

Abstract
The perylenequinones are a novel class of natural products characterized by pentacyclic conjugated chromophore giving rise to photoactivity. Potentially useful light-activated biological activity, targeting protein kinase C (PKC), has been identified for several of the natural products. Recently discovered new members of this class of compound, as well as several related phenanthroperylenequinones, are reviewed. Natural product modifications that improve biological profiles, and avenues for the total synthesis of analogs, which are not available from the natural product series, are outlined. An overview of structure/function relationships is provided.
Keywords: Perylenequinone, Chiral Biaryl, Binaphthol, Photodynamic Therapy, Natural Products
Introduction
Perylenequinone natural products have been used in traditional Chinese herbal medicine for centuries. They are dark colored pigments; isolated from diverse sources such as mold species, aphids and recently, from plant species. These structures have been studied extensively due to their interesting biological activity, notably for their use as anti-cancer agents. This review will describe the isolation and characterization of perylenequinones, including some recently identified novel structures and the related discoveries of new phenanthroperylenequinones, the preparation and biological activity of some derivatives prepared from the parent natural products, total synthesis of the natural products, and the synthesis of novel perylenequinones followed by a summary of the biological activity of these new derivatives.
The perylenequinones comprise a family of natural products characterized by an oxidized pentacyclic core as represented by the parent perylenequinone 1 (Figure 1). i Dihydroxy 1 was first isolated in 1956 from the fungus Daldinia concentrica as blue-black crystals and is the simplest of the naturally occurring perylenequinones.ii Since then, many more perylenequinones have been discovered in the extracts of a variety of species. Among these vibrantly colored natural products, most can be divided into one of three classes (examples of each in Figure 1): A) C20 compounds without carbon substituents, B) perylenequinones like cercosporin (2), usually isolated from mold, containing carbon substituents, and C) erythroaphins like rhodoaphin-be (3), isolated from aphids. Of the three, the most prevalent is class B. As such, the unique structural, biological and photochemical properties of the class B pigments have been extensively studied, and many of these have been the subject of reviews.i Phenanthroperylenequinones such as hypericin (4) have also been thoroughly reviewed.iii
Figure 1.

Classes of perylenequinone natural products.
Section 1: The Perylenequinone Family of Natural Products, Isolation and Characterization
The section covers the recent isolation and characterization of several new perylenequinone natural products of class B. In addition, some novel phenanthroperylenequinone compounds, including the gymnochromes will be described.
Perylenequinones
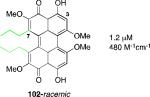
The class B perylenequinone natural products that have been reviewed previously include cercosporin (2, Figure 2), isolated from the fungus Cercospora kikuchii4; calphostins A-D (5a-d), isolated from Cladosporium cladosporioides 5, phleichrome (6), isolated from Cladosporium phlei6, and the elsinochromes (7)7, isolated from several species of the genus Elsinoe. One class of perylenequinones that has been extensively studied is the hypocrellin family (8-10), isolated from the species Hypocrellin Bambusae 8,9. The hypocrellins have been used for centuries in Chinese folk medicine for the treatment of vitiligo, psoriasis, and other diseases. There have been extensive studies of the hypocrellins and their derivatives for their use in photodynamic therapy for cancer treatment.
Figure 2.

Naturally occurring mold perylenequinones.
The architecturally similar compounds 2 and 5-6 are characterized by a chiral helical oxidized core and stereogenic C7,C7’-2-hydroxypropyls. Upon structural elucidation, it was discovered that many of these pigments, excluding the hypocrellins, are C2-symmetric, having C7,C7’-substitution with centrochiral stereocenters of the same absolute configuration. While the structures of 5a-d and 6 were assigned in conjunction with their isolation,5,6 it was not until the 1970s that the structure of 2 (isolated in 1957) was elucidated.4,10,11 This determination was accomplished through a series of chemical transformations and subsequent spectroscopic analysis.10-12 The crystal structure of a cercosporin derivative (wherein the 2-hydroxypropyl group was esterified) was used to assign the absolute and relative configuration of 2.13 Correlation of the circular dichroism (CD) and NMR spectra allowed determination of the helical configurations and relative stereochemistries of the remaining perylenequinone natural products. These data continue to be used by researchers to assign axial chirality to new perylenequinones as they have been isolated and characterized.
The stability of the axial chirality of the perylenequinones is variable depending on the nature of the C2 and C7 substituents. Calphostins 5 undergo atropisomerization at 110 °C, but cercosporin 2, with its 7-membered ring bridge at C2 and C2’, has a much lower barrier and isomerizes at 40 °C. The hypocrellins 8, with their 7-membered ring bridging at C7 and C7’, exist as a mixture of rapidly interconverting diastereomers at room temperature, the predominant isomer of which is depicted in Figure 2.
Recently a new orange-red colored pigment was isolated from the fruiting bodies of Shiraia bambusicola, and named hypocrellin D (Figure 3).14 Its structure was elucidated with MS, 13C NMR, 1H NMR, and 2D NMR (HMBC, ROESY) studies. It is not a perylenequinone, as it does not have the fused pentacyclic core of the other hypocrellins, but it contains the same 7-membered ring motif of hypocrellin A (8). This material likely forms via an oxidative cleavage of hypocrellin A, a process mediated by the material itself which acts as a photosensitizer delivering singlet oxygen. It has been shown that prolonged irradiation of hypocrellin generates bis-para-naphthoquinones like 11, presumably via cycloaddition of singlet oxygen to form an endoperoxide followed by rearrangement.15
Figure 3.
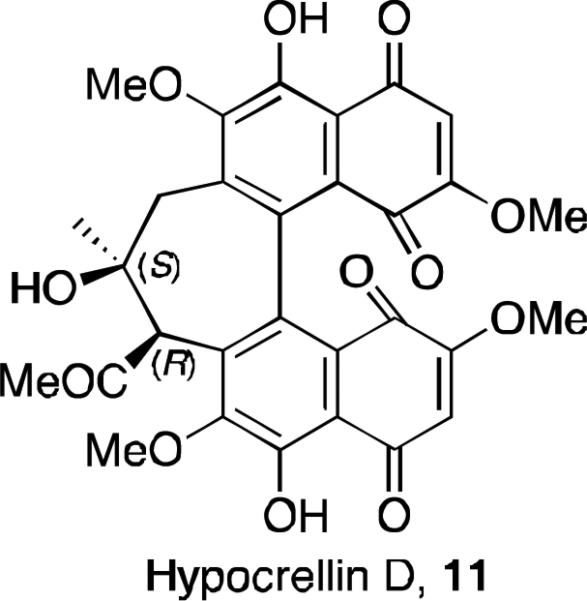
The structure of hypocrellin D.
A novel type of perylenequinone, hypomycin A (Figure 4, 12), was isolated from the fungus Ascomycetes hypocreaceas hypomyces.16 Its structure was reported as shown in Figure 4 and was elucidated by means of the 1H and 13C NMR, HMQC, and HMBC spectra. Correlations in the NOESY led to the assignment of relative stereochemistry. This structure is unique in that the typical conjugated system of the perylenequinones has been disrupted. It is optically active, but its absolute stereochemistry was not assigned by the authors. It is likely that this structure arises from oxidative cyclization of hypocrellin A (8) or hypocrellin (ent-8). A second natural product isolated from the same species, hypomycin B (Figure 4, 13), was reported and the structure elucidated. 17 This compound likely arises from an oxidative insertion of one of the phenols into the adjacent methoxy C–H bond. It is structurally distinct from the related perylenequinones in that it is planar, not C2-symmetric, and has only one intramolecular hydrogen bond. It was initially characterized and its structure elucidated from HMBC and NOESY spectra. This unique perylenequinone with only one hydroxyl group and one intramolecular H-bond has proven useful to study photochemical processes of excited state intramolecular hydrogen atom transfer.18
Figure 4.
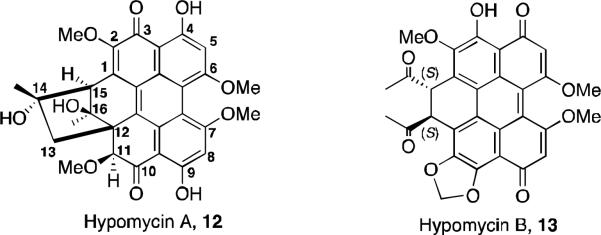
The hypomycins.
Recently, perylenequinones were isolated from a plant source instead of mold or aphid species. Scutiaquinones A (14) and B (15) are deep red solids and were isolated from the root of Scutia myrtina19 (Figure 5). These compounds show anthelmintic activity against the parasitic worm Haemonchus contortus. The structures in Figure 5 were determined by 1H NMR and 13C NMR, DEPT, COSY, HSQC, HMBC, and ROESY. Both scutiaquinones have similar spectra, indicating the presence of the perylenequinone core with two dihydropyran rings and having C2-symmetry. However, ROESY correlations for 15 are different than for 14 and indicate that the dihydropyran rings are on the opposite side of the system in 15. In the CD spectra for both compounds, there is a much greater magnitude of molar ellipticities for 14, suggesting that the perylenequinone nucleus is twisted farther out of plane. This degree of the helicity supports the proposed structure for 14, which would have greater steric interactions between the dihydropyran side chains. The stereochemistry of the perylenequinone core was inferred by comparing the CD spectra with several known perylenequinone natural products, having axial chirality similar to that of hypocrellin A. At the time of publication, the absolute stereochemistry of C-9 and C-11 were not known.
Figure 5.
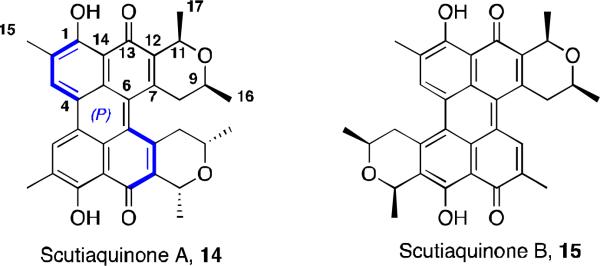
Scutiaquinones.
Hypericins
Hypericin 4 (Figure 1) is a well studied natural product, one of the principle active constituents of Hypericum (Saint John's Wort).20 It has a similar structure to the perylenequinones but with a fused octacyclic core. It possesses antiviral and antitumor activity. Gymnochromes A-D and isogymnochrome D (Figure 6) are five violet colored pigments isolated from the stalked crinoid Gymnocrinus richeri, a “living fossil” located in deep sea water.21 The presence of these natural products in this species lends support to the theory that quinoid pigments were biosynthesized by crinoids many millions of years ago.22 The gymnochromes posses the core structure of hypericin, but differ in the substitution flanking the central core and also contain bromo-substitution. Due to steric crowding around the central core, these compounds possess helical chirality.
Figure 6.

Gymnochromes A-D and isogymnochrome D.
The structure and stereochemistry of the gymnochromes were determined by FABMS, UV-Vis, IR, 1H NMR, 13C NMR, and CD spectra. Gymnochromes A and B are not C2-symmetric, although the other three reported products are. At the time of the first report, the location of the third bromo-substituent on either the C9 or C12 carbon of gymnochrome B was still under investigation. The helical configuration of the gymnochromes is inferred by comparison of the CD spectra to that of cercosporin, with gymnochrome B and isogymnochrome D having the opposite helical configuration of the other gymnochromes. The absolute configuration on alkyl substituents was predicted by observing the shielding effects in the 1H NMR spectra of the helical aromatic core on the terminal methyl protons, and comparing the shifts with those observed in perylenequinone natural products of known structure. To support this predicted absolute configuration, Horeau's method was applied to the hexamethyl aromatic ether derivative of gymnochrome D, and the anticipated R stereochemistry was observed.
Recently, gymnochromes E and F (Figure 7) were isolated as brown oils from the crinoid Holopus rangii.23 Their structures were determined by MS, 1H NMR, 13C NMR, and CD spectra. The alkyl substituent structures were determined by COSY and HMBC correlations. Gymnochrome E is a non-C2-symmetric, dibrominated phenanthroperylenequinone with a very similar structure to gymnochrome A, although with the opposite helical configuration. Gymnochrome F is a desulfated version of isogymnochrome D where one of the alkyl substituents is also truncated.
Figure 7.
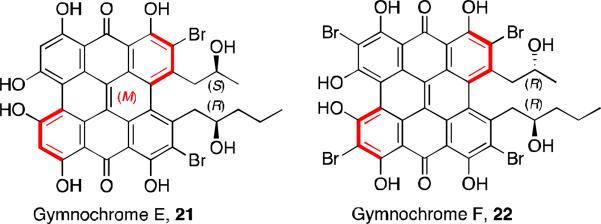
Gymnochromes E and F.
Section 2: Synthesis of Natural Product Derivatives
Hypocrellin Derivatives
The hypocrellin natural products are of intense interest due to their potential anticancer properties, as well as their possible use to treat skin conditions such as psoriasis and acne.24 There have been numerous studies to establish their use for PDT (photodynamic therapy), as well as SDT (sonodynamic therapy). 25 The mechanism of action of these photosensitizers is still under investigation, but the generation of reactive oxygen species upon irradiation is considered an important factor in their photodynamic activity. Due to the water insolubility of the hypocrellins, much work has been done on the formulation of hypocrellins and their derivatives. Modifications to the hypocrellin structure that have been reported include sulfonation26, amination27, halogenation28, and metal ion-chelation. 29 Most of the water-soluble perylenequinone (PQ) derivatives that have been made have significantly less photodynamic activity compared to the activity of the parent natural product. This review will focus on some examples of synthetic modifications of the perylenequinone natural products that have improved their pharmacokinetic properties and their photodynamic activity.30
Photosensitizers for use in PDT have been limited in part due to the poor extinction coefficient in the wavelength of light in the therapeutic window (600-900 nm) needed to penetrate deep tissue. Analogs with a stronger absorption in this window (600-900 nm) are therefore desirable. Amine-substituted hypocrellins that shown promise have been prepared by amine addition to the enone followed by extrusion of methoxide. Thus, the synthesis of 2-BADMHA 23 (Figure 8) was achieved from the reaction of hypocrellin A (8, Figure 2) with butylamine in pyridine. This analog has a strong absorption shoulder at 628 nm and was found to be cytotoxic to the gastric adenocarcinoma MGC803 cell line. Its photopotentiation factor (LD50 dark/LD50 light) was found to be more than 200-fold, compared to hypocrellin A which was 4-fold under the same conditions. 31 Another amine derivative was prepared from the reaction of hypocrellin B (10, Figure 2) and butylamine. Both butylamino-substituted hypocrellins were found to have increased ability to generate O2.- as compared to their parent hypocrellins, and both had increased in vitro activity (HeLa, MGC803, and the Capan-1 human pancreatic tumor cell line) and in vivo activity (mouse model of human pancreatic cancer).
Figure 8.
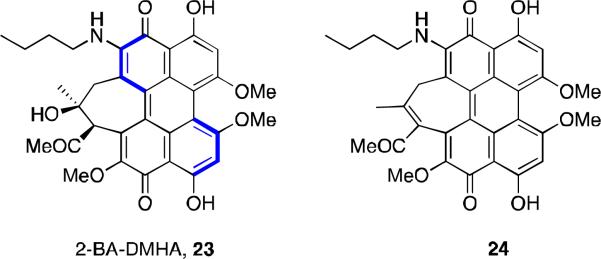
Butylamino-substituted hypocrellins.
To further improve the pharmacokinetic properties of hypocrellin derivatives, compounds that are more amphiphilic were prepared. The hypocrellin B (10, Figure 2) ketone was modified by treatment with either 3-methoxypropylamine in refluxing pyridine to provide 25 (Figure 9) or the appropriate amino acid and a solution of DMFH2O (pH 13) under reflux to provide 26 and 27.32 These enamine compounds were characterized using MS, 1H NMR, and IR spectra; comparison with hypocrellin B confirmed the substitution of the ketone as well as replacement of the C11-methoxy with a hydroxyl group. The three new derivatives all showed an enhanced red absorption compared to hypocrellin B, but quantum yields for generation of singlet oxygen were lower. The amphiphilicities of the three derivatives, as measured by their partition coefficient (PC) in octanol and 0.1 mol L-1 phosphate buffer pH 7, were greatly improved. While hypocrellin B has a PC of 46.4, compounds 25, 26, and 27 have PC values of 9.3, 5.9, and 4.9, respectively. Evaluation against the human oral cavity epithelial carcinoma KB cell line revealed undesirable dark toxicity, with compound 27 showing some improvement with a photopotentiation factor 10-fold better than that of hypocrellin B.
Figure 9.
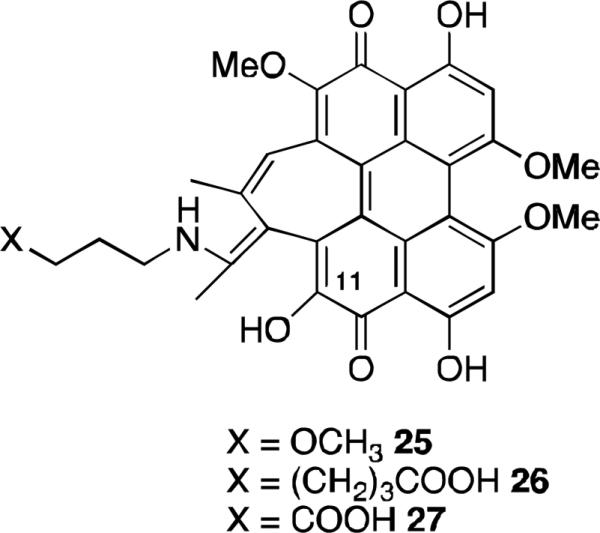
Scutiaquinones.
Compounds 28 and 29 (Figure 10), hypocrellin B derivatives substituted with a benzyl amine at the C6-position, were prepared by reaction of the natural product with phenmethylamine and THF at 50-55 °C in the dark. 33 The two main products were purified by TLC and characterized by 1H NMR, UV-Vis, IR, and mass spectra. Both compounds showed a new absorption band at 628 nm. These compounds along with hypocrellin B were tested for dark and light toxicity against the cancer cell line MGC803 and the photopotentiation factors for 28 and 29 were 200 and 80, compared with 15 for hypocrellin B.
Figure 10.
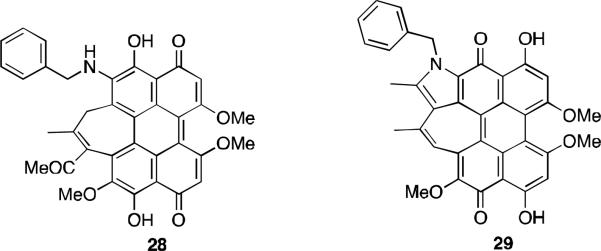
Phenmethylamine derivatives of hypocrellin B.
An additional amine substituted hypocrellin B derivative 30 combining the C6-amine substitution and imine substitution (Figure 11) was found to be active against MGC803 cancer cells. As with the previously reported amine derivatives, this compound showed a strong absorption peak in the therapeutic window, but had reduced quantum yield of singlet oxygen as compared to hypocrellin B. When tested against the MGC803 cells, 30 had a photopotentiation factor 4-fold greater than that of hypocrellin B.34
Figure 11.
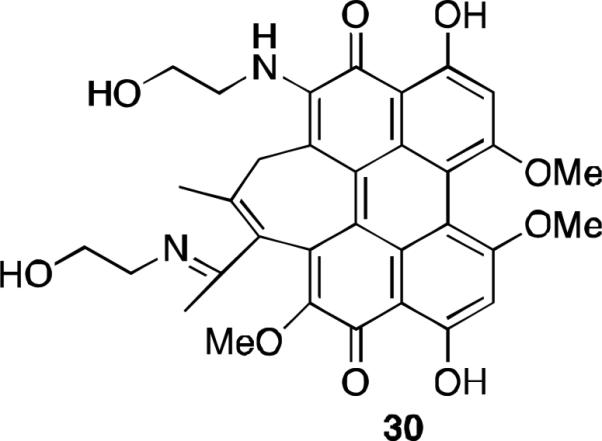
Ethanolamine substitution of hypocrellin B.
Further progress in the synthesis of hypocrellin B derivatives with desirable properties was made with the synthesis of C6-amino substituted hypocrellin B compounds 31 and 32 (Figure 12).35 These derivatives were prepared by an addition/elimination sequence using a diamine to displace the C6-methoxy of hypocrellin B. As with most of the amine derivatives, these compounds exhibited enhanced absorption in the phototherapeutic window. Both were active against the colon cancer cell line HCT 116 and both had enhanced hydrophilicity compared to hypocrellin B as measured by chromatography Rf values.
Figure 12.
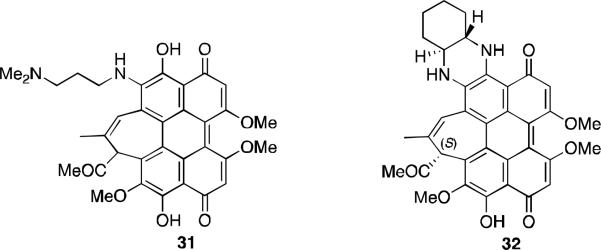
Amine substituted derivatives of hypocrellin B.
Compound 31, SL017, was chosen as a clinical candidate for both PDT and SDT. The mechanisms of action for both treatments are still under investigation. A recent study reports that it is activated with either light or ultrasound and targets mitochondria by disrupting the membrane potential and generating reactive oxygen species (ROS). 36 This compound is under clinical evaluation for three indications: actinic keratosis, acne vulgaris, and for permanent hair removal.37
The cyclic diamine derivative 32, SL052, was also chosen as a clinical candidate, and is currently in the pipeline for prostate cancer.37 Although this derivative does not have adequate water solubility, formulation with nanoclusters of polyvinylpyrrolidones is one of the methods being studied to improve physicochemical properties and has been successful in several in vivo mouse tumor models with both PDT and SDT.38
Several other hypocrellin B derivatives have been prepared in order to improve solubility. In general, increased hydrophobicity is often accompanied by reduced photodynamic activity and this obstacle has been the subject of a number of studies. The amphiphilic sulfonic acid 33 was prepared (Figure 13) and tested for photodynamic activity and PK properties. It is soluble in PBS (6.6 mg/mL) at therapeutically relevant concentrations for administration by IV injection, and a partition coefficient of 5:1 indicated its potential bioavailability. It has a 12.5 times higher quantum yield of singlet oxygen under red light (600-700 nm) than hypocrellin B, and it has a comparable activity against human lung cancer A549 cells.39
Figure 13.
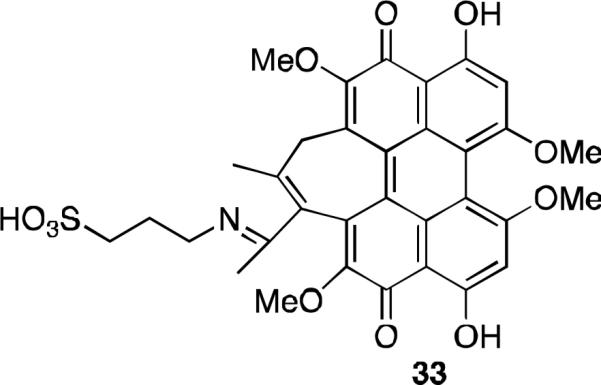
Water soluble derivative of hypocrellin B.
Further investigation into improvements on bioavailability involved the preparation of two surfactant-like derivatives. Carboxylate salts 34 and 35, abbreviated as SAHB and DMHB respectively (Figure 14), were synthesized and both demonstrated increased solubility. DMHB was more active against human breast carcinoma MCF-7 cells than hypocrellin B by PDT. 40 An interesting feature of both these analogs is that their measured singlet oxygen quantum yield is identical to their PDT activity against the MCF-7 cells. This indicates that their cellular affinity is independent of their hydrophilicity and that singlet oxygen generation is an important mechanism of PDT.
Figure 14.
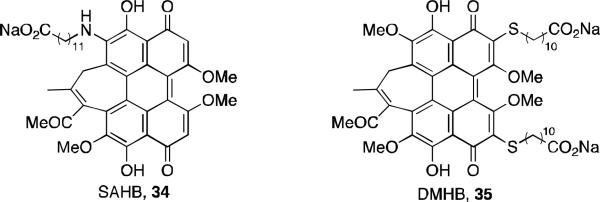
Hypocrellin B derivatives with surfactant-like properties.
Analogs of Other Perylenequinones and Phenanthroperylenequinones
Elsinochrome A (7a, Figure 2) possesses the highest singlet oxygen quantum yield amongst the perylenequinones but there have been few studies into its medicinal applications in part due to its low solubility. There have been extensive studies into preparing hypocrellin B analogs in order to improve water solubility as well as photodynamic activity. With these studies in mind, the preparation of a sulfonated elsinochrome derivative (36, Figure 15) that includes an extended aliphatic chain with a sulfate group was intended to maintain photodynamic activity and cell permeability while improving solubility.41 However, this material is sparingly soluble and has a log P of 7. Its quantum yield of singlet oxygen generation is 0.73, and the photodynamic activity against human lung cancer cell line A549 is 60% of the parent elsinochrome A, a clinically acceptable result as this is still higher than most photosensitizers.
Figure 15.
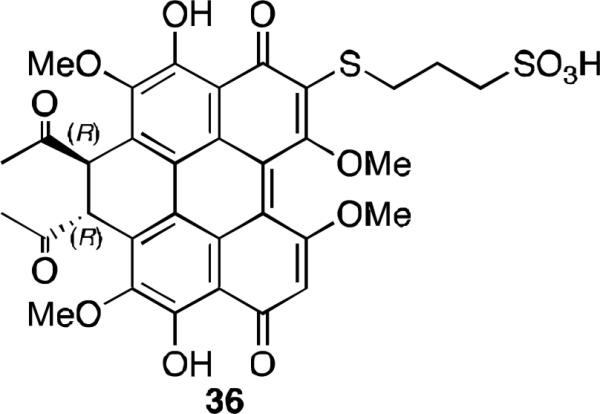
Water soluble derivative of elsinochrome A.
There have been some preparation and studies of derivatives of hypericin and gymnochromes to compare their biological activity to that of the parent natural products. Several brominated hypericins were prepared by the treatment of hypericin with the appropriate number of equivalents of bromine in pyridine to prepare the di-, tri- and tetrabrominated compounds 37-39 (Figure 16).42 Dibromohypericin 39 was found in later studies to have anti-viral activity; and it was more active against herpes simplex virus (in light, MIC = 0.9 nM) and influenza virus (in light, MIC < 5 nM) than hypericin or gymnochrome B.43
Figure 16.
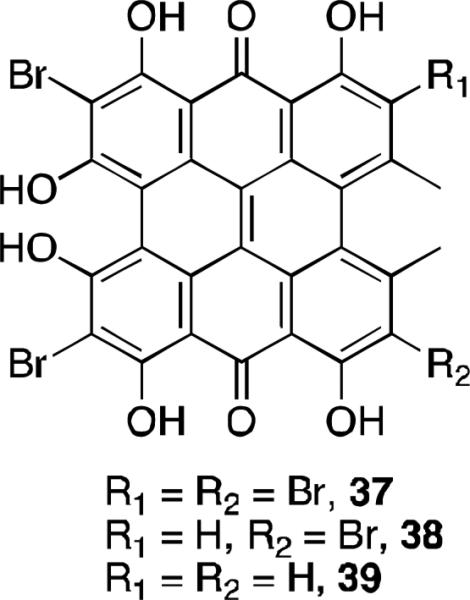
Brominated hypericins.
While investigating the biosynthesis of the natural product actinorhodin 40, a novel perylenequinone-type shunt product named actinoperylene 41 was identified (Figure 17). 44 Actinorhodin is an aromatic polyketide natural product from the benzoisochromanequinone family of antibiotics and is isolated from Streptomyces coelicolor. The investigation began with the proposal that the proteins actVA-ORF5 and –ORF6 were involved in the C6-oxidation step in the biosynthetic pathway. A ΔactVA-5,6 mutant was developed, and this produced a yellow-brown pigment. After culturing and growing this mutant, the supernatant of the culture was purified by HPLC to yield a new compound. The structure was determined to be a symmetrical dimer by LC/MS analysis, 1H NMR, and UV-Vis spectrosopy. NOe experiments revealed the pyran ring has a trans configuration, and correlation between the protons on C4 and C9’ led to the assignment of the antiparallel connection between the two tricyclic units.
Figure 17.
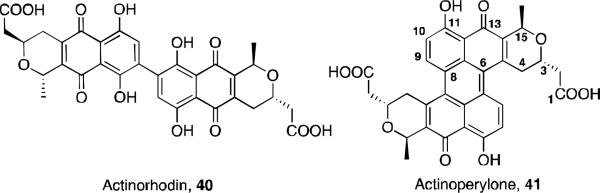
Actinorhodin and actinoperylene.
The formation of this novel product supports the hypothesis that genes involved in the biosynthesis of benzoisochromanequinone antibiotics, specifically in the C6-oxidation of intermediate 43 (Scheme 1), were knocked out. Interruption of the oxidation allows for tautomerization, then oxidative dimerization to form actinoperylone 41 through another enzymatic pathway.
Scheme 1.

Proposed shunt pathway to actinoperylone 41.
Section 3: Perylenequinone Total Synthesis Efforts
Syntheses of the Perylenequinone Core Structure
Given their unique architecture and desirable photo-activated biological profiles, the perylenequinones have attracted a considerable amount of attention from a synthesis standpoint. The most challenging physical aspect of these compounds is the extended conjugated pentacyclic core. In the past three decades, work on the generation of the core ring system has flourished.45,46,47,48,49,50,51,52,53,54,55,56,57,58,59
Prior to these achievements, the first reported synthesis of parent perylenequinone 46 was published by Zinke and coworkers in 1929 via oxidation of tetranitroperylene 47 with sulfuric acid at 140 °C under air (Scheme 2).60 However, the correct structure was not elucidated until 1954.61 Due to the atypical stability of the product 46 proposed by Zinke, Calderbank and coworkers repeated the procedure, which led to the structural reassignment of the product as parent perylenequinone 1.
Scheme 2.
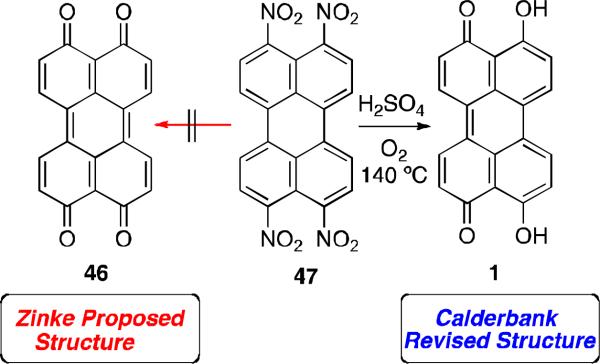
Synthesis of parent perylenequinone 1.
Further progress on perylenequinone syntheses was not made until 1972 when Weisgraber and Weiss attempted to synthesize perylenequinone 51, containing the desired oxygenation pattern present in the natural products.54 They utilized a one-pot, two-step cationic cyclization/oxidative dimerization sequence to obtain the biaryl precursor (Scheme 3). Unfortunately, the initial assignments of both the biaryl precursor 50 and the subsequent perylenequinone 51 were found to be incorrect,62 and the final cyclized product was reassigned as dinaphthofuranedione 55.
Scheme 3.

Weiss attempted synthesis of perylenequinone 51 (PPA = polyphosphoric acid).
Several years later Dallacker and Leidig, reported a different approach to synthesize 2,2’,4,4’-tetramethoxyperylenequinone 59 starting from a bisphenyl substrate 56 (Scheme 4).55 While novel, the synthesis was lengthy (14 steps) and lacked the necessary C6,C6’-oxygenation.
Scheme 4.

Dallacker perylenequinone synthesis.
The perylenequinones from these early efforts are optically inactive. While some of the compounds may display a helical conformation, none contain the sufficiently large substituents flanking the perylenequinone core to prevent rapid interconversion of the helical stereochemistry. As a result, Zhang45 and Lown46 developed preparations of racemic C7,C7’-substituted perylenequinone derivatives. The syntheses involved the oxidative biaryl coupling of the highly oxygenated substrates 60 and 61, to provide the perylenequinone 62 directly (Scheme 5).
Scheme 5.
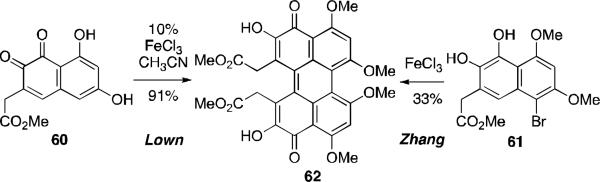
Zhang and Lown: Racemic syntheses of perylenequinone 62.
The First Stereoselective Syntheses of Perylenequinones
The first stereoselective synthesis of a perylenequinone natural product was the generation of (–)-calphostin A (ent-5a), (–)-calphostin D (ent-5d), and phleichrome by Broka in 1991.56 Since then, the Hauser,57 Coleman,58 and Merlic59 research groups have also published alternate routes to provide the calphostins. A common theme of these total syntheses is the diastereoselective biaryl coupling of stereogenic naphthalenes (i.e., 63 and 64 in Scheme 6), the stereochemistry of which is retained during perylenequinone formation (e.g., 66 to 5a in Figure 18). The key difference among these syntheses is the way in which the C7,C7’-stereoarrays were generated.
Scheme 6.
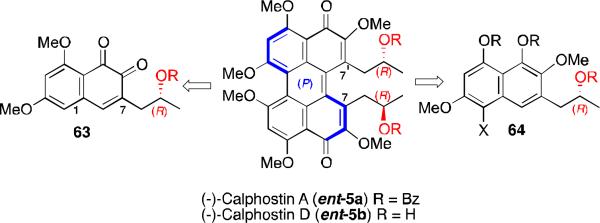
Retrosynthesis of first stereoselective syntheses of perylenequinones.
Figure 18.

Diastereoselective dimerizations en route to the calphostins.
The centrochiral C7-substitution provided modest stereocontrol during the dimerizations. Presumably, the distance between the newly formed C1,C1’-biaryl bond and the C7,C7’-substituents prevents effective stereochemical communication. Furthermore, the predominant diastereomer does not usually correspond to the calphostin array, and Mitsunobu inversion of the stereocenter or thermal isomerization of the perylenequinone core was needed to access these natural products.56,57,58,59
Synthesis of a Perylenequinone Containing Only a Helical Chiral Stereochemical Element
Despite the multiple total syntheses of perylenequinone natural products,56,57,58,59 there were no reports of an enantiopure perylenequinone having to rely on helical chirality as the only stereochemical element. Later work by our group showed that such helical chiral perylenequinones (78, Scheme 7) can be produced and are atropisomerically stable.47,50 The key elements of this synthetic venture included a catalytic enantioselective biaryl coupling with copper catalyst 74, a phenyliodosobistrifluoroacaete-induced C5,C5’-hydroxylation, and finally oxidation to the perylenequinone with MnO2. Transfer of the axial stereochemistry to the perylenequinone helical stereochemistry proceeded with good fidelity. This synthesis venture represented the first example an enantioselective preparation of a perylenequinone containing only an axial chiral element. In addition, this strategy allows easy access to both enantiomers of the perylenequinone backbone without having relying on other stereochemical elements for control.47,50
Scheme 7.

Asymmetric synthesis of homochiral perylenequinone 78.
Total Synthesis of Hyprocrellin A
As installation of perylenequinone axial chirality in the absence of any other stereochemical element was now obtainable,47,50 syntheses of the more complex perylenequinone natural products became the focus of synthetic effort. Notably, a synthesis of 8, which contained the additional complexity of a seven membered ring, could be addressed.48,53 The biosynthesis of 8 calls for the formation of the seven-membered ring via a transannular intramolecular aldol reaction of 79 (Scheme 8). Relying on the prior methods of perylenequinone construction would require oxidation of C7,C7’-alcohol stereocenters, which had required significant effort to generate in enantiomerically pure form. As an alternative we pursued an approach in which the helical stereochemistry would be established with complete stereofidelity first, followed a potentially biomimetic, dynamic stereochemistry transfer (DST) aldol reaction.48,53
Scheme 8.

Retrosynthesis analysis of hypocrellin A.
The stereochemistry transfer was complicated by the dynamic state of the helical axis of 8.9 The helical stereochemistry of perylenequinone 79 is stable at room temperature and should be able, in principle, to direct the stereochemistry of the intramolecular aldol reaction. After formation of the seven-membered ring, however, the integrity of the helical axis is lost as seen in the rapid room temperature atropisomerization of 8 ((M):(P) = 1:4, Scheme 8).9
The synthesis of the desired aldol perylenequinone precursor 79 is outlined in Scheme 9.48,53 Enantioselective oxidative biaryl coupling of 81 further demonstrated the utility of our copper 1,5-diaza-cis-decalin catalyst system, providing 80 in 81% ee (>99% ee after one trituration) and good yield. Deacylation of the C4,C4'-positions and subsequent protection of the free naphthols was followed by a highly efficient Suzuki coupling to install the C7, C7’-allyl moeities, affording 84. After C5,C5’-hydroxylation using phenyliodosobistrifluoroacetate, the allyl groups were converted to the desired diketone via a Wacker oxidation. Other tactical steps to the key aldol substrate 79 included a palladium-mediated decarboxylation of 86, oxidative cyclization where the axial stereochemistry was transferred to the helical stereochemistry of perylenequinone 88, and PdCl2 deprotection of the bisketal 88.48,53
Scheme 9.

Synthesis of hypocrellin A chiral perylenequinone retron.
With a practical synthesis for 79 developed, the intramolecular diketone aldol cyclization was investigated.48,53 Although an aldol reaction has been proposed for the biosynthesis pathway of the hypocrellins, precedent for this type of 1,8-dicarbonyl aldol reaction is unknown. The only examples of such diketone aldol cyclizations involved multicyclic or bridged bicyclic systems,63 and no such examples exist for 1,8-diketones forming seven-membered rings.64 MM2 calculations indicated that a (Z)-enolate of 79 would give rise to the syn aldol product corresponding to hypocrellin A via a closed chair-like transition state that favors exposure of one face of the ketone (Transition State A) by about 1.5 kcal/mol (Scheme 10).65
Scheme 10.

Dynamic stereochemistry transfer.
Silazide bases are known to give predominately (Z)-enolates.66 After screening, it was found that the aldol cyclization of 79 with LiN(SiMe2Ph)2 at –105 °C provided the desired seven-membered ring (Scheme 11).48,53 The product was immediately subjected to selective deprotection the C4, C4’ methyl ethers with MgI2, yielding the natural form of hypocrellin A as the major product. As predicted, the two newly formed chiral stereocenters in the 7-membered ring were dictated by the helical (P)-stereochemistry and the (Z)-enolate geometry. A small amount of the (E)-enolate led to the anti aldol product affording the diastereomeric natural product shiraiachrome (9). This culminated in the completion of the first total syntheses of hypocrellin A and shiraiachrome A (syn:anti = 10:1; syn diastereomer, 92% ee).48,53
Scheme 11.

First total synthesis of hypocrellin A.
Total Syntheses Phleichrome, Calphostins, and Cercosporin
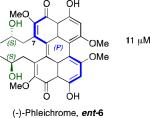
Our group continued synthesis efforts with the phleichrome, calphostin, and cercosporin natural products.49,51,52 In addition to synthesizing the natural products themselves, a high priority was the synthesis derivatives that had not been accessible previously and testing of their biological activity. Key to entry into all the possible isomers was introducing the C7,C7’-stereochemistry from an external source already containing the target absolute stereochemistry. Notably, 5d and 6 contain the same stereogenic C7,C7’-2-hydroxypropyl groups, but they differ in the stereochemistry of the perylenequinone backbone. Double alkylation via a copper-mediated epoxide-opening was selected for installation of the 2-hydroxypropyl groups onto the highly functionalized bisiodides 80 and ent-80 (Figure 19). The requisite enantiomeric biaryls 80 and ent-80 48,49,51,52,53 were readily generated using our 1,5-diaza-cis-decalin copper catalyst (Scheme 9).67
Figure 19.

Common homochiral intermediate to the perylenequinone natural products.
This effort entailed formation of a double cuprate of unprecedented complexity and reaction with two epoxide units.49,51,52 Even though such epoxide openings are difficult, rigorous control of the reaction parameters provided a high yielding and reproducible process. Key subsequent transformations to complete syntheses of (+)-calphostin D (5d) and (+)-phleichrome (6) (Scheme 12) include C5,C5’-hydroxylation via palladium catalyzed coupling of the hindered aryl chloride, C3,C3’-decarbonylation, and oxidative cyclization.49,51
Scheme 12.
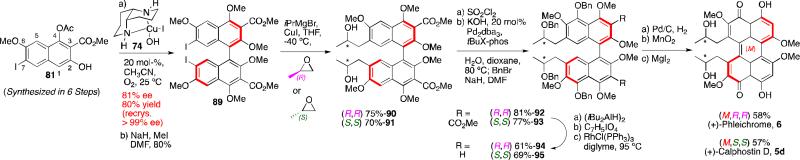
Total syntheses of phleichrome and calphostin D.
For cercosporin, the main challenge lays in the instability of the natural product (atropisomerizes to a mixture of diastereomers at 37 °C). The atropisomerization barrier of phleichrome (29.4 kcal/mol)6 allows retention of the perylenequinone helicity up to 60 °C, but the addition of the methylidene bridge lowers the barrier in cercosporin to 28.2 kcal/mol (27.4 kcal/mol in benzene)10. By designing a synthesis that withheld perylenequinone formation to the latest stage possible, we were able to exploit the much greater atropisomeric stability of the binaphthyl series (Scheme 13). Key methods that were developed to enable this enantioselective synthesis included, chlorination followed by Buchwald-Hartwig coupling to selectively hydroxylate the C5,C5’-positions of the binaphthol intermediate without atropisomerization, formation of the seven-membered ring containing the methylidene bridge, aromatic decarbonylation without accompanying C-H insertion into the adjacent methylidene bridge, and formation of the perylenequinone without concomitant atropisomerization. After considerable screening, it was discovered that that BrCH2Cl was the best electrophile for forming the seven-membered ring providing good yield with no atropisomerization. Selective removal of the C4,C4’-methyl ethers completed the first total synthesis of cercosporin (2).49,52
Scheme 13.
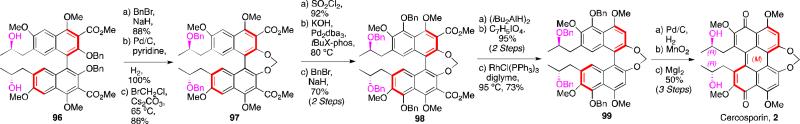
First total synthesis of cercosporin.
As a consequence of the copper catalyzed enantioselective biaryl coupling, the two enantiomers of a key precursor (bisiodides 80 and ent-80, Scheme 9, could be prepared in pure form. With these compounds in hand, selective syntheses of cercosporin (2), epicercosporin (epi-2), calphostin D (5d), phleichrome (6), and entphleichrome (ent-6) were achieved following the strategy outlined in Figure 19.49,51,52
Section 4: Biological Activity of Analogs from Total Synthesis
Perylenequinones exhibit light induced biological activity and are possible photodynamic therapeutic candidates. Photodynamic therapy involves several key components: a photosensitizer (drug), oxygen, and a light source.68 The mechanism of action involves the production of an excited photosensitizer species that transfers its energy to molecular oxygen to produce reactive oxygen species that are cytotoxic. Any tissues that have absorbed the photosensitizer and have been exposed to light undergo chemical damage.
When exposed to light, perylenequinones generate singlet oxygen from atmospheric oxygen with high quantum efficiency and are viable photosensizers for PDT.69 The most promising indication is cancer, and as discussed previously, light-induced activity has been seen with the natural products against several tumor lines. 70, 71, 72 In addition, antiviral72, 73 and immunotherapeutic 74 properties have also been reported. For perylenequinones to be furthered as drug candidates for PDT it was imperative that derivatives of the natural products be designed to be activated by longer wavelengths light, as longer wavelength light (650-800 nm) would penetrate further into tissue. As such, development of new classes of perylenequinones, with longer absorption wavelengths, greater stability, and better selectivity against cancer cells is an exciting area of research. Perylenequinones have an advantage over other photosensitizers as they also cause cancer cell apoptosis via a second mode. Specifically, perylenequinones selectively bind the C1 regulatory domain of protein kinase C (PKC).5,75,76 The various protein kinase C isoforms phosphorylate serine and threonine residues on proteins that are involved in tumor formation and progression. PKC over-expression has been shown to contribute to cell transformation and PKCs are considered tumor promoters that enhance multiple cellular signaling pathways.77,78 Since PKC has been recognized as a significant target for anticancer and antiviral drugs, considerable attention has been devoted to the development of potent and specific PKC inhibitors.77 Catalytic domain-acting PKC inhibitors, such as staurosporine (PKC, IC50 = 9 nM) and other indolocarbazoles, are highly potent but are also unselective inhibitors since they also bind to other protein kinases, which possess similar active sites.79 By targeting the unique regulatory domain of PKC, perylenequinones have been demonstrated to be both potent and specific PKC inhibitors (calphostin C (5c): PKC, IC50 = 0.05 – 0.46 mM; PKA, IC50 = > 100 mM; PPK, IC50 = > 100 mM; Cercosporin (2): PKC, IC50 = 0.6 – 1.3 mM; PKA, IC50 = > 500 mM; PPK, IC50 = > 180 mM).75,76 Due to their small molecular weight and specific binding to PKC, the perylenequinones are ideal PKC inhibitors. Until recently, however, evaluation of the perylenequinones as PKC inhibitors has been confined to derivatives of the natural products due to limitations of available synthetic methods.69,70,76,80 With the advent of new synthetic routes to novel perylenequinones, determination of the structural features necessary for PKC inhibition as well as generation of structures with improved photodynamic properties has begun.49,52
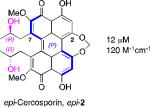
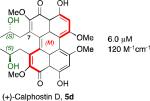
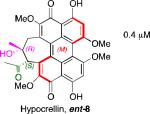
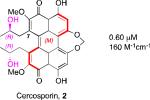
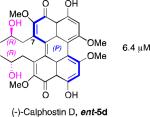
A systematic evaluation of perylenequinone architecture and PKC inhibition revealed several interesting trends.49,52 First, the helical stereochemistry was found to impact inhibition. Namely, all the (M)-perylenequinones were more potent than the corresponding (P)-isomers with the same C7,C7’-stereochemistry ((+)-5d vs (–)-ent-6, (+)-6 vs (–)-ent-5d, and 2 vs epi-2, Table 1) In some cases the effect was as much as a 20-fold greater potency for the-(M) over the (P)-isomers. For the hypocrellins, the greatest PKC inhibition was observed for the compound where the (M)-helicity predominated (ent-8). Particularly noteworthy is that the unnatural isomers of the calphostins (+)-5d and (+)-6 (M-helicity) had greater affinity for PKC than the natural products (–)-ent-5d and (–)-ent-6 (P-helicity). The stereochemistry of the C7,C7’-2-hydroxypropyl groups also had an impact on potency with the (R,R)-series showing marginally better (1.7-fold) activity than the (S,S)-array ((+)-5d vs (+)-6, (–)-ent-5d vs (–)-6).49,52
Table 1.
Perylenequinone PKC IC50 values.
| PKC IC50, ε670 | PKC IC50, ε670 | PKC IC50, ε670 | PKC IC50, ε670 | ||||
|---|---|---|---|---|---|---|---|
|
3.5 μM 225 M-1cm-1 |
|
12 μM 120 M-1cm-1 |
|
1.5 μM 360 M-1cm-1 |
|
4.0 μM 5180 M-1cm-1 |
|
6.0 μM 120 M-1cm-1 |
|
4.5 μM |
|
0.60 μM 160 M-1cm-1 |
|
3.5μM 1430 M-1cm-1 |
|
6.4 μM |
|
4.5 μM |
|
1.2 μM 480 M-1cm-1 |
|
0.40 μM 480 M-1cm-1 |
|
11 μM |
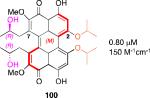
|
0.80 μM 150 M-1cm-1 |
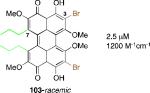
|
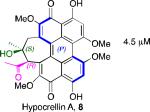
2.5 μM 1200 M-1cm-1 |
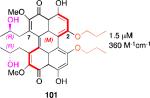
Models based on a cocrystal of PKCδ isozyme with a phorbol ester indicated that perylenequinones with larger hydrophobic groups at the C2,C2’-positions would fill a binding pocket in the C1 regulatory domain. To determine whether such substitution would correspond to greater activity, analogs of the most active (M,R,R)-stereochemical array corresponding to 6 were constructed. In accord with the modeling, the bisisopropyl ether 100 (0.8 μM) and bispropyl ether 101 (1.5 μM) were more potent than the parent compound (+)-6 (3.5 μM) (Table 1).49,52
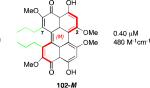
Prior reports from derivatives of the natural products suggested that acylation of the C7,C7’-hydroxypropyl group provided better potency for PKC (i.e. calphostin A, 5a, R1 = R2 = COPh, 0.25 μM vs calphostin D, 5d, R1 = R2 = H, 6.4 μM). To determine if this resulted from a simple increase in hydrophobicity due to removal of the two hydroxyl H-bond donors, our group synthesized the C7,C7’-n-propyl analogs. Notably, racemic 102 exhibited an IC50 of 1.2 μM. This finding indicated that hydrophobic C7,C7’-groups are indeed optimal for PKC binding. As previously stated, (M)-helicity provided superior inhibition, therefore enantiopure (M)-102 was also generated. Following the expected trends, this (M) derivative was more potent than the racemic compound and with an IC50 of 0.40 μM. In addition, (P)-102 was a poorer inhibitor (2.0 μM) supporting the prior conclusions regarding helicity. Furthermore, enantiopure (M)-102 is a 9-25 times stronger inhibitor than the nearest structural relatives, (+)-5d and (+)-6, and 16-28 times more potent than the corresponding natural products, (–)-ent-5d and (–)-ent-6.49,52
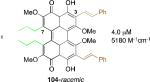
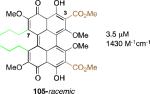
After finding that the stereogenic C7,C7’-substitution was not necessary for potency, examination of C3,C3’-analogs of architecturally simple 102 became the focus (Table 1). In doing so, perylenequinones (103-105) with methyl ester, bromo, and styryl groups as the C3,C3’-positions were designed to alter the chromophore to improve the absorption profile. Analogs 103-105 displayed increased absorption in the 600-800 nm range compared to the natural products. While better inhibitors from a photodynamic therapy point of view, 103 and 105 were found to be 2-4 times less potent in terms of PKC inhibition than the parent analog 102, which had the same architecture but was not substituted at the C3,C3’-positions.49,52
To determine whether the structural effects on PKC inhibition translate to anticancer activity, cancer cell lines were screened against the most potent (M)-isomers: phleichrome ((+)-6), cercosporin (2), and hypocrellin (ent-8). Finally, the racemic form of potent analog, 102, was included to assess the importance of C7,C7’-stereochemistry to cytotoxicity. All of the compounds exhibited micromolar or submicromolar CC50 values for growth inhibition (Table 2).49,52 In organisms, PKC binding is expected to localize the perylenequinones to cancer cells whereupon directed light irradiation would initiate the phototoxic response. Here, the cells were uniformly subjected to medium containing the perylenequinones. These tests would indicate which compounds possess the greatest phototoxicity and which cell lines were most susceptible to photodamage. Upon evaluation, differences were seen between the compounds and between different cell lines.
Table 2.
| Compound | NCI-H460 | PC3 | SK-MEL-5 | SN 12C | U251 | A2780 | MC F7 |
|---|---|---|---|---|---|---|---|
| Lung | Prostate | Skin | Colon | Brain | Ovary | Breast | |
| Phleichrome 6 | 0.52 | 0.71 | 0.86 | 0.66 | 0.49 | 0.28 | 1.1 |
| Cercosporin 2 | 0.13 | 0.26 | 0.15 | 0.17 | 0.15 | 0.12 | 0.20 |
| Hypocrellin ent-8 | 0.49 | 0.51 | 0.57 | 1.0 | 0.57 | 0.41 | 0.84 |
| racemic-102 | 0.51 | 0.71 | 0.55 | 0.58 | 0.50 | 0.22 | 0.84 |
All assays conducted with a 30 min exposure to a 32 W light at 5 cm.
Cercosporin (2) was the most active perylenequinone in every cell line tested, exhibiting CC50 values of 0.12-0.27 μM (Table 2). The remaining compounds showed similar activity among the cell lines although analog 102 was a superior inhibitor of A2780 (Table 2) and JHU-012 (Table 3) relative to the other perylenequinones. Given the selectivity of cercosporin (2) and simple analog 102 for the head and neck cancer line JHU-012, the compounds were further examined without light exposure in this line (Table 3)). Among the natural products, activity without light exposure versus activity with light exposure was highest with cercosporin (4.9-fold). Furthermore, this effect was even more marked for simple analog 102 (5.8-fold).49,52
Table 3.
Effect of light on perylenequinone cytotoxicity in the JHU-012 (head and neck cancer) cell line lines (see Table 1 for structures).
| Perylenequinone | Light, CC50/μM[a] | Dark, CC50/μM[b] | Photopotentiation |
|---|---|---|---|
| Phleichrome 6 | 0.81 | 2.8 | 3.5 |
| Cercosporin 2 | 0.16 | 0.78 | 4.9 |
| Hypocrellin ent-8 | 1.0 | 1.9 | 1.9 |
| racemic- 102 | 0.23 | 1.3 | 5.8 |
All assays conducted with a 30 min exposure to a 32 W light (570 nm) at 5 cm.
Assays conducted without 30 min light exposure.
Conclusion
The perylenequinones are a structural class characterized by a vinylogous quinone chromophore containing helical stereochemistry. Potentially useful light-activated biological activity has been identified for several of the natural products. New members of this class of compounds, with light-activated properties continue to be discovered. The natural products can be modified to improve biological profiles. In addition, flexible avenues are now available for the total synthesis of the natural product isomers and analogs, which are providing insight into how these molecules function. With the diverse structures available, the community is positioned to learn more about how these molecules interact with their biological targets including the different PKC isozymes.
Acknowledgments
We thank our co-workers Xiaolin Li and Sangeeta Dey who also contributed the experimental and intellectual advances described in this review. Funding for this research was provided by the National Science Foundation (CHE-0616885 and CHE-0911713) and the National Institutes of Health (CA-109164).
Biography
 Carol Mulrooney was born in Connecticut, USA in 1971. She obtained her Bachelor's degree in Chemistry from the University of Connecticut 1993 and was employed by Boehringer Ingelheim Pharmaceuticals, Inc. as an associate chemist until returning to graduate school. She received her doctorate in 2006 from the University of Pennsylvania in the lab of Prof. Marisa Kozlowski, developing asymmetric syntheses of natural product scaffolds. She subsequently joined the pharmaceutical company PolyMedix, Inc. as a medicinal chemist, and in 2008 moved to the Diversity Oriented Synthesis group at the Broad Institute, Cambridge, MA working in the design and production of structurally complex and diverse chemical libraries.
Carol Mulrooney was born in Connecticut, USA in 1971. She obtained her Bachelor's degree in Chemistry from the University of Connecticut 1993 and was employed by Boehringer Ingelheim Pharmaceuticals, Inc. as an associate chemist until returning to graduate school. She received her doctorate in 2006 from the University of Pennsylvania in the lab of Prof. Marisa Kozlowski, developing asymmetric syntheses of natural product scaffolds. She subsequently joined the pharmaceutical company PolyMedix, Inc. as a medicinal chemist, and in 2008 moved to the Diversity Oriented Synthesis group at the Broad Institute, Cambridge, MA working in the design and production of structurally complex and diverse chemical libraries.
 Erin O'Brien was born in Gander, NL, Canada in 1979. She obtained her Bachelor of Science degree with honors from Mount Allison University in 2001, followed by her doctorate in Chemistry in 2007 from the University of Pennsylvania. During her doctorate, Erin worked on asymmetric syntheses of perylenequinone natural products, culminating in the first total synthesis of Hypocrellin A. She subsequently joined Roche, Palo Alto LLC as a research scientist in the chemical synthesis group and moved to 2009 to Millennium: The Takeda Oncology Company working as a process chemist.
Erin O'Brien was born in Gander, NL, Canada in 1979. She obtained her Bachelor of Science degree with honors from Mount Allison University in 2001, followed by her doctorate in Chemistry in 2007 from the University of Pennsylvania. During her doctorate, Erin worked on asymmetric syntheses of perylenequinone natural products, culminating in the first total synthesis of Hypocrellin A. She subsequently joined Roche, Palo Alto LLC as a research scientist in the chemical synthesis group and moved to 2009 to Millennium: The Takeda Oncology Company working as a process chemist.
 Barbara Morgan obtained her Bachelor of Arts degree in Chemistry from Kenyon College and Doctorate degree in Organic Chemistry from the University of Pennsylvania working in the lab of Professor Marisa Kozlowski. She subsequently moved to the Broad Institute of Harvard and MIT where she worked on the identification of small molecule probes for the study of genes and biochemical pathways in health and disease. She currently serves as an Investment Associate, supporting North Coast Angel Fund's investment process and ventures. Prior to joining NCAF, Barbara was a Science Advisor in the Intellectual Property Transactions & Strategies group at Goodwin Procter, LLP, in Boston, MA.
Barbara Morgan obtained her Bachelor of Arts degree in Chemistry from Kenyon College and Doctorate degree in Organic Chemistry from the University of Pennsylvania working in the lab of Professor Marisa Kozlowski. She subsequently moved to the Broad Institute of Harvard and MIT where she worked on the identification of small molecule probes for the study of genes and biochemical pathways in health and disease. She currently serves as an Investment Associate, supporting North Coast Angel Fund's investment process and ventures. Prior to joining NCAF, Barbara was a Science Advisor in the Intellectual Property Transactions & Strategies group at Goodwin Procter, LLP, in Boston, MA.
 Marisa Kozlowski received an AB in Chemistry from Cornell University in 1989 and a PhD from the University of California at Berkeley in 1994. After a NSF postdoctoral fellowship with David A. Evans at Harvard University, she joined the faculty at the University of Pennsylvania in 1997 and is currently Professor of Chemistry. The Kozlowski group research focuses on the design of asymmetric catalysts and transformations. She has also coauthored the book “Fundamentals of Asymmetric Catalysis” with Patrick Walsh.
Marisa Kozlowski received an AB in Chemistry from Cornell University in 1989 and a PhD from the University of California at Berkeley in 1994. After a NSF postdoctoral fellowship with David A. Evans at Harvard University, she joined the faculty at the University of Pennsylvania in 1997 and is currently Professor of Chemistry. The Kozlowski group research focuses on the design of asymmetric catalysts and transformations. She has also coauthored the book “Fundamentals of Asymmetric Catalysis” with Patrick Walsh.
References
- i.Reviews, see: Weiss U, Merlini L, Nasini G. Prog. Chem. Org. Nat. Prod. 1987;52:1–71. doi: 10.1007/978-3-7091-8906-1_1.Bringmann G, Günther C, Ochse M, Schupp O, Tasler S. Prog. Chem. Org. Nat. Prod. 2001;82:1–249. doi: 10.1007/978-3-7091-6227-9_1.
- ii.Anderson JM, Murray J. Chem. and Ind. 1956:376–376. [Google Scholar]
- iii.Reviews, see Falk H. Ang. Chem. Int. Ed. 1999;39:3116–3136.Waser M, Falk H. Curr. Org. Chem. 2011;15:3894–3907.
- 4.Lousberg RJJ, Weiss U, Salemink CA, Arnone A, Merlini L, Nasini G. Chem. Commun. 1971:1463–1464. [Google Scholar]
- 5.a Kobayashi E, Ando K, Nakano H, Iida T, Ohno H, Morimoto M, Tamaoki T. J. Antibiotics. 1989;42:1470–1474. doi: 10.7164/antibiotics.42.1470. [DOI] [PubMed] [Google Scholar]; b Iida T, Kobayashi E, Yoshida M, Sano H. J. Antibiotics. 1989;42:1475–1481. doi: 10.7164/antibiotics.42.1475. [DOI] [PubMed] [Google Scholar]
- 6.Yoshihara T, Shimanuki T, Araki T, Saramura S. Agr. Biol. Chem. 1975;39:1683–1684. [Google Scholar]
- 7.Weiss U, Ziffer H, Batterham TJ, Blumer M, Hackeng WHL, Copier H, Salemink CA. Can. J. Microbiol. 1965;11:57–66. doi: 10.1139/m65-008. [DOI] [PubMed] [Google Scholar]
- 8.Hypocrellin (ent-1.8): Chen WS, Chen YT, Wang XY, Friedrichs E, Puff H, Breitmaier E. Liebigs Ann. Chem. 1981:1880–1885.
- 9.Hypocrellin A (8) and Hypocrellin B (10): Kishi T, Tahara S, Tsuda M, Tanaka C, Takahashi S. Planta Med. 1991;57:376–379. doi: 10.1055/s-2006-960121. (shiraiachrome A is called hypocrellin B here) Wu H, Lao XF, Wang QW, Lu RR. J. Nat. Prod. 1989;52:948–951. (hypocrellin A is called shiraiachrome B here)
- 10.Yamazaki S, Ogawa T. Agr. Biol. Chem. 1972;36:1707–1718. [Google Scholar]
- 11.Yamazaki S, Okube A, Akiyama Y, Fuwa K. Agric. Biol. Chem. 1975;39:287–288. [Google Scholar]
- 12.Arnone A, Camarda L, Nasini G. J. Chem. Soc. Perkin Trans. 1. 1985:1387–1392. [Google Scholar]
- 13.Nasini G, Merlini L, Andreetti GD, Bocelli G, Sgarabotto P. Tetrahedron. 1982;38:2787–2796. [Google Scholar]
- 14.Fang L-Z, Qing C, Shao H-J, Yang Y-D, Dong Z-J, Wang F, Zhao W, Yang W-Q, Liu J-K. J. Antibiot. 2006;59:351–354. doi: 10.1038/ja.2006.49. [DOI] [PubMed] [Google Scholar]
- 15.Diwu Z, Lown JW. Photosensitization with anticancer agents 16. The photo-oxidation of hypocrellin A. A mechanism study using 18O labeling. J. Photochem. Photobiol. B: Biol. 1993;18:145–154. doi: 10.1016/1011-1344(93)80056-f. [DOI] [PubMed] [Google Scholar]
- 16.Liu WZ, Shen YX, Liu XF, Chen YT, Xie JL. Chinese Chemical Letters. 2001;12:431–432. [Google Scholar]
- 17.a Liu WZ. M.D. thesis. Yunnan University; 2000. Fermentation and Structure Determination of Hypomycin B. [Google Scholar]; b Liu WZ, Chen YT, Xie JL. J. Yunnan Univ., Nat. Sci. Ed. 2000;22:389–391. [Google Scholar]; c Liu WZ, Ma LY, Li C, Chen YT, Xie JL. Acta Pharmacol. Sin. 2001;36:313–314. [PubMed] [Google Scholar]
- 18.Choudhury PK, Das K, Datta A, Liu WZ, Zhang H-Y, Petrich JW. J. Photochem and Photobiol. A: Chemistry. 2002;154:107–116. [Google Scholar]
- 19.Ayers S, Zink DL, Mohn K, Powell JS, Brown CM, Murphy T, Brand R, Pretorius S, Stevenson D, Thompson D, Singh SB. J. Nat. Prod. 2007;70:425–427. doi: 10.1021/np0604937. [DOI] [PubMed] [Google Scholar]
- 20.For a review: Miskovsky P. Curr. Drug Targets. 2002;3:55–84. doi: 10.2174/1389450023348091.
- 21.De Riccardis F, Iorizzi M, Minale L, Riccio R, Richer de Forges B, Debitus C. J. Org. Chem. 1991;56:6781–6787. [Google Scholar]
- 22.Hypericin has been discovered in the fossil record from the Upper Jurassic to the Middle Triassic: Wolkenstein K, Gross JH, Falk H, Schöler HF. Proc. R. Soc. B. 2006;273:451–456. doi: 10.1098/rspb.2005.3358.
- 23.Kemami Wangun HC, Wood A, Fiorilla C, Reed JK, McCarthy PJ, Wright AE. J. Nat. Prod. 2010;73:712–715. doi: 10.1021/np900526y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.For a review of hypocrellins in photodynamic therapy: Diwu Z, Lown JW. Photochemistry and Photobiology. 1990;52:609–616. doi: 10.1111/j.1751-1097.1990.tb01807.x.
- 25.For a review of the use of sonodynamic therapy to treat cancer: Umemura S, Kawabata K, Sasaki K;, Yumita N, Umemura K, Nishigaki R. Ultrasonics Sonochemistry. 1996;3:S187–S191.
- 26.a Hu YZ, An JY, Jiang LJ. J. Photochem. Photobiol. B: Biol. 1993;17:195–201. [Google Scholar]; b Hu YZ, Jiang LJ. J. Photochem. Photobiol. B: Biol. 1996;33:51–59. [Google Scholar]
- 27.a Xia WL, Zhang MH, Jiang LJ. Youji Huaxue. 1992;12:618–623. [Google Scholar]; b Diwu ZJ, Zhang CL, Lown JW. Anticancer Drug Des. 1993;8:129–143. [PubMed] [Google Scholar]
- 28.a Jiang Y, An JY, Liang LJ. Chin. Sci. Bull. 1993;28:797–800. [Google Scholar]; B Diwu ZJ, Zhang CL, Lown JW. J. Photochem. Photobiol. A: Chem. 1992;66:99–112. [Google Scholar]
- 29.Hu YZ, An JY, Jiang LJ. J. Photochem. Photobiol. B: Biol. 1994;22:219–227. doi: 10.1016/1011-1344(93)06973-7. [DOI] [PubMed] [Google Scholar]
- 30.For a Chinese language review: Liu Y, Wang X, Zhang B. Huaxue Jinzhan. 2008;20:1345–1353.
- 31.a Zhang W-G, Weng M, Pang S-Z, Zhang M-H, Yang H-Y, Zhao H-X, Zhang Z-Y. J. Photochem. And Photobiol. B: Biology. 1998;44:21–28. doi: 10.1016/S1011-1344(98)00101-8. [DOI] [PubMed] [Google Scholar]; b Xu S, Chen S, Zhang M, Shen T, Xhao Y, Liu Z, Wu Y. Biochimica et Biophysica Acta. 2001;1537:222–232. doi: 10.1016/s0925-4439(01)00074-6. [DOI] [PubMed] [Google Scholar]
- 32.Tang Y-J, Liu H-Y, An J-Y, Han R. Photochem. And Photobiol. 2001;74:201–205. doi: 10.1562/0031-8655(2001)074<0201:SCAPAO>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 33.Xu S, Chen S, Zhang M, Shan T. J. Photochem. And Photobiol. B: Biology. 2003;72:61–67. doi: 10.1016/j.jphotobiol.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 34.Lee H-Y, Zhou Z-X, Chen S, Zhang M-H, Shen T. Dyes and Pigments. 2006;68:1–10. [Google Scholar]
- 35.Bibbon PT, Babu MS, Santhoshkumar TR, Karunagaran D, Selvam GS, Brown K, Woo T, Sanjay S, Selvaraj N, Ramachandran M. J. Photochem. And Photobiol. B: Biology. 2009;94:38–44. doi: 10.1016/j.jphotobiol.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 36.El-Sikhry HE, Miller GG, Madiyalakan MR, Seubert JM. Invest. New Drugs. 2011;29:1328–1336. doi: 10.1007/s10637-010-9495-2. [DOI] [PubMed] [Google Scholar]
- 37.Quest Pharmatech URL: www.questpharmatech.com/sl017.htm.
- 38.Meng Y, Zou C, Madiyalakan R, Woo T, Huang M, Yang X, Swanson E, Chen J, Xing JZ. Nanomedicine. 2010;5:1559–1569. doi: 10.2217/nnm.10.91. [DOI] [PubMed] [Google Scholar]
- 39.Liu X, Xie J, Zhang L, Chen H, Gu Y, Zhao J. J. Photochem. Photobiol. B: Biology. 2009;94:171–178. doi: 10.1016/j.jphotobiol.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Y, Song L, Xie J, Qiu H, Gu Y, Zhao J. Photochem. Photobiol. 2010;86:667–672. doi: 10.1111/j.1751-1097.2010.00711.x. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Y, Xie J, Zhang L, Li C, Chen H, Gu Y, Zhao J. Photochem. Photobiol. Sci. 2009;8:1676–1682. doi: 10.1039/b9pp00046a. and references therein. [DOI] [PubMed] [Google Scholar]
- 42.Falk H, Schmitzberger W. Monatshefte für Chemie. 1993;124:77–81. [Google Scholar]
- 43.Hudson JB, Delaey E, de Witte PA. Photochem. Photobiol. 1999;70:820–822. [PubMed] [Google Scholar]
- 44.Taguchi T, Okamoto S, Itoh T, Ebizuka Y, Ochi K, Ichinose K. Tetrahedron Lett. 2008;49:1208–1211. [Google Scholar]
- 45.Chao C, Zhang P. Tetrahedron Lett. 1988;29:225–226. [Google Scholar]
- 46.a Diwu Z, Lown JW. Tetrahedron. 1992;48:45–54. [Google Scholar]; b Liu J, Diwu Z, Lown JW. Tetrahedron. 1993;49:10785–10792. [Google Scholar]
- 47.Mulrooney CA, Li X, DiVirgilio ES, Kozlowski MC. J. Am. Chem. Soc. 2003;125:6856–6857. doi: 10.1021/ja027745k. [DOI] [PubMed] [Google Scholar]
- 48.O'Brien EM, Morgan BJ, Kozlowski MC. Angew. Chem. Int. Ed. 2008;47:6877–6880. doi: 10.1002/anie.200800734. [DOI] [PubMed] [Google Scholar]
- 49.Morgan BJ, Dey S, Johnson SW, Kozlowski MC. J. Am. Chem. Soc. 2009;131:9413–9425. doi: 10.1021/ja902324j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mulrooney CA, Morgan BJ, Li X, Kozlowski MC. J. Org. Chem. 2010;75:16–29. doi: 10.1021/jo9013832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morgan BJ, Mulrooney CA, O'Brien EM, Kozlowski MC. J. Org. Chem. 2010;75:30–43. doi: 10.1021/jo901384h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morgan BJ, Mulrooney CA, Kozlowski MC. J. Org. Chem. 2010;75:44–56. doi: 10.1021/jo9013854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.O'Brien EM, Morgan BJ, Carroll PJ, Kozlowski MC. J. Org. Chem. 2010;75:57–68. doi: 10.1021/jo901386d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.a Weisgraber KH, Weiss U. J. Chem. Soc. Perkin Trans. 1. 1972:83–88. doi: 10.1039/p19720000083. [DOI] [PubMed] [Google Scholar]; b Weiss U, Fales HM, Weisgraber KH. Liebigs Ann. Chem. 1979:914–919. [Google Scholar]
- 55.Dallacker F, Leidig H. Chem. Ber. 1979;112:2672–2679. [Google Scholar]
- 56.Broka CA. Tetrahedron Lett. 1991;32:859–862. [Google Scholar]
- 57.Hauser FM, Sengupta D, Corlett SA. J. Org. Chem. 1994;59:1967–1969. [Google Scholar]
- 58.a Coleman RS, Grant EB. J. Am. Chem. Soc. 1994;116:8795–8796. [Google Scholar]; b Coleman RS, Grant EB. J. Am. Chem. Soc. 1995;117:10889–10904. [Google Scholar]
- 59.a Merlic CA, Aldrich CC, Albaneze-Walker J, Saghatelian A. J. Am. Chem. Soc. 2000;122:3224–3225. doi: 10.1021/ja994313+. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Merlic CA, Aldrich CC, Albaneze-Walker J, Saghatelian A, Mammen J. J. Org. Chem. 2001;66:1297–1309. doi: 10.1021/jo0014663. [DOI] [PubMed] [Google Scholar]
- 60.Zinke A, Hirsch W, Brozek E. Monatsh. Chem. 1929;51:205–220. [Google Scholar]
- 61.Calderbank A, Johnson AW, Todd AR. J. Chem. Soc. 1954:1285–1289. [Google Scholar]
- 62.The proposed structure of 51 is the keto-enol tautomer of the mold perylenequinones seen in Figures 2 and 4, with exception of hypomycin B 13.
- 63.For selected examples see: Miyahara Y. J. Org. Chem. 2006;71:6516–6521. doi: 10.1021/jo060889n.Davies S, Sheppard RL, Smith AD, Thomson JE. Chem. Commun. 2005:3802–3804. doi: 10.1039/b506780b.Piers E, Skupinska KA, Wallace DJ. Synlett. 1999;12:1867–1870.Sasai H, Suzuki T, Arai S, Arai T, Shibasaki M. J. Am. Chem. Soc. 1992;114:4418–4420.
- 64.There is one report of an acyclic intramolecular aldol to yield the 7-membered ring via a reductive aldol of the enone aldehyde: Baik TG, Luis AL, Wang LC, Krische MJ. J. Am. Chem. Soc. 2001;123:5112–5113. doi: 10.1021/ja0040971.
- 65.MM2 Calculations: Still WC. MacroModel. Columbia University; V4.0, V5.0, V6.0, V6.5.
- 66.a Gaudemer M, Bellassoued M. Tetrahedron Lett. 1989;30:2779–2782. [Google Scholar]; b Masamune S, Ellingboe J, Choy W. J. Am. Chem. Soc. 1982;104:5526–5528. [Google Scholar]
- 67.Hewgley JB, Stahl SS, Kozlowski MC. J. Am. Chem. Soc. 2008;130:12232–12233. doi: 10.1021/ja804570b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.a McCaughan JS., Jr. Drugs & Aging. 1999;15:49–68. doi: 10.2165/00002512-199915010-00005. [DOI] [PubMed] [Google Scholar]; b Detty MR. Expert Opinion on Therapeutic Patents. 2001;11:1849–1860. [Google Scholar]; c Lane N. Sci. Am. 2003;288:38–41. 43–45. doi: 10.1038/scientificamerican0103-38. [DOI] [PubMed] [Google Scholar]; d Dolmans DEJGJ, Fukumura D, Jain RK. Nature Reviews Cancer. 2003;3:380–387. doi: 10.1038/nrc1071. [DOI] [PubMed] [Google Scholar]; e Triesscheijn M, Baas P, Schellens JHM, Stewart FA. Oncologist. 2006;11:1034–1044. doi: 10.1634/theoncologist.11-9-1034. [DOI] [PubMed] [Google Scholar]; f Kessel D. J. Porphyrins Phthalocyanines. 2008;12:877–880. doi: 10.1142/s1088424608000273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lown JW. Can. J. Chem. 1997;75:99–119. [Google Scholar]
- 70.Paul BT, Babu MS, Santhoshkumar TR, Karunagaran D, Selvam GS, Brown K, Woo T, Sharma S, Naicker S, Murugesan R. J. Photochem. Photobiol. B: Biol. 2009;94:38–44. doi: 10.1016/j.jphotobiol.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 71.a Chiarini A, Whitfield JF, Pacchiana R, Armato U, Dal Pra I. Biochimica et Biophysica Acta. 2008;1783:1642–1653. doi: 10.1016/j.bbamcr.2008.03.014. [DOI] [PubMed] [Google Scholar]; b Olivo M, Ali-Seyed M. Int. J. Oncology. 2007;30:537–548. [PubMed] [Google Scholar]; c Guo B, Hembruff SL, Villeneuve DJ, Kirwan AF, Parissenti AM. Breast Canc. Res. Treat. 2003;82:125–141. doi: 10.1023/B:BREA.0000003969.21267.81. [DOI] [PubMed] [Google Scholar]; d Ma L, Tai H, Li C, Zhang Y, Wang Z-H, Ji W-Z. World J. Gastroenterology. 2003;9:485–490. doi: 10.3748/wjg.v9.i3.485. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Ali SM, Olivo M, Yuen GY, Chee SK. Int. J. Oncology. 2001;19:633–643. doi: 10.3892/ijo.19.3.633. [DOI] [PubMed] [Google Scholar]; f Dubauskas Z, Beck TP, Chumura SJ, Kovar DA, Kadkhodaian MM, Shrivastav M, Chung T, Stadler WM, Rinker-Schaeffer CW. Clinical Cancer Res. 1998;4:2391–2398. [PubMed] [Google Scholar]; g Vandenbogaerde AL, Delaey WS, Vantieghem AM, Himpens BE, Merlevede WS, De Witte PA. Photochem. Photobiol. 1998;67:119–125. [PubMed] [Google Scholar]; h Zhang J, Cao E-H, Li J-F, Zhang T-C, Ma W-J. J. Photochem. Photobiol. B. 1998;43:106–111. doi: 10.1016/S1011-1344(98)00092-X. [DOI] [PubMed] [Google Scholar]; i Pollack IF, Kawecki S. J. Neuro-oncology. 1997;31:255–66. doi: 10.1023/a:1005729626354. [DOI] [PubMed] [Google Scholar]; j Diwu Z. Photochem. Photobiol. 1995;61:529–539. doi: 10.1111/j.1751-1097.1995.tb09903.x. [DOI] [PubMed] [Google Scholar]
- 72.Wang HK, Xie JX, Chang JJ, Hwang KM, Liu SY, Ballas LM, Jiang JB, Lee KH. J. Med. Chem. 1992;35:2717–2721. doi: 10.1021/jm00093a001. [DOI] [PubMed] [Google Scholar]
- 73.a Hudson JB, Imperial V, Haugland RP, Diwu Z. Photochem. Photobiol. 1997;65:352–354. doi: 10.1111/j.1751-1097.1997.tb08570.x. [DOI] [PubMed] [Google Scholar]; b Hirayama J, Ikebuchi K, Abe H, Kwon K-W, Ohnishi Y, Horiuchi M, Shinagawa M, Ikuta K, Kamo N, Sekiguchi S. Photochem. Photobiol. 1997;66:697–700. doi: 10.1111/j.1751-1097.1997.tb03209.x. [DOI] [PubMed] [Google Scholar]
- 74.Leveugle B. 2002 U. S. Patent 2001-264677.
- 75.Bruns RF, Miller FD, Merriman RL, Howbert JJ, Heath WF, Kobayashi E, Takahashi I, Tamaoki T, Nakano H. Biochem. Biophys. Res. Commun. 1991;176:288–293. doi: 10.1016/0006-291x(91)90922-t. [DOI] [PubMed] [Google Scholar]
- 76.Diwu Z, Zimmermann J, Meyer T, Lown JW. Biochem. Pharmacol. 1994;47:373–385. doi: 10.1016/0006-2952(94)90029-9. [DOI] [PubMed] [Google Scholar]
- 77.a Mackay HJ, Twelves CJ. Nature Rev. Cancer. 2007;7:554–562. doi: 10.1038/nrc2168. [DOI] [PubMed] [Google Scholar]; b Gonzalez-Guerrico AM, Meshki J, Xiao L, Benavides F, Conti CJ, Kazanietz MG. J. Biochem. Mol. Biol. 2005;38:639–645. doi: 10.5483/bmbrep.2005.38.6.639. [DOI] [PubMed] [Google Scholar]; c da Rocha AB, Mans DRA, Regner A, Schwartsmann G. Oncologist. 2002;7:17–33. doi: 10.1634/theoncologist.7-1-17. [DOI] [PubMed] [Google Scholar]
- 78.Reviews on Protein Kinase C: Nishizuka Y. Nature. 1984;308:693–698. doi: 10.1038/308693a0.Nishizuka Y. Science. 1986;233:305–312. doi: 10.1126/science.3014651.
- 79.Tamaoki T, Nomoto H, Takahashi I, Kato Y, Morimoto M, Tomita F. Biochem. Biophys. Res. Commun. 1986;135:397–402. doi: 10.1016/0006-291x(86)90008-2. Reviews on Indolocarbazoles: Nakano H, Omura S. J. Antibiotics. 2009;62:17–26. doi: 10.1038/ja.2008.4.Prudhomme M. Curr. Pharm. Design. 1997;3:265–290.
- 80.Kishi T, Saito H, Sano H, Takahashi I, Tamaoki T. Eur. Pat. Appl. EP-390181. 1990