown in Figure 1A, which show common mutations that generally accompanies the SG insertion at the fingers domain, which includes the T69S and T215Y TAM known to render viral resistance to multiple NRTIs 8; 16; 23. The T215Y mutation is persistently found in the Zidovudine (AZT) resistant RT populations and have shown to enhance primer unblocking in the presence of a dipeptide insertion18; 21; 25.
FIG. 1. Sequence comparison of fingers domain dipeptide insertion HIV-1 RT variants during NRTI therapy and their effects on AZTTP sensitivity.
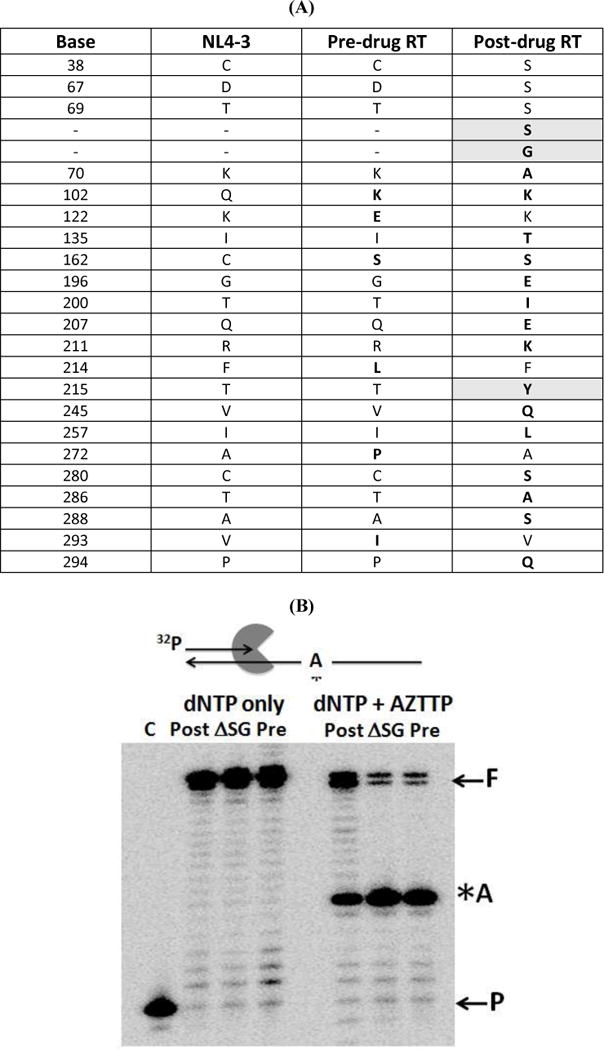
(A) The full lengths RT genes isolated from a same individual before (pre-drug RT) and after NRTI treatment (post-drug RT) were sequenced and compared to WT RT of NL4-3. Only the RT polymerase domain is shown. The shaded boxes are NRTI multiple drug resistant mutations. The bold amino acids represent mutations that are different from NL4-3 sequence (B) AZTTP terminated primer unblocking. A 5’ end labeled 20 mer primer annealed to 38 mer DNA template with only one dTTP/AZTTP incorporation site shown as “*A” was extended by the post: post-drug RT, pre: pre-drug RT, and ASG: post-drug ASG RT. In the absence of AZTTP the reaction was incubated at 37°C for 5 minutes. In presence of AZTTP, the reaction was incubated at 37°C for 30 minute with all dNTPs (100 μM) except dTTP and dATP. An additional 100 μM dTTP and 3.2mM ATP were added to the reaction and was incubated for an additional 30 min at 37°C and terminated.
Next, we confirmed the effect of the SG dipeptide insertion of the post-drug RT on the biochemical AZT resistance using the AZTTP inhibition assay. For this test, we additionally constructed an SG-deletion mutant form of the post-drug RT (Post-Drug ∆SG RT). We then determined how to set the three proteins to equal polymerase activity using a primer extension reaction with a 5’ end 32P-labeled 23-mer primer annealed to a 38-mer DNA template, carried out at 250 μM dNTPs (Supplementary figure 2). As shown in Supplementary figure 2, all three RT proteins displayed a similar level of primer extension reaction in 5 min reactions as estimated by the percentage of the 38 nt long full length product (“F”).
Next, we repeated the same experiment as the control experiment with a longer incubation time (30 min), which generated the complete extension of the primer to the full length product in the absence (Fig. 1B: “(−) AZTTP”) and presence of 25 mM AZTTP (Fig. 1B: “(+) AZTTP”). In this reaction, the template sequence harbors a single TTP incorporation site, which is also the AZTTP incorporation site (“A” site in Fig. 1B). AZTTP incorporation by RT at this site terminates the primer extension. However, RT proteins carrying AZT resistance will remove the incorporated AZTMP by pyrophosphorolysis excision (or AZTMP unblocking) and continue the primer extension, generating the full length product. As shown in Fig. 1B, upon the addition of AZTTP to the reaction, the post-drug RT was able to extend past the AZTTP incorporation site (“A” site) and continue primer extension, generating a significant amount of the full length product. In contrast, pre-drug RT was unable to overcome the primer block by AZTTP beyond the “A” site as evident from little generation of the full lengths extension product (“F” in Fig. 1B). This is consistent with previous findings, in which RT with dipeptide insertion and T215Y enhanced primer unblocking 21; 22; 23; 25. Significantly, when the dipeptide insertion was removed from post-drug RT, the primer unblocking activity diminished to levels similar to the pre-drug RT. This is consistent with previous findings in which RTs with the dipeptide insertion and a background T215Y TAM greatly renders AZT resistance by enhanced primer unblocking 22; 23. TAM T215Y mutation has been known to aid in positioning the ATP pyrophosphate donor during AZT mediated excision 25. However, T215Y mutation alone has greatly diminish primer unblocking abilities due to its inability to synthesize beyond the AZTTP incorporation site upon removal of the SG insertion.
Strand transfer activity of Pre-drug and Post-drug HIV-1 RT proteins
The structures of HIV-1 RT binary (RT-T/P) and ternary (RT-T/P-dNTP) polymerization complexes showed that the fingers domain of HIV-1 RT, where the dipeptide insertion was found, directly interacts with the template 16; 27; 28. In addition, the stable interaction of HIV-1 RT with template, which is essential for efficient RNA template cleavage by the RNase H activity of RT, plays a key role in the template switching (recombination) activity of HIV-1 RT between two homologous RNA strands 4; 29; 30; 31. Thus, we hypothesized that the SG dipeptide insertion found in the fingers domain affects the strand transfer activity of HIV-1 RT. To test hypothesis, we examined strand transfer activity of the pre-drug RT and post-drug RT.
Figure 2 is a diagram depicting the strand transfer system used in our previous study 29; 30. In this system, a 5’ 32P-labeled 20 nt DNA primer is annealed to an 80 nt “donor” non-viral RNA sequence template 26. The acceptor RNA strand is 119 nt and contains a 64 nt homology region shared with the donor template to allow internal strand transfer events. In addition, the acceptor template has a 19 nt insert (INT. Seq.) near its 5’ end that is not homologous with the donor, and then an identical 16 nt sequence also found at the 5’ end of the donor. The primer 3′ terminus has a 3 nt mismatch with the acceptor RNA to prevent extension of any primers that equilibrate to the acceptor before performing some synthesis on the donor. This system was designed to monitor template switches only between the 64 nt homology regions of donor and acceptor templates, generating a 119 nt long transfer product. This is because any strand transfer from the 5’ end 16 nt region of the donor template beyond the 64 nt homology region will jump to the 16 nt complementary region of the acceptor, generating the same smaller size (80 nt) final extension product as the product of full length extension only on the donor 26. Thus, the strand transfer efficiency can be measured by quantifying the 119 nt long internal transfer product as a percentage of all products of primer extension.
FIG. 2. Strand transfer efficiency of the pre-drug RT and post-drug RT proteins.
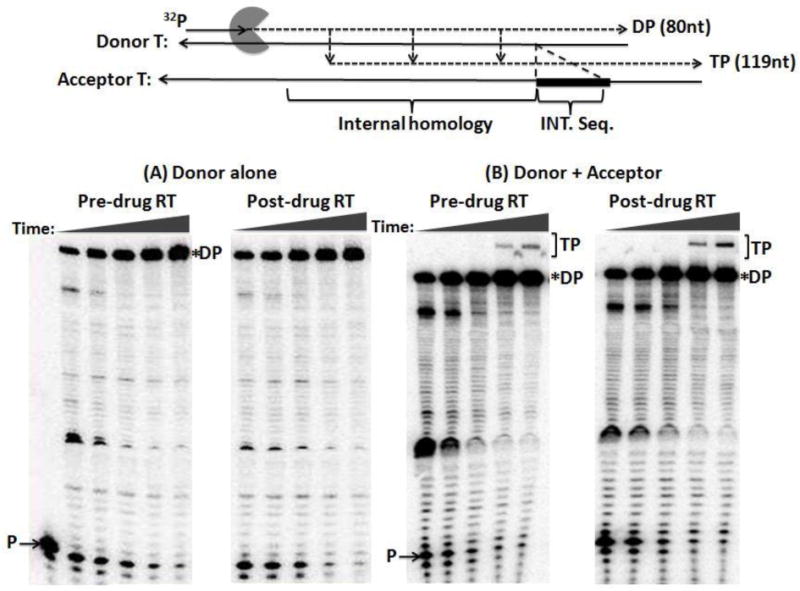
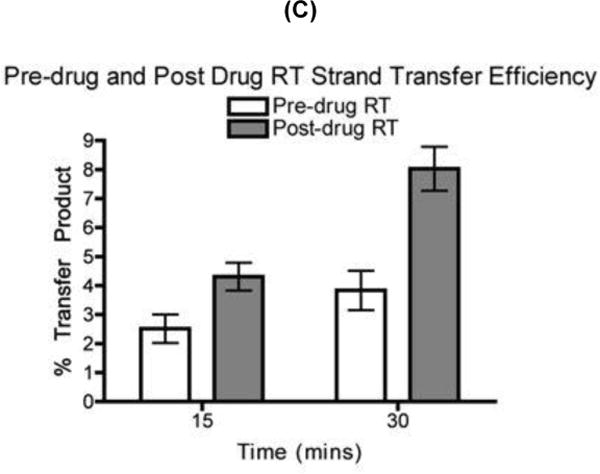
(A) Diagram for the strand transfer assay substrates. Donor and acceptor RNAs are 80 nt and 119 nt long respectively. Both templates share a 64 nt homology region for internal strand transfer, with the acceptor template carrying 19 nt disrupted homology region (INT. seq). Primer extension shows similar amounts of pre-drug RT and post-drug RT activity place into each transfer reactions. P= unextended primer DP= donor product fully extended 80 nt (B) Strand transfer efficiency of the pre-drug and post-drug RT variants. 32P 5’ end labeled 20 nt DNA primer is annealed to the 80 nt donor RNA for primer extension with the addition of the 119 nt acceptor RNA in this time course assay, with earlier time points showing the RT primer extension activity placed into each reaction (.5, 1, 3, 15, & 30 minutes) P= unextended primer DP= donor product fully extended 80 nt TP= 119 nt transfer product. C) Quantitative analysis of strand transfers efficiency of the pre-drug and post-drug RT variants at the 15 and 30 min time points. The experiments were repeated at least in triplicates with p-value <0.05 at 30 min time points
Strand transfer reactions were carried out as described in Experimental Procedures. In making a comparison between RTs, it was important to perform the assay at equivalent polymerization activity and to compare samples at equivalent times. This is because transfer efficiency measured in vitro rises with both RT concentration and time. First, the total DNA polymerase activity of pre-drug and post-drug RTs was normalized by quantitation of the amount of the 80 nt fully extended product in the time-course reactions only with the donor template (Fig. 2A). Next, identical donor extension reactions were repeated, but in the presence of the acceptor template with the two RT proteins displaying similar RT activity. As shown in Fig. 2B at the 30 min time point, the post-drug RT yielded more strand transfer products (“TP”) than the pre-drug RT. Strand transfer efficiencies of these two RT proteins were determined as previously described and compared at 15 and 30 min time points. Percent of transfer products were calculated using the equation [TP/(TP+F)] 31, in which TP is the amount of transfer product and F is the amount of full-length extension product only on the donor template. Indeed, as shown in Fig. 2C, the post-drug RT at 30 min showed two-fold higher transfer efficiency than pre-drug RT. These results indicate that the post-drug RT containing the SG dipeptide insertion along with T215Y is more effective at executing a template switch during reverse transcription than pre-drug RT.
Is enhanced strand transfer a direct result of the dipeptide insertion?
Since the post-drug RT that we employed in this study contains the T215Y TAM and dipeptide insertion, we tested whether the enhanced strand transfer efficiency observed in post-drug RT (Fig. 3) derives from the dipeptide insertion. For this test, we removed the SG between positions 69 and 70 from the post-drug RT, leaving other RT mutations, including T215Y, unchanged. Using the same template as in Fig. 2, first the activities of all three RTs, pre-drug RT, post-drug RT and post-drug ∆SG RT, were normalized in an extension reaction in Supplementary figure 3 and quantified in Fig. 3A. Then, at comparable polymerase activities of the three RT proteins, strand transfer efficiencies of the three RT proteins were measured as described above in Fig. 2. Figure 3B and 3C show the strand transfer assay results: indeed, the post-drug RT produced the higher percentage of transfer products at 15 and 30 min than pre-drug RT, confirming the data in Fig. 2. Importantly, post-drug ∆SG RT showed reduced strand transfer efficiency, compared to the post-drug RT, and has similar strand transfer efficiency with pre-drug RT. To further validate our results with a more biologically relevant form of RT, we utilized a heterodimer pre-drug RT and post-drug RT for the strand transfer assay. Supplementary figure 4 shows quantified data of a time course primer extension assay use for normalizing RT activity. The purpose was to normalize the heterodimer pre-drug RT and post-drug RT activity used in the strand transfer assay. Supplementary figure 5 shows that heterodimer post-drug RT contains higher strand transfer activity than the heterodimer pre-drug RT even when less RT activity was placed into the reaction. The results above support the conclusion that the SG dipeptide insertion is mainly responsible for the elevated strand transfer activity of post-drug RT. Other post-drug RT specific mutations including T215Y (Fig. 1) do not significantly contribute to the elevated strand transfer efficiency.
FIG. 3. Strand transfer efficiency of the pre-drug, post-drug RT and post-drug ΔSG RT proteins.
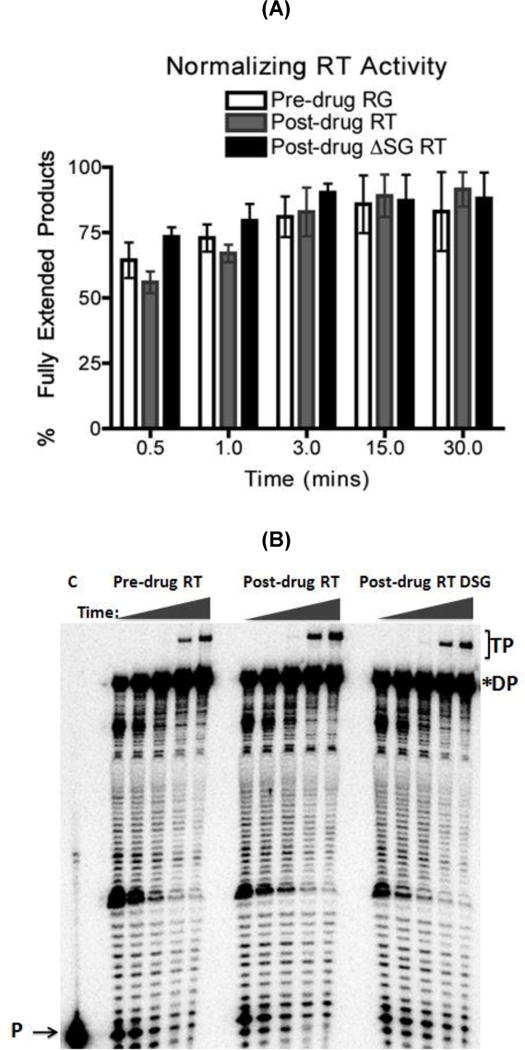
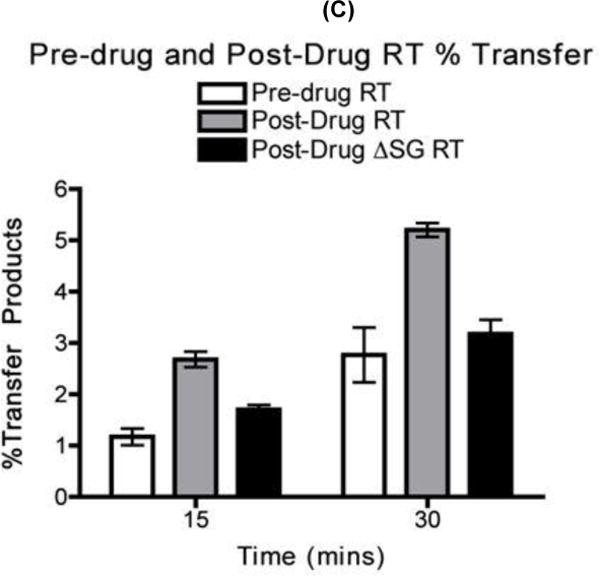
(A) The pre-drug RT, post-drug RT and post-drug ΔSG RT proteins activities from Fig. 3A were quantified with primer extension assay using the same T/P substrate used in the strand transfer reaction represented in Fig. 3B. The activity of all three RT proteins were normalized and quantified at each time points for fully extended 80 nt donor product starting at 0.5, 1, 3, 15, and 30 minutes. Percent of fully extended product was calculated by (F/F+P) × 100. Error bars represents results in triplicates (B) Strand transfer efficiency of the pre-drug RT, post-drug RT, and post-drug ASG RT. (C) Quantitative analysis of strand transfers efficiency of the pre-drug RT and post-drug RT variants from Fig. 3C. Statistical analysis using paired T-test resulted in a p-value <0.05 at 30 mins for the strand transfer efficiency bar graph. P= Primers, F= Fully extended 80nt Donor, TP= Transfer products 119nt
Confirmation of the elevated strand transfer efficiency of post-drug RT using another strand transfer system
To ensure that the enhanced strand transfer efficiencies that we measured for post-drug RT were not biased because of the specific sequence template employed, we used another strand transfer system previously described 31: this system employs a 184 nt donor template and a 227 nt acceptor RNA encoding a portion of the HIV Pol gene as described in the diagram at the top of Fig. 4 31; 32. In this system the internal transfer during the donor template replication generates a (227 nt) long product while the donor extension without transfer generates a short product (184 nt). First, RT activities were normalized and quantified in Fig. 4A to ensure that similar amounts of RT activity were placed into each strand transfer reaction. The transfer assay was then performed with both donor and acceptor RNA templates. As shown in Fig. 4B, post-drug RT showed the highest strand transfer efficiency at 30 min. These results confirmed that post-drug xcvRT containing SG has higher strand transfer activity than pre-drug RT and post-drug ∆SG RT using a second template system.
FIG. 4. Strand transfer efficiency of the pre-drug RT, post-drug RT and post-drug ΔSG RT proteins using a HIV-1 pol derived sequence tempate.
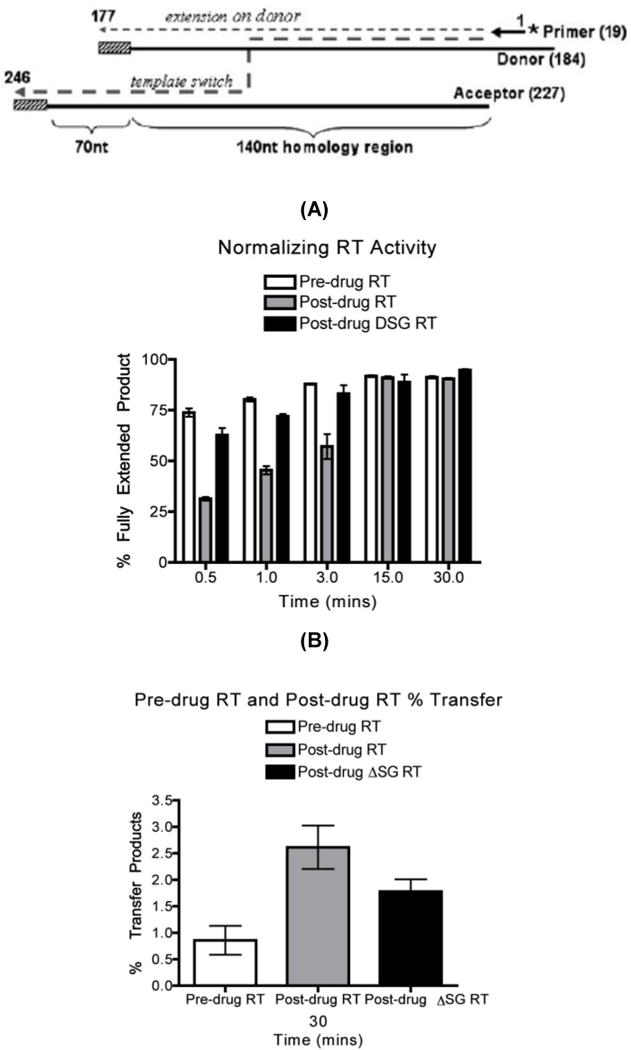
A) Diagram for the strand transfer assay substrates 31. Donor and acceptor RNAs are 184 nt and 227 nt long respectively. Both templates share a 140 nt homology region for internal strand transfer, with the donor template carrying 16 nt disrupted homology (slash box) region to prevent end transfer. The 32P 5’ end labeled 20 nt DNA primer is annealed to the donor RNA. Normalized activities of the pre-drug, post-drug RT and post-drug RT ΔSG were established through 184 nt donor extension assay and quantified to ensure the same amount of RT activities were used for each reactions. (B) Quantitative analysis of strand transfers efficiency at the 30 mintute time point of all three RT variants using this strand transfer system. Strand transfer experiments were repeated at least in triplicates.
Processivity of the Pre-drug and Post-drug RT
Low processivity has been shown to contribute to elevating strand transfer efficiency 33. Although previous study has shown that HIV-1 RT containing the T69S, SG insertion, and K79R did show a slight decrease in processivity compared to WT HIV-1 RT, our base substitution mutations are slightly different from previous work and needs to confirm 21. This will deduce whether processivity plays a mechanistic role in the enhanced stand transfer observe for the post-drug RT. The processivity assay was carried out utilizing the 184 nt donor RNA substrate in the presence of a poly (rA)- oligo (dT) trap. Equal pre-drug RT and post-drug RT activity was normalized through a primer extension assay in the absence of the poly (rA)- oligo (dT) trap (Supplementary figure 6 lanes 1 and 5 respectively) using the substrate described in materials and methods. In the presence of the trap, RT will only be able to undergo one round of primer extension. Once the RT dissociates, it will bind to the excess trap and no longer be able to reinitiate primer extension. 1000-fold excess of trap was added to the reaction mix before the addition of RT to produce a trap control shown in lane 3 and lane 7 (Supplementary figure 6). Processivity of the pre-drug RT and post-drug RT is shown in (Supplementary figure 6) lane 4 and 8 respectively. Substrate was pre-incubated with RT before the initiation of the reaction with dNTP, MgCl2, and trap. Pre-drug RT and post-drug RT exhibit similar processivity since both were able to extend the primers to full lenth products. Lower processivity is not the mechanism for the enhanced strand transfer seen in post-drug RT.
Donor Template Degradation During Primer Extension
During proviral DNA synthesis, HIV-1 RT degrades the RNA template during and after DNA synthesis 4; 34. The RNA template degradation is an essential step for the transfer of the first strong stop DNA from the 5’ end of the viral genome to the 3’ end of the genome because the first strong stop DNA must be freed from the RNA template to execute the strand transfer 4; 35. The degradation of donor RNA template is catalyzed by two different types of RNase H activity of RT 31; 34; 36; 37: RT exhibits RNase H activity while it is polymerizing, which is called polymerization-dependent RNase H activity, and RT also binds in a 5’ orientation to segments of RNA annealed to DNA where it can cleave the RNA, which is called polymerization-independent RNase H activity34; 38. Thus, to measure the essential determinant of strand transfer efficiency, donor RNA degradation executed by both RNase H DNA polymerization dependent and independent processes were analyzed.
We tested whether strand transfer activity differences between the pre-drug and post-drug RTs were a consequence of differences in RNase H activities. More specifically, we postulated that the post-drug RT degrades the donor template more than the pre-drug RT. To examine the donor template degradation profile comparing these two RT proteins during the strand transfer reaction, we employed the 5’ 32P end labeled donor 80 nt RNA template instead of the 20-mer primer to monitor the donor RNA degradation during the strand transfer assay. The total RT polymerization activity of the two RT proteins used in this reaction was the same as used for the transfer reaction in Fig. 3. Since the reactions included dNTPs, the template degradation should have been catalyzed by both DNA polymerization-dependent and independent RNase H activities of RT. As shown in Fig. 5, post-drug RT had less template degradation through the initial three time points, but showed no difference in template degradation through the last two time points when compared to pre-drug RT. The same experiment was repeated with the post-drug ∆SG RT and results showed no major differences in template degradation than the pre-drug and the post-drug RT. This shows that the elevated strand transfer events seen with the post-drug RT do not appear to be a consequence of the overall donor template degradation, which is catalyzed by both polymerization-dependent and -independent RNase H activities.
FIG. 5. Donor template degradation of the pre-drug RT, post-drug RT, and post-drug ΔSGRT.
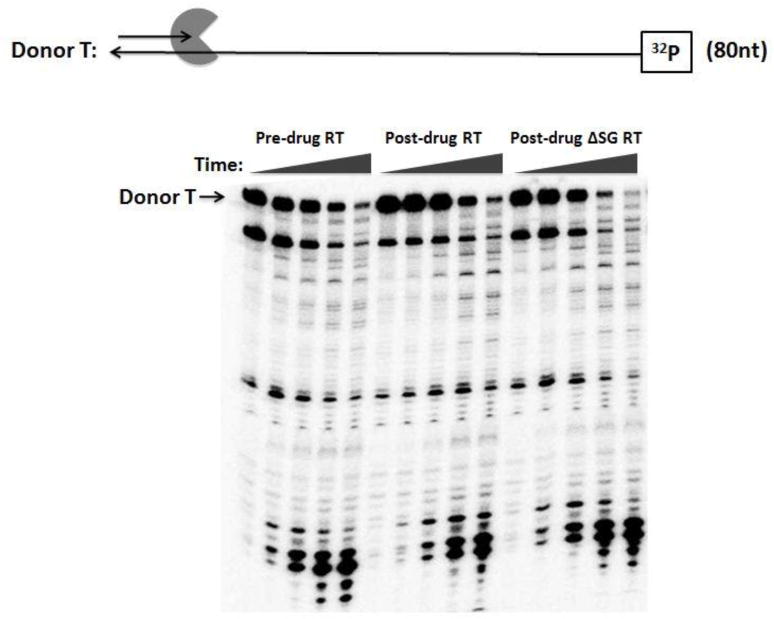
(A) 32P labeled donor RNA 80 nt template was annealed to cold 20 nt DNA primers at a 1:2 ratio respectively. Pre-drug RT, post-drug RT, and post-drug ΔSG RT was pre-incubated with the T/P complex for 3 min before the addition of 6 mM/dNTP 5 μM for initiating the reaction. Reactions were quenched with EDTA in the following time course assay at (0.5, 1, 3, 15, 30 minutes). Donor T: 32P labeled donor RNA 80 nt template. The RT activities were similar to Fig. 3.
Assessing the Individual Polymerization-dependent and Polymerization independent RNase H activities of the pre-drug and post- drug RT proteins
Since the donor template degradation described in Fig. 5 was catalyzed by both polymerization-dependent and -independent RNase H activities of RT, we examined the polymerization dependent RNase H activities of the pre-drug, post-drug and post-drug ∆SG RT. For this assay, a 32 nt long DNA primer was annealed to a 5’ end 32P-labeled 38 nt long RNA template, and this RNA/DNA hybrid substrate (see diagram at Fig. 6) was incubated separately with the three RT proteins showing an equal RNA-dependent DNA polymerase activity in the absence of dNTPs. As shown in Fig. 6, the post-drug RT showed faster degradation of the 38 nt full lengths RNA substrate than the pre-drug and post-drug ∆SG RT. In this polymerization-dependent RNase H assay, RT molecules, which are engaged in DNA synthesis, execute the two sequential cleavage events: 1) 1° cleavage of the donor RNA template, generating the 25 nt long products, and 2) 2° cleavage for further degradation of the 1° cleavage product, generating the 13 nt long products 31. Indeed, as shown in the quantified time-course degradation profile Fig. 6B, the post-drug RT protein displayed faster accumulation of both 1° and 2° cleavage products. These data suggest that the post-drug-RT polymerization- dependent RNase H activity is faster, which can contribute to its higher strand transfer activity compared to the pre-drug RT and the post-drug ∆SG RT. Importantly, the lower RNase H activity of the post-drug ∆SG RT suggests that the dipeptide insertion is responsible for not only the elevated strand transfer activity, but also enhanced polymerization-dependent RNase H activity of the post-drug RT. Next, we examined the polymerization-independent RNase H activities of the pre-drug, post-drug and post-drug ∆SG RT as described in Experimental Procedures (Supplementary figure 7). Interestingly, we found the post-drug RT actually has reduced polymerization-independent RNase H activity, compared to the pre-drug RT and post-drug ∆SG RT. This suggests that the delayed donor template degradation by the post-drug RT (Fig. 5) could be due to its reduced polymerization-independent RNase H activity, and also the polymerization-independent RNase H activity is not the mechanism involved in the elevated strand transfer efficiency of the post-drug RT.
FIG. 6. RNase H assay consisted of 5’ 32P end labeled 38-mer RNA annealed to a 32-nt unlabeled DNA primer.
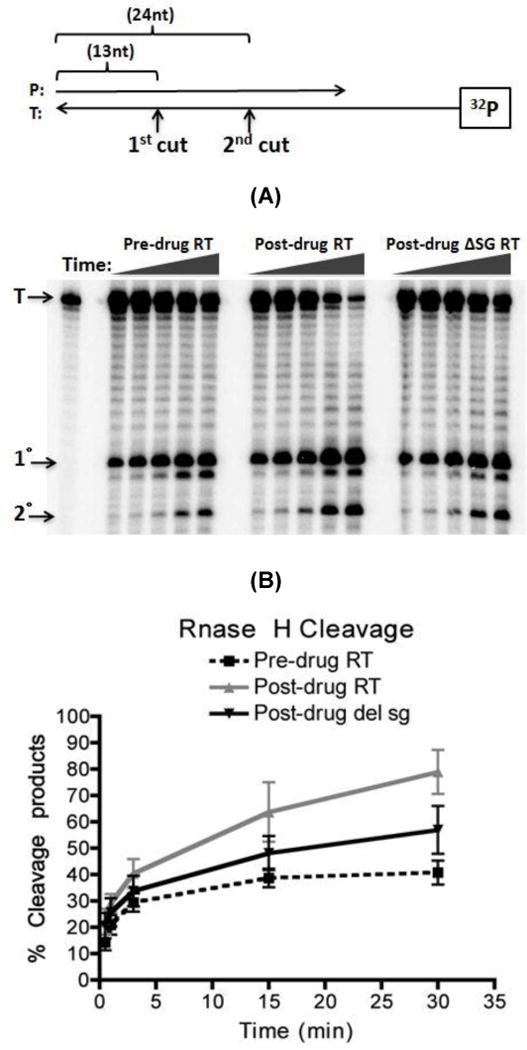
(A) The same amount of RT activities was used as in Fig. 3 for this assay. The reaction was carried out in a time course assay (0.5, 1, 3, 15, 30 minutes) in the absence of dNTP and quenched with the addition of EDTA. RNase H cleavages yielded a 24 nt primary cut product and a 13 nt secondary cut product. (B) Quantification of RNase H 5’ 32P end labeled 38-mer RNA degradation from Fig. 6A shows percent of degraded RNA over time. Percent degraded was calculated base on amount of (saturated volume from 1 □ + 2 □ cleavages)/(saturated volume from 1 □ + 2 □ + Uncut) × 100
Determining the template/ primer binding affinity (KD), on-rate (kon) and off-rate (koff) of the pre-drug RT, post-drug RT, and post-drug ∆SG RT
To complete the mechanistic investigation of the elevation of strand transfer activity of the post-drug RT, we determined the KD value of HIV-1 RT. This parameter numerically represents steady state equilibrium events between the rate of RT initial binding to T/P (kon) and the rate of RT release from the T/P (koff). Since the fingers domain of HIV-1 RT harboring the dipeptide insertion directly interacts with the template, we reasoned that the higher strand transfer activity and elevated polymerization-dependent RNase H activity of the post-drug RT may result from altered binding affinity of the RT to template/primer complex (T/P). It is possible that the increased T/P binding affinity of the post-drug RT may be due to either faster initial T/P binding rate or slower releasing rate from T/P.
First, we measured the binding affinity (KD) of the pre-drug RT and post-drug RT proteins to the T/P (19 nt primer annealed to 184 nt RNA template) using a double filter binding blot assay, which uses both protein and nucleic acid binding filters (refs: see experimental procedures), and the percents of bound DNA vs. concentrations of RT were plotted on a sigmoidal binding curve 39. As summarized and shown in Table 1, the post-drug RT displayed a lower KD (equilibrium binding constant) value than pre-drug RT and post-drug ∆SG RT (Pre-drug RT KD= 5.9 × 10−7 M Post-drug RT KD= 7.8 × 10−8 M Post-drug ∆SG RT KD= 1.7 × 10−7 M). From these data we conclude that the post-drug RT has 7 times higher binding affinity to the T/P than the pre-drug RT and 3 times higher than post-drug ∆SG RT. Second, we measured the koff of the RTs using the RT-off rate assay previously reported 33. Using a primer extension assay with the addition of greater than 1000-fold molar excess of the poly(rA) – oligo(dT) trap, we determined the amount of RT bound to template based on primer extension. Since RT dissociation from the substrate is slower than dNTP incorporation, the amount of primer extension was representative of RT bound to substrate 33; 40. The trap was incubated at increasing time points with RT pre-bound to the T/P (2 nM). In this system, RT molecules that dissociated from the T/P will be captured by the poly(rA)-oligo(dT) trap (8000 nM) and are no longer able to extend the primer in the presence of dNTP and MgCl2. Therefore, this experiment measured a single round of primer extension. Table 1 is a summary of our koff data extrapolated from the exponential decay curve base on the equation Y=e−koff(t), where Y=relative rate of incorporation and t=time 33. From these exponential decay equations we calculated the koff of all three RTs to be (Pre-drug RT koff = 6.1 × 10−4 s−1 Post-drug RT koff = 3.1 × 10−4 s−1 Post-drug ∆SG RT koff = 2.6 × 10−4 s−1). Basically, the koff rate difference between the two RT proteins was only two fold. Finally, using the KD values and the koff rates, which were experimentally measured, we are able to calculate the on-rate (kon) of the RTs using the equation KD= koff/kon. The calculated Kon values of the three RT proteins were roughly (Pre-drug Kon = 1.9 × 104 M−1s−1 Post-drug RT Kon = 3.5 × 105 M−1s−1 Post-drug ∆SG RT Kon = 1.5 × 105 M−1s−1) (Table 1). These kon values data imply that the post-drug RT is able to associate with its T/P substrate 18 times faster than the pre-drug RT and 8 times faster than the post-drug ∆SG RT. Mechanistically this may contribute to the tight binding affinity of the T/P.
Table 1.
Determining the KDKoffKon of pre-drug RT and post-drug RT
| KDa | Koffb | Konc | |
|---|---|---|---|
| Pre-drug RT | 5.9 × 10−7 M ± 0.0616 | 6.1 × 10−4s−1 ± 0.0105 | 1.9 × 104 M−1s−1 |
| Post-drug RT | 7.8 × 10−8 M ± 0.2761 | 3.1 × 10−4 ± 0.0167 | 3.5 × 105 M−1s−1 |
| Post-drug ASG RT | 1.7 × 10−7 M ± 0.2560 | 2.6 × 10−4 s−1 ± 0.0217 | 1.5 × 105 M−1s−1 |
DNA binding study summarized. The KD of each RT isolate was extrapolated from a sigmoid binding curve.
Koff was calculated from the exponential decay equation (Y=e−Koff(t)) where Y=relative rate of incorporation and t=time. Each data point represents value from three independent experiments with standard error.
Quantification of the percent bound DNA of all three RT protein isolates. Kon rate of the three RTs were calculated using the equation KD = Koff/Kon.
Discussion
Inter-strand homologous recombination of HIV-1 occurs mainly during minus strand proviral DNA synthesis. Recombination enhances viral diversity and facilitates HIV-1 escape from antiviral selective pressures including HAART 5; 9; 41. The dipeptide fingers domain insertion are positioned at the end of the HIV-1 RT fingers domain and in the cleft where the primer-template hybrid binds, but far from the polymerase active site 27; 42. Interestingly, although they are located outside of the polymerase active site, these mutations are associated with viral resistance to multiple NRTIs such as AZT, which bind to the polymerase active site 8; 17.
HIV-1 RT interactions with the double stranded region of the template annealed to the primer were extensively studied 43. However, RT interactions with the single stranded portion of the template are less well understood. Moreover, the structure of the tertiary complex of HIV-1 RT and a template with extended single stranded portion is not available. Positioning of the single stranded part of the template at the cleft near the tip of the fingers domain has been predicted but not proven.
Significantly, the fingers domain of HIV-1 RT undergoes a large conformational change during T/P binding (RT-T/P binary complex formation) 27, and it is highly likely that the β3–β4 loop at the tip of the fingers domain is structurally dynamic during DNA synthesis. The β3–β4 loop is likely the section of the replicating RT molecule that first contacts RNA template secondary structures. It has been shown that stable local structures of the RNA template mechanistically induce RT pausing 44. Pausing during synthesis in turn facilitates the RNA template degradation by RNase H activity of RT, which is an essential step for strand transfer 29; 30. Thus we hypothesized that the dipeptide insertion in HIV-1 RT, which lies at the tip of the fingers domain, may alter the RT-RNA template interaction and thus strand transfer efficiency of HIV-1 RT.
The biochemical data in this report demonstrate that the SG insertion RT at the β3–β4 loop has altered RNA binding kinetics of HIV-1 RT, which also is likely to relate to the enhancement of the strand transfer activity. More importantly, the post-drug RT displayed a faster initial binding rate (kon) than the pre-drug RT, leading to 7X time higher template binding affinity (KD) than the pre-drug RT and 3X higher than the post-drug ∆SG RT. Thus, these biochemical data supports the idea that the SG fingers domain insertion elevates the transfer activity presumably by enhanced interaction to the RNA templates.
Enhanced binding of post-drug RT to the T/P could be also related to a mechanism previously proposed for AZT resistant mutations found at the connection domain of HIV-1 RT: the connection mutations stabilize the RT-template complex yielding more time for the bound RT molecule to remove the incorporated AZTMP at the 3’ end of the primer 45. Similarly, the dipeptide insertion mutations may also confer AZT resistance by enhancing the binding affinity, which lengthens the time span for HIV-1 RT to remain bound to the 3’ end of the primer for pyrophosphorolysis of AZTMP. We also envision that the increased frequency of binding events of post-drug RTs to the T/P can increase the degradation capability of the RNA template by the RT polymerization-dependent and –independent RNase H activity, which in turn would enhance the strand transfer activity. Importantly, our data demonstrate the removal of the dipeptide insertion from the mutant concomitantly induces loss of both the AZTMP pyrophosphorolysis and elevated strand transfer. This supports the idea that AZT resistance and elevated strand transfer share a common mechanism, which is the enhanced interaction with the RNA template.
It is well established that the efficient RNA template degradation by RNase H increases the strand transfer of RT 29; 30; 36; 37; 42; 46. As shown in Fig. 6B, the pre-drug RT showed a higher polymerization-dependent RNase H activity and a faster binding rate (kon) to the template (Table 1), compared to pre-drug RT. These two biochemical alterations by the dipeptide insertion can mechanistically contribute to the enhanced strand transfer efficiency of the post-drug RT: the post-drug RT creates 1° cleavage of the donor template more efficiently, followed by faster 2° cleavage of the donor template, compared to the pre-drug RT. These elevated 1° and 2° cleavages during DNA synthesis will generate more gaps, which serve as evasion sites for the acceptor RNA template during the template switch, and ultimately increase the strand transfer activity. Importantly, the fast binding rate (kon) of the post-drug RT can facilitate the 2° cleavage of the donor template, which likely requires the rebinding of RT after the 1° cleavage. Importantly, the overall delayed template degradation of the post-drug RT (Fig. 5) is the result of both polymerization-dependent and independent RNase H activities. As shown in Supplementary figure 7, the post-drug RT actually has reduced polymerization-independent RNase H activity, compared to the pre-drug RT. Thus, these two sets of data suggest that the polymerization-independent RNase H activity, which could be responsible for the delayed template degradation, is not mechanistically involved in the elevated strand transfer activity of the post-drug RT. Instead, the polymerization-dependent RNase H activity, which creates the invasion sites for the acceptor during DNA synthesis, is more likely the mechanistic reason for the elevated strand transfer activity of the post-drug RT.
In summary, the multiple drug resistant post-drug RT displayed altered biochemical behaviors including elevated strand transfer activity, increased polymerization-dependent RNase H activity and tighter binding affinity to the T/P substrate. We believe that with these simultaneous mechanistic changes made by the dipeptide insertion RT, a follow-up virological experiment to test the actual impact of the post-drug RT mutations on the recombination efficiency of the viruses should be conducted in the future.
Materials and Methods
HIV-1 RT proteins
Two HIV-1 RT clinical clones were kindly provided from Dr. Jaap Goudsmit (Amstredam Medical Center, Netherland) named pre-drug RT and post-drug RT. These RT genes were cloned into an E. coli overexpression plasmid (pHis) and encode a protein with a fused 6-histidine tag at its N-terminal end used for protein purification of homodimer RTs. The clones were transformed into BL-21 competent E. coli for protein expression and purified through a nickel column (Novagen). Protein purification methods were done accordingly base on reference 31. The post-drug RT with the ΔSG dipeptide insert was generated using a Quikchange mutagenesis kit (Stratagene) with the post-drug RT clones as template. The primers designed to introduce the SG dipeptide deletion were (Deleted SG-Forward GAAGAAAAGCAGTAGCGCTTGGAGAAAATTAGTAGATTTC) (Deleted SG-Reverse GAAATCTACTAATTTTCTCCAAGCGCTACTGCTTTTCTTC). The Post-Drug ∆SG RT clone was sequenced and all protein purification methods were the same as above. Heterodimer HIV-1 RT was prepared by cloning the pre-drug or post-drug RT p66 subunit into a pet28a expression plasmid containing a Kanamycin selection marker. The p51 subunit derived from HIV-1 RT HXB2 strain was cloned into a pCDF plasmid containing a Spectinomycin antibiotic selection marker and a fused 6-histidine tag at its N-terminal. The clones were transformed into BL-21 competent E. coli for protein expression selecting for both antibiotics marker and purified through a nickel column (Novagen).
Generation of RNA Templates
RNA templates were generated as described previously 29; 30. Briefly, PCR product was generated using primers PCIS5 and PCIS6 and the pD0 plasmid as a template. The PCR product was then purified by agarose gel electrophoresis and Qiaquick gel extraction kit (Qiagen). T7 RNA polymerase Megashortscript kit (Ambion) was implemented for synthesis of Donor 80 nucleotide (nt) RNA using the purified PCR product as a template and the primer PCIS 14 (TGGTAAACATTCTTGAGTGC). The 119 nt acceptor RNA template were generated from T7 RNA polymerase Megashortscript kit (Ambion) using the previously described plasmid pAM2 30. The 184 nt donor and 227 nt acceptor substrate encodes a portion of the HIV-1 Pol gene sequence and were generated as previously described 31; 32.
AZTTP Terminated Primer Unblocking
A 5’ end-labeled 20 nt primer was annealed to a 38 nt DNA template that had only one dATP downstream of the primer 3’ terminus where a dTTP or AZTTP could be incorporated (Figure 1B, shown by arrow). Primer extension by pre-drug or post-drug RT was performed in the absence or presence of 25 uM AZTTP. Synthesis reactions in absence of AZTTP were carried out at 37° C for 5 minutes. In presence of AZTTP, reactions were incubated at 37° C for 30 min with all dGTP and dCTP (100 μM). Then 100 μM dTTP and 3.2 mM ATP was added and the reaction incubated for an additional 30 min at 37° C. All reactions were terminated with addition of 40 mM EDTA, 99% formamide 22.
Strand Transfer Assay
DNA primer 20 nt (0.64 nM) with the 5’ terminus 32P-radiolabeled was annealed to donor RNA 80 nt template (4 nM) in RT reaction buffer (50 mM Tris pH 8, 50 mM KCl, 1 mM DTT, 1 mM EDTA) at 95° C for 5 minutes, and slowly cooled to room temperature. RT was incubated with the substrate for 3 minutes before the reaction was started with the addition of MgCl2 (6 mM), dNTP (50 μM) and acceptor RNA 119 nt template (8 nM). All reactions were done in a 37.5 μL master mix volume at 37°C. Reactions were terminated at various time points by transferring an aliquot of 6.25 μL from the master mix volume into 6.25 μL 40 mM EDTA, 99% formamide. The second substrate used for this study was a 19 nt 5’ 32P radiolabeled DNA primer annealed to a 184 nt RNA template. The acceptor template was a 227 nt RNA template. Both the 184 nt donor and 227 nt acceptor RNA template encode a portion of the HIV-1 Pol gene. The exact experimental conditions were applied towards this set of substrate as described above. Reaction products were resolved in a 10% polyacrylamide-urea denaturing gel and scanned on a PhosphoImager (BioRad). Percent transfer was calculated [(Transfer product/ 1st time point extension + Transfer product) × 100%] based on saturation volume quantifed by Quantity One software. Background saturation volume was subtracted toward all quantifications 31; 26. The primer extension assay methodology was performed exactly as the strand transfer assay described above with the exception that initiation of the reaction did not include the acceptor RNA.
Donor degradation Assay
The same donor RNA 80 nt template (50 nM) used for the strand transfer assay was 5’ end radiolabeled with 32P and annealed with the unlabeled 20 nt DNA primer (100 nM) in RT reaction buffer by heating at 95°C for 5 min and slowly cooled to room temperature. All reactions were carried out with the same procedures as in the strand transfer assay, except the initiation of the reaction was with MgCl2 (6 mM) and dNTPs (5 μM).
RNase H Assay
To measure polymerization-dependent RNase H activity, an RNA template of 38 nt (8 nM) was 5’ 32P radiolabeled and annealed to a 32 nt DNA primer (32 nM). To measure polymerization-independent RNase H activity, an RNA template of 38 nt (8 nM) was 5’ 32P radiolabeled and annealed to a 53 nt DNA primer (32 nM). Both polymerization-dependent and – independent RNase H activity reaction conditions and time points were the same as in the strand transfer assay except the initiation of the reaction was done with just MgCl2 (6 mM). The products were fractionated on a 14% polyacrylamide-urea denaturing gel and analyzed with the same equipment and methods described previously 31.
Processivity Assay
A 19 nt 5’ 32P radiolabeled DNA primer was annealed a 184 nt RNA template previously described in the strand transfer experiment as the HIV Pol template at a 2:1 (8nM: 4nM final concentration) ratios, respectively. RT with equal activity was pre-bound to the substrate for 3 minutes at 37°C in RT reaction buffer (50 mM Tris pH 8, 50 mM KCl, 1 mM DTT, 1 mM EDTA). The reaction was initiated with the addition of MgCl2 (6 mM), dNTP (25 μM), and 1000-fold excess of poly(rA) – oligo(dT) trap (IDT) (annealed at a 1:2 ratio respectively) over substrate concentration. The final reaction mixture volume was 12.5 μL. The primer extension assay was incubated for 5 minutes at 37°C before quenching with 12.5 μL 40 mM EDTA, 99% formamide. The products were fractionated on a 10% polyacrylamide-urea denaturing gel 33.
koff Measurements (Dissociation Rate Constant)
The 19 nt 5’ 32P radiolabeled DNA primer was annealed with the 184 nt RNA template at a 2:1 ratio. RT was prebound to the template in the presence of a 1000-fold excess of poly(rA) – oligo(dT) from (IDT) were annealed at a 1:2 ratio respectively and incubated at increasing time points with the reaction mixture before the addition of MgCl2 (6 mM) and dNTP (50 μM). RT that fell off the labeled substrate will bind to the poly(rA) – oligo(dT) trap. RT that remains bound to the labeled substrate after the addition of MgCl2 and dNTP was quantitated by primer extension. The final reaction mixture volume was 12.5 μL containing 50 mM Tris pH 8, 50 mM KCl, 1 mM DTT, and 1 mM EDTA. Reaction products were resolved on a 10 % polyacrylamide-urea denaturing gel and scanned with a BioRad PhosphorImager. The relative incorporation values were calculated with the equation: relative incorporation = [(extended product/1st time point extension at 10 sec) × 100%]. Saturation volumes used in the above equation to determine relative incorporation were obtained by analysis of scanned gel images using Quantity One software. All values were normalized with subtracted background values. The dissociation rate was calculated based on an exponential decay graph in which total a relative incorporation versus time curve was fitted to an exponential decay equation, Y=e−koff(t) 33.
KD Measurements (Equilibrium Dissociation Constant)
The double filter blot assay uses a nitrocellulose membrane that binds RT proteins and a nucleic acid binding nylon membrane (Biodyne nylon membrane from VWR). The nylon membrane was washed for 10 minutes with 0.1M EDTA and had three 10 minute washes in 1M NaCl, followed by a quick wash in 0.5M NaOH then dH20. The nitrocellulose membrane was washed for 10 minutes with 0.4M KOH at room temperature and then washed with dH2O. The membrane was then soaked in 1X dialysis buffer at 4°C for one hour before being placed on top of the nylon membrane to create the double filter blot. Using the same substrate as described in the koff experiment, the DNA primer of 19 nt was 5’ 32P radiolabeled and annealed to the 184 nt RNA template used in the strand transfer assay mentioned above. The radiolabeled hot primers to non-radiolabeled cold primers were mixed at 8 nM to 42 nM respectively during annealing to the RNA template. The concentration of primer to template for annealing was 1:6 ratios respectively. The premixed solutions had fixed template/primer (T/P) concentration containing increasing concentrations of the RT proteins for each reaction. Each DNA binding reactions were carried out at 37°C for 3 min before the reactions were terminated by being vacuumed through the double membrane filters. The RT that is bound to the T/P will bind to the nitrocellulose membrane, while T/P substrates without bound proteins will pass the nitrocellulose membrane and bind to the nylon membrane. The blot was wash with 100 μL of 1x dialysis buffer. The reactions were carried out in the absence of MgCl2 and dNTP. The blots were exposed with a phosphor screen for 20 min and quantify using a PhosphorImager from (BioRad). The percent of RT proteins bound to DNA was quantified based on saturation volume from (protein blot / protein blot + DNA blot) × 100%, all subject to subtracted background values) by Quantity One software 47. The KD was extrapolated from a sigmoidal curve.
Supplementary Material
Acknowledgments
This study was supported by grants NIH AI049781 (B.K.) and NIH GM049573 (R.A.B)
Abbreviations
- SG
Ser Gly
- Post-Drug ∆SG RT
post-drug deleted SG RT
- T/P
template/primer
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Volberding PA, Deeks SG. Antiretroviral therapy and management of HIV infection. Lancet. 2010;376:49–62. doi: 10.1016/S0140-6736(10)60676-9. [DOI] [PubMed] [Google Scholar]
- 2.Preston BD, Poiesz BJ, Loeb LA. Fidelity of HIV-1 reverse transcriptase. Science. 1988;242:1168–71. doi: 10.1126/science.2460924. [DOI] [PubMed] [Google Scholar]
- 3.Mansky LM, Temin HM. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J Virol. 1995;69:5087–94. doi: 10.1128/jvi.69.8.5087-5094.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Basu VP, Song M, Gao L, Rigby ST, Hanson MN, Bambara RA. Strand transfer events during HIV-1 reverse transcription. Virus Res. 2008;134:19–38. doi: 10.1016/j.virusres.2007.12.017. [DOI] [PubMed] [Google Scholar]
- 5.Onafuwa-Nuga A, Telesnitsky A. The remarkable frequency of human immunodeficiency virus type 1 genetic recombination. Microbiol Mol Biol Rev. 2009;73:451–80. doi: 10.1128/MMBR.00012-09. Table of Contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shriner D, Rodrigo AG, Nickle DC, Mullins JI. Pervasive genomic recombination of HIV-1 in vivo. Genetics. 2004;167:1573–83. doi: 10.1534/genetics.103.023382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roberts JD, Bebenek K, Kunkel TA. The accuracy of reverse transcriptase from HIV-1. Science. 1988;242:1171–3. doi: 10.1126/science.2460925. [DOI] [PubMed] [Google Scholar]
- 8.Tamalet C, Yahi N, Tourres C, Colson P, Quinson AM, Poizot-Martin I, Dhiver C, Fantini J. Multidrug resistance genotypes (insertions in the beta3-beta4 finger subdomain and MDR mutations) of HIV-1 reverse transcriptase from extensively treated patients: incidence and association with other resistance mutations. Virology. 2000;270:310–6. doi: 10.1006/viro.2000.0261. [DOI] [PubMed] [Google Scholar]
- 9.Carvajal-Rodriguez A, Crandall KA, Posada D. Recombination favors the evolution of drug resistance in HIV-1 during antiretroviral therapy. Infect Genet Evol. 2007;7:476–83. doi: 10.1016/j.meegid.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kellam P, Larder BA. Retroviral recombination can lead to linkage of reverse transcriptase mutations that confer increased zidovudine resistance. J Virol. 1995;69:669–74. doi: 10.1128/jvi.69.2.669-674.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moutouh L, Corbeil J, Richman DD. Recombination leads to the rapid emergence of HIV-1 dually resistant mutants under selective drug pressure. Proc Natl Acad Sci USA. 1996;93:6106–11. doi: 10.1073/pnas.93.12.6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belec L, Piketty C, Si-Mohamed A, Goujon C, Hallouin MC, Cotigny S, Weiss L, Kazatchkine MD. High levels of drug-resistant human immunodeficiency virus variants in patients exhibiting increasing CD4+ T cell counts despite virologic failure of protease inhibitor-containing antiretroviral combination therapy. J Infect Dis. 2000;181:1808–12. doi: 10.1086/315429. [DOI] [PubMed] [Google Scholar]
- 13.Shafer RW. Genotypic testing for human immunodeficiency virus type 1 drug resistance. Clin Microbiol Rev. 2002;15:247–77. doi: 10.1128/CMR.15.2.247-277.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eggink D, Huigen MC, Boucher CA, Gotte M, Nijhuis M. Insertions in the beta3-beta4 loop of reverse transcriptase of human immunodeficiency virus type 1 and their mechanism of action, influence on drug susceptibility and viral replication capacity. Antiviral Res. 2007;75:93–103. doi: 10.1016/j.antiviral.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 15.Imamichi T, Murphy MA, Imamichi H, Lane HC. Amino acid deletion at codon 67 and Thr-to-Gly change at codon 69 of human immunodeficiency virus type 1 reverse transcriptase confer novel drug resistance profiles. J Virol. 2001;75:3988–92. doi: 10.1128/JVI.75.8.3988-3992.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Curr K, Tripathi S, Lennerstrand J, Larder BA, Prasad VR. Influence of naturally occurring insertions in the fingers subdomain of human immunodeficiency virus type 1 reverse transcriptase on polymerase fidelity and mutation frequencies in vitro. J Gen Virol. 2006;87:419–28. doi: 10.1099/vir.0.81458-0. [DOI] [PubMed] [Google Scholar]
- 17.Larder BA, Bloor S, Kemp SD, Hertogs K, Desmet RL, Miller V, Sturmer M, Staszewski S, Ren J, Stammers DK, Stuart DI, Pauwels R. A family of insertion mutations between codons 67 and 70 of human immunodeficiency virus type 1 reverse transcriptase confer multinucleoside analog resistance. Antimicrob Agents Chemother. 1999;43:1961–7. doi: 10.1128/aac.43.8.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cases-Gonzalez CE, Franco S, Martinez MA, Menendez-Arias L. Mutational patterns associated with the 69 insertion complex in multi-drug-resistant HIV-1 reverse transcriptase that confer increased excision activity and high-level resistance to zidovudine. J Mol Biol. 2007;365:298–309. doi: 10.1016/j.jmb.2006.09.073. [DOI] [PubMed] [Google Scholar]
- 19.Winters MA, Coolley KL, Girard YA, Levee DJ, Hamdan H, Shafer RW, Katzenstein DA, Merigan TC. A 6-basepair insert in the reverse transcriptase gene of human immunodeficiency virus type 1 confers resistance to multiple nucleoside inhibitors. J Clin Invest. 1998;102:1769–75. doi: 10.1172/JCI4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Antoni A, Foli A, Lisziewicz J, Lori F. Mutations in the pol gene of human immunodeficiency virus type 1 in infected patients receiving didanosine and hydroxyurea combination therapy. J Infect Dis. 1997;176:899–903. doi: 10.1086/516511. [DOI] [PubMed] [Google Scholar]
- 21.Boyer PL, Sarafianos SG, Arnold E, Hughes SH. Nucleoside analog resistance caused by insertions in the fingers of human immunodeficiency virus type 1 reverse transcriptase involves ATP-mediated excision. J Virol. 2002;76:9143–51. doi: 10.1128/JVI.76.18.9143-9151.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mas A, Parera M, Briones C, Soriano V, Martinez MA, Domingo E, Menendez-Arias L. Role of a dipeptide insertion between codons 69 and 70 of HIV-1 reverse transcriptase in the mechanism of AZT resistance. EMBO J. 2000;19:5752–61. doi: 10.1093/emboj/19.21.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meyer PR, Lennerstrand J, Matsuura SE, Larder BA, Scott WA. Effects of dipeptide insertions between codons 69 and 70 of human immunodeficiency virus type 1 reverse transcriptase on primer unblocking, deoxynucleoside triphosphate inhibition, and DNA chain elongation. J Virol. 2003;77:3871–7. doi: 10.1128/JVI.77.6.3871-3877.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.White KL, Chen JM, Margot NA, Wrin T, Petropoulos CJ, Naeger LK, Swaminathan S, Miller MD. Molecular mechanisms of tenofovir resistance conferred by human immunodeficiency virus type 1 reverse transcriptase containing a diserine insertion after residue 69 and multiple thymidine analog-associated mutations. Antimicrob Agents Chemother. 2004;48:992–1003. doi: 10.1128/AAC.48.3.992-1003.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matamoros T, Franco S, Vazquez-Alvarez BM, Mas A, Martinez MA, Menendez-Arias L. Molecular determinants of multi-nucleoside analogue resistance in HIV-1 reverse transcriptases containing a dipeptide insertion in the fingers subdomain: effect of mutations D67N and T215Y on removal of thymidine nucleotide analogues from blocked DNA primers. J Biol Chem. 2004;279:24569–77. doi: 10.1074/jbc.M312658200. [DOI] [PubMed] [Google Scholar]
- 26.Gu Z, Gao Q, Faust EA, Wainberg MA. Possible involvement of cell fusion and viral recombination in generation of human immunodeficiency virus variants that display dual resistance to AZT and 3TC. J Gen Virol. 1995;76(Pt 10):2601–5. doi: 10.1099/0022-1317-76-10-2601. [DOI] [PubMed] [Google Scholar]
- 27.Huang H, Chopra R, Verdine GL, Harrison SC. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science. 1998;282:1669–75. doi: 10.1126/science.282.5394.1669. [DOI] [PubMed] [Google Scholar]
- 28.Liu S, Abbondanzieri EA, Rausch JW, Le Grice SF, Zhuang X. Slide into action: dynamic shuttling of HIV reverse transcriptase on nucleic acid substrates. Science. 2008;322:1092–7. doi: 10.1126/science.1163108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rigby ST, Van Nostrand KP, Rose AE, Gorelick RJ, Mathews DH, Bambara RA. Factors that determine the efficiency of HIV-1 strand transfer initiated at a specific site. J Mol Biol. 2009;394:694–707. doi: 10.1016/j.jmb.2009.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rigby ST, Rose AE, Hanson MN, Bambara RA. Mechanism analysis indicates that recombination events in HIV-1 initiate and complete over short distances, explaining why recombination frequencies are similar in different sections of the genome. J Mol Biol. 2009;388:30–47. doi: 10.1016/j.jmb.2009.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Operario DJ, Balakrishnan M, Bambara RA, Kim B. Reduced dNTP interaction of human immunodeficiency virus type 1 reverse transcriptase promotes strand transfer. J Biol Chem. 2006;281:32113–21. doi: 10.1074/jbc.M604665200. [DOI] [PubMed] [Google Scholar]
- 32.Balakrishnan M, Roques BP, Fay PJ, Bambara RA. Template dimerization promotes an acceptor invasion-induced transfer mechanism during human immunodeficiency virus type 1 minus-strand synthesis. J Virol. 2003;77:4710–21. doi: 10.1128/JVI.77.8.4710-4721.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao L, Hanson MN, Balakrishnan M, Boyer PL, Roques BP, Hughes SH, Kim B, Bambara RA. Apparent defects in processive DNA synthesis, strand transfer, and primer elongation of Met-184 mutants of HIV-1 reverse transcriptase derive solely from a dNTP utilization defect. J Biol Chem. 2008;283:9196–205. doi: 10.1074/jbc.M710148200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wisniewski M, Balakrishnan M, Palaniappan C, Fay PJ, Bambara RA. Unique progressive cleavage mechanism of HIV reverse transcriptase RNase H. Proc Natl Acad Sci USA. 2000;97:11978–83. doi: 10.1073/pnas.210392297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peliska JA, Benkovic SJ. Mechanism of DNA strand transfer reactions catalyzed by HIV-1 reverse transcriptase. Science. 1992;258:1112–8. doi: 10.1126/science.1279806. [DOI] [PubMed] [Google Scholar]
- 36.Purohit V, Roques BP, Kim B, Bambara RA. Mechanisms that prevent template inactivation by HIV-1 reverse transcriptase RNase H cleavages. J Biol Chem. 2007;282:12598–609. doi: 10.1074/jbc.M700043200. [DOI] [PubMed] [Google Scholar]
- 37.Purohit V, Balakrishnan M, Kim B, Bambara RA. Evidence that HIV-1 reverse transcriptase employs the DNA 3′ end-directed primary/secondary RNase H cleavage mechanism during synthesis and strand transfer. J Biol Chem. 2005;280:40534–43. doi: 10.1074/jbc.M507839200. [DOI] [PubMed] [Google Scholar]
- 38.Wisniewski M, Balakrishnan M, Palaniappan C, Fay PJ, Bambara RA. The sequential mechanism of HIV reverse transcriptase RNase H. J Biol Chem. 2000;275:37664–71. doi: 10.1074/jbc.M007381200. [DOI] [PubMed] [Google Scholar]
- 39.Lin Y, Nieuwlandt D, Magallanez A, Feistner B, Jayasena SD. High-affinity and specific recognition of human thyroid stimulating hormone (hTSH) by in vitro-selected 2′-amino-modified RNA. Nucleic Acids Res. 1996;24:3407–14. doi: 10.1093/nar/24.17.3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeStefano JJ, Bambara RA, Fay PJ. Parameters that influence the binding of human immunodeficiency virus reverse transcriptase to nucleic acid structures. Biochemistry. 1993;32:6908–15. doi: 10.1021/bi00078a014. [DOI] [PubMed] [Google Scholar]
- 41.Yusa K, Kavlick MF, Kosalaraksa P, Mitsuya H. HIV-1 acquires resistance to two classes of antiviral drugs through homologous recombination. Antiviral Res. 1997;36:179–89. doi: 10.1016/s0166-3542(97)00053-3. [DOI] [PubMed] [Google Scholar]
- 42.Sarafianos SG, Marchand B, Das K, Himmel DM, Parniak MA, Hughes SH, Arnold E. Structure and function of HIV-1 reverse transcriptase: molecular mechanisms of polymerization and inhibition. J Mol Biol. 2009;385:693–713. doi: 10.1016/j.jmb.2008.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beard WA, Bebenek K, Darden TA, Li L, Prasad R, Kunkel TA, Wilson SH. Vertical-scanning mutagenesis of a critical tryptophan in the minor groove binding track of HIV-1 reverse transcriptase. Molecular nature of polymerase-nucleic acid interactions. J Biol Chem. 1998;273:30435–42. doi: 10.1074/jbc.273.46.30435. [DOI] [PubMed] [Google Scholar]
- 44.Roda RH, Balakrishnan M, Hanson MN, Wohrl BM, Le Grice SF, Roques BP, Gorelick RJ, Bambara RA. Role of the Reverse Transcriptase, Nucleocapsid Protein, and Template Structure in the Two-step Transfer Mechanism in Retroviral Recombination. J Biol Chem. 2003;278:31536–46. doi: 10.1074/jbc.M304608200. [DOI] [PubMed] [Google Scholar]
- 45.Delviks-Frankenberry KA, Nikolenko GN, Boyer PL, Hughes SH, Coffin JM, Jere A, Pathak VK. HIV-1 reverse transcriptase connection subdomain mutations reduce template RNA degradation and enhance AZT excision. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:10943–8. doi: 10.1073/pnas.0804660105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raja A, DeStefano JJ. Interaction of HIV reverse transcriptase with structures mimicking recombination intermediates. J Biol Chem. 2003;278:10102–11. doi: 10.1074/jbc.M210201200. [DOI] [PubMed] [Google Scholar]
- 47.Weiss KK, Bambara RA, Kim B. Mechanistic role of residue Gln151 in error prone DNA synthesis by human immunodeficiency virus type 1 (HIV-1) reverse transcriptase (RT). Pre-steady state kinetic study of the Q151N HIV-1 RT mutant with increased fidelity. J Biol Chem. 2002;277:22662–9. doi: 10.1074/jbc.M200202200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.