

Abstract
Structural optimizations of the prior lead 1a led to the discovery of a series of N-aryl-6-methoxy-1,2,3,4-tetrahydroquinoline derivatives as a novel class of tubulin polymerization inhibitors targeted at the colchicine binding site. The most active compound 6d showed extremely high cytotoxicity against a human tumor cell line panel (A549, KB, KBvin, and DU145) with GI50 values ranging from 1.5 to 1.7 nM, significantly more potent than paclitaxel, especially against the drug-resistant KBvin cell line, in the same assays. Analogues 5f, 6b, 6c, and 6e were also quite potent, with a GI50 range of 0.011–0.19 μM. In further studies, active compounds 6b–6e and 5f significantly inhibited tubulin assembly, with IC50 values of 0.92 to 1.0 μM and strongly inhibited colchicine binding to tubulin, with inhibition rates of 75–99% (at 5 μM), comparable with or more potent than combretastatin A-4 (IC50 0.96 μM). Current studies included design, synthesis, and biological evaluations of 24 new compounds (series 3–6). Related SAR analysis, molecular modeling, and evaluation of essential drug-like properties, i.e. water solubility, log P, and in vitro metabolic stability, were also performed.
Keywords: N-aryl-6-methoxy-1,2,3,4-tetrahydroquinolines; cytotoxicity; tubulin polymerization inhibitors; colchicine binding site
1.Introduction
Microtubules, formed by a dynamic polymerization and depolymerization of α- and β-tubulin heterodimers, play important roles in cellular activities [1]. For many years, certain drugs that bind to the taxol or vinca site on tubulin, such as paclitaxel and vinblastine, have been widely used in the clinic to treat cancers [2]. However, their narrow therapeutic windows and the emergence of drug resistance have encouraged continued efforts to discover safer and more effective agents capable of treating resistant cancer phenotypes. Another natural product colchicine (Figure 1) binds to a unique site on tubulin, distinct from the above two binding sites, and_can effectively inhibit tubulin assembly. Even though colchicine is not used clinically because of its high toxicity, other small molecules bind at the colchicine site, e.g., N-deacetyl-N-(2-mercaptoacetyl)-colchicine (DAMA-colchicine) and combretastatin A-4 (CA-4), and some have exhibited high potency, lower toxicity, and greater efficacy against cancers resistant to existing tubulin-targeting drugs [3]. More attractively, some of these compounds exhibit a vascular disrupting effect on vascular endothelial cells and have entered into clinical development as vascular disrupting agents (VDAs) for cancer treatment. Examples of this novel class of anticancer agents are CA-4P and ZD6126 (Figure 1) [4,5].
Fig. 1.
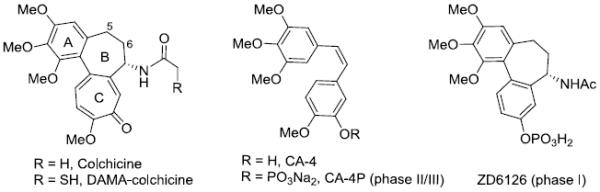
Colchicine, DAMA-colchicine, and clinical trial candidates targeted at the colchicine site of tubulin.
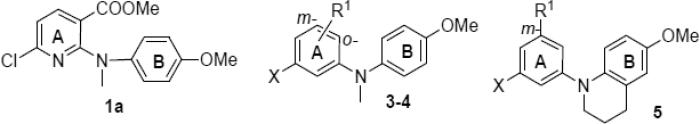
In our prior studies on new anticancer agents [6,7], we discovered methyl 6-chloro-2-(N-(4-methoxyphenyl)-N-methyl)aminonicotinate (1a, Figure 2) as a lead compound with potent cytotoxic activity (GI50 0.20–0.26 μM) against A549, KB, KBvin, and DU145 human tumor cell lines. Its biological target was identified as tubulin, specifically the colchicine site. Previous related SAR studies indicated that a para-methoxyphenyl moiety (B-ring) is an essential pharmacophore, that a tertiary amine linker can provide a feasible binding conformation, and that the substituted pyridine ring (A-ring) can be modified to improve potency. Based on these known SAR findings, we pursued optimization of lead 1a, following the strategies shown in Figure 2, to identify additional tubulin inhibitors with new scaffolds and high potency. Because the o-chloropyridine moiety could potentially lead to metabolic toxicity resulting from in vivo nucleophilic replacement [8], we initially replaced the pyridine ring (A-ring) of 1a with a benzene ring and also changed the identities and positions of substituents R1 and X on the A-ring (series 3-4 compounds) to investigate how these modifications impacted inhibitory activity against tumor cell growth. Previously, we also postulated that the degree of torsional angle between the two aryl rings (A- and B-rings) might be crucial for activity; therefore, our next modification focused on the N-linkage. By applying a conformation restriction strategy [9], we connected a propyl group on the N-linker to the neighboring benzene ring (B-ring) to form a six-membered fused tetrahydropyridine ring (series 5 compounds). By constraining the flexible conformation, we could better explore feasible binding conformations and possible ligand-target interactions. We next postulated that introducing a fused aromatic ring onto the A-ring might make the molecular binding conformation of new series 6 compounds more similar to that of colchicine and enhance affinity with the binding site on tubulin, leading to better antitumor activity. All newly synthesized compounds were evaluated by cellular screening against a human tumor cell line (HTCL) panel, and selected potent compounds were also assayed for tubulin inhibition. Molecular modeling was used to illustrate the interaction between tubulin and the new ligand(s) as well as differences among ligands. Furthermore, certain essential parameters related to drug-like properties, such as water solubility, log P, and metabolic stability, were determined for highly potent compounds. Chemical synthesis, biological evaluation, molecular modeling, and SAR analysis are presented in this paper.
Fig. 2.
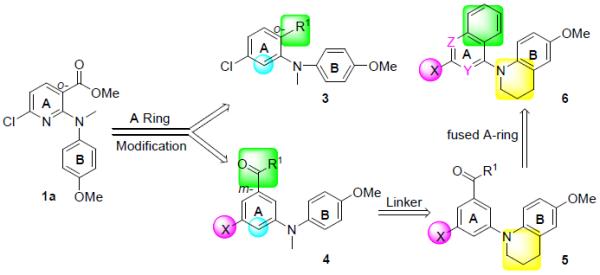
Modification strategies and new target compounds 3–6 series
2. Chemistry
The syntheses of target compounds 3–6 are outlined in Schemes 1 and 2. Intermediate 2a was prepared by Ullmann condensation reaction [10] of commercially available 4-methoxyaniline and 2,4-dichlorobenzoic acid in the presence of Cu/Cu2O in 2-ethoxyethanol, following esterification with DMF-DMA (two equivalents) in toluene at reflux for 24 h. Intermediates 2b-d were obtained by a Buchwald-Hartwig coupling reaction between 4-methoxyaniline and methyl 5-substituted-3-bromo (or iodo) benzoate in the presence of catalyst Pd(OAc)2 and Cs2CO3 either with X-phos [11] under microwave (mw) irradiation at 120–150 °C (Method A for 2b and 2c) or with BINAP and traditional heating in toluene at reflux under nitrogen protection (Method B for 2d). Subsequently, secondary amines 2a-d were treated with methyl iodide in the presence of sodium hydride to afford corresponding tertiary diarylamines 3a and 4a-c. When using LiAlH4 in THF at 0 °C for 1 h, the ester group of 3a was reduced to a hydroxymethyl moiety, affording 3b in nearly quantitative yield. Compound 3b was further methylated with methyl iodide to give ether 3c. Alternatively, the ester group in 3a and 4a-c was converted to an N-methylcarbamoyl group by treatment with methylamine in MeOH under mw irradiation at 100–120 °C to produce corresponding compounds 3d and 4d-f, respectively, in high yields (79–89%). Methylation of 4f with methyl iodide yielded compound 4g with a N,N-dimethylcarbamoyl group on the A-ring. Ester compound 4c was hydrolyzed under basic conditions to produce carboxylic acid compound 4h. Compound 4h was then treated with 1-hydroxybenzotriazole (HOBt) in the presence of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) hydrochloride salt, followed by reaction with cyclopropylamine or cyclopentylamine to produce corresponding N-cyclopropylamide and N-cyclopentylamide compounds 4i and 4j, respectively [12]. By using Buchwald-Hartwig coupling reactions and the same conditions, compounds 5a-5b and 6a-6b were synthesized from intermediate 6-methoxy-1,2,3,4-tetrahydroquinoline (7) [13] and an aryl halide, such as phenyl, naphthyl, and quinolyl halides, in 68–75% yields. Similarly to series 3 and 4, the ester group in 5a and 5b was converted into an N-alkyl carbamoyl or carboxy group to provide corresponding compounds 5c-f by using the same methods described above. Differently from 6a and 6b, the preparation of 6c and 6d involved coupling 7 with a 2-substituted 4-haloquinazoline under EtOH reflux in the presence of NaHCO3, resulting in 79–87% yields. Using the same method for preparing 5c, compound 6d was converted to 6e. All new series 3-6 compounds were identified by 1H NMR and MS spectroscopic data, with purity determined by HPLC.
Scheme 1.
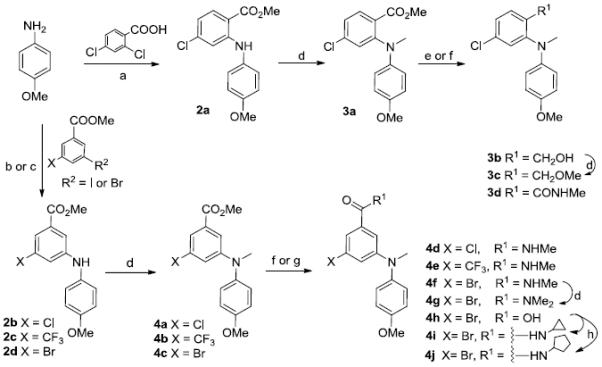
a) i. K2CO3, Cu, Cu2O, 2-ethoxyethanol, N2, 130 °C, 24 h, 67%; ii. DMF-DMA in toluene, reflux, 2 h, 85%; b) i. Cs2CO3, Pd(OAc)2, X-Phos, Celite, toluene/t-BuOH, N2, mw, 120-150 °C, 30-60 min, 74% for 2b and 2c; c) Cs2CO3, Pd(OAc)2, BINAP, toluene, N2, reflux 12 h, 64% for 2d; d) MeI/NaH (60% oil suspension), 0 °C, 1 h; e) LiAlH4/THF, 0 °C, 1 h, 97%; f) 30% NH2Me/MeOH, 100-120 °C, mw, 1-2 h for 3d, 4d-f; g) MeOH/THF, 3N NaOH aq, reflux, 5 h, 90% for 4h; h) EDCI, HOBt, cyclopropylamine or cyclopentylamine, CH2Cl2, rt, 24 h for 4i and 4j.
Scheme 2.
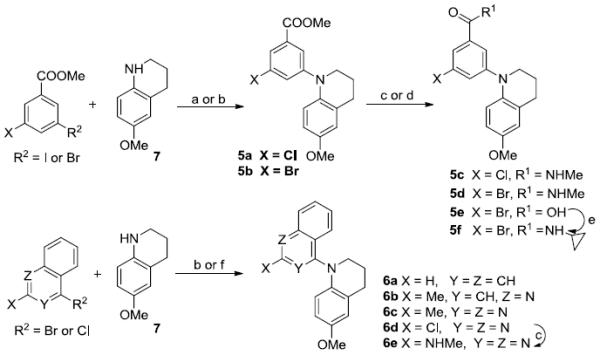
a) Cs2CO3, Pd(OAc)2, X-Phos, Celite, N2, mw, 120 °C, 30 min, 77% for 5a; b) Cs2CO3, Pd(OAc)2, BINAP, toluene, N2, reflux, 12 h, 68-75% for 5b and 6a-6b; c) 30% NH2Me/MeOH, 100 °C, mw, 1 h for 5c, 5d, and 6e; d) MeOH/THF, 3 N NaOH aq, reflux, 5 h, 82% for 5e; e) EDCI, HOBt, cyclopropylamine, CH2Cl2, rt, 24 h, 90%; f) NaHCO3/EtOH, reflux, 3 h for 6c and 6d.
3. Results and Discussion
3.1 Evaluation of Cytotoxicity and Tubulin Inhibition Activity in Vitro
The 24 newly synthesized tertiary arylamines (series 3–6) were evaluated in cellular cytotoxicity assays against a HTCL panel, including A549 (lung carcinoma), KB (epidermoid carcinoma of the mouth), KBvin, a P-gp-expressing multidrug-resistant cell line (vincristine-resistant KB)[14, 15], and DU145 (prostate cancer) with paclitaxel as a reference compound. The in vitro anticancer activity (GI50) was determined using the established sulforhodamine B (SRB) method [16]. The cytotoxicity data of all new compounds in HTCL assays are listed in Tables 1 and 2. Subsequently, selected compounds with high potency in cellular assays were further tested in tubulin assays to determine biological target and binding site.
Table 1.
Cytotoxicity of series 3–5 compounds against human tumor cell lines (HTCL)
| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| X | R1 | GI50 (μM)a |
||||
| A549 | KB | KBvin | DU145 | |||
| 1a | 0.23±0.01 | 0.26±0.04 | 0.20±0.03 | 0.21±0.04 | ||
| 3a | Cl | o-COOMe | 3.36±0.49 | 1.98±0.37 | 1.89±0.17 | 1.68±0.04 |
| 3b | Cl | o-CH2OH | 21.7±2.53 | 19.5±1.33 | 18.1±2.24 | 16.0±2.73 |
| 3c | Cl | o-CH2OMe | 4.53±0.84 | 5.24±0.42 | 2.85±0.62 | 3.13±0.35 |
| 3d | Cl | o-CONHMe | 43.2±4.81 | 188±33.8 | 61.0±5.13 | 98.9±4.22 |
| 4a | Cl | m-COOMe | 1.60±0.36 | 1.61±0.08 | 1.57±0.10 | 1.46±0.16 |
| 4b | CF3 | m-COOMe | 15.1±2.6 | 13.5±1.21 | 13.1±1.41 | 11.5±1.71 |
| 4c | Br | m-COOMe | 1.24±0.34 | 1.36±0.17 | 1.20±0.39 | 0.97±0.08 |
| 4d | Cl | m-CONHMe | 0.27±0.01 | 0.30±0.04 | 0.23±0.04 | 0.23±0.03 |
| 4e | CF3 | m-CONHMe | 14.7±2.23 | 12.3±1.72 | 12.6±1.81 | 11.9±2.20 |
| 4f | Br | m-CONHMe | 0.25±0.07 | 0.21±0.03 | 0.14±0.01 | 0.16±0.07 |
| 4g | Br | m-CONMe2 | 2.83±0.59 | 6.52±1.16 | 2.38±0.37 | 3.34±0.54 |
| 4h | Br | m-COOH | NAb | NA | NA | NA |
| 4i | Br |
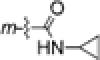
|
0.17±0.02 | 0.17±0.02 | 0.17±0.02 | 0.17±0.02 |
| 4j | Br |
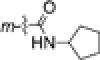
|
0.12±0.02 | 0.12±0.01 | 0.12±0.01 | 0.11±0.01 |
| 5a | Cl | m-COOMe | 2.23±0.23 | 2.33±0.47 | 1.78±0.31 | 1.51±0.46 |
| 5b | Br | m-COOMe | 1.12±0.14 | 1.37±0.31 | 1.27±0.22 | 1.28±0.25 |
| 5c | Cl | m-CONHMe | 0.74±0.08 | 1.04±0.11 | 0.43±0.03 | 1.02±0.10 |
| 5d | Br | m-CONHMe | 0.37±0.08 | 0.63±0.34 | 0.93±0.02 | 0.56±0.09 |
| 5f | Br |
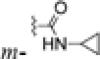
|
0.17±0.01 | 0.16±0.03 | 0.19±0.01 | 0.15±0.02 |
|
| ||||||
| Paclitaxel c | 0.0076±0.0017 | 0.0064±0.0014 | 1.21±0.19 | 0.006±0.001 | ||
Concentration of compound that inhibits 50% human tumor cell growth, presented as mean ± standard deviation (SD), performed at least in triplicate.
Not active; test compound (60 μM) did not reach 50% inhibition.
Positive control.
Table 2.
Cytotoxicity of 6 series against human tumor cell lines
| |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Fused A-ring | GI50 (μM)a |
||||||
|
| |||||||
| X | Y | Z | A549 | KB | KBvin | DU145 | |
| 6a | H | CH | CH | 17.8±2.6 | 15.6±1.3 | 17.6±2.5 | 14.8±1.7 |
| 6b | Me | CH | N | 0.045±0.008 | 0.091±0.009 | 0.055±0.010 | 0.038±0.008 |
| 6c | Me | N | N | 0.018±0.001 | 0.011±0.001 | 0.018±0.004 | 0.015±0.005 |
| 6d | Cl | N | N | 0.0017±0.0003 | 0.0017±0.0008 | 0.0017±0.0003 | 0.0015±0.0003 |
| 6e | NHMe | N | N | 0.027±0.005 | 0.025±0.002 | 0.029±0.005 | 0.023±0.003 |
| Paclitaxel b | 0.0076±0.0017 | 0.0064±0.0014 | 1.21±0.19 | 0.006±0.001 | |||
Concentration of compound that inhibits 50% human tumor cell growth
Positive control.
In series 3 compounds with a phenyl A-ring and substituent X equal to m-chloro, two compounds 3a (R1 = o-COOMe) and 3c (R1 = o-CH2OMe) showed promising potency with low micromolar GI50 values of 1.68 to 5.24 μM, indicating that replacement of the pyridine A-ring in lead 1a is tolerated and R1 on the A-ring is modifiable. When the COOMe group (R1) was moved to the meta-position on the A-ring, the resulting compound 4a exhibited somewhat improved activity (GI50 1.46–1.61 μM) compared with 3a (R1 = o-COOMe, GI50 1.68–3.36 μM). This finding prompted us to synthesize compounds 4b-f, in which the meta-R1 was either maintained as an ester (COOMe) or changed to a N-methylcarbamoyl (CONHMe) group, while substituent X was present as either a chloro, trifluoromethyl or bromo group. Interestingly, compounds 4a, 4c, 4d, and 4f where X is either chloride or bromide were more potent than 3a. In addition, compounds 4d and 4f with a meta-N-methylcarbamoyl group (R1) showed significantly improved potency with a GI50 range of 0.14–0.30 μM, compared with related meta-ester compounds 4a and 4c. In contrast, 4b and 4e with a trifluoromethyl (X) group were less active than corresponding chloro- and bromo-compounds in the same assays, Next, the cytotoxicity results for bromo-substituted compounds 4g-j revealed the impact of the m-alkanoyl group (R1). When the meta-N-methylcarbamoyl (4f) or meta-ester (4c) was converted to a more bulky N,N-dimethylcarbamoyl (4g) or more polar carboxy (4f) group, potency decreased or was abolished completely, respectively. But when R1 was an N-cyclopropylcarbamoyl or N-cyclopentylcarbamoyl group, the corresponding compounds 4i and 4j showed improved potency with GI50 values of 0.11 to 0.17 μM, more potent than the other series 3 and 4 compounds, as well as 1a. Because corresponding series 4 compounds (4a, 4d) were more potent than series 3 compounds (3a, 3d), we postulated that the meta-substitution on the A-ring might favor an appropriate torsional angle between the two aromatic rings, due to decreased steric hindrance around the N-linker, which would be consistent with our prior hypothesis. Moreover, the high potency of 4d, 4f, 4i, and 4j demonstrated that a hydrophobic meta-N-alkylcarbamoyl group with a suitable volume could enhance antitumor potency.
When the N-linker between the A- and B-rings was incorporated into a six-membered 1,2,3,4-tetrahydropyridine ring fused with the B-ring, all five compounds 5a-d,f exhibited high cytotoxicity with GI50 values of 0.15 to 2.33 μM (Table 1). Thus, the ring-restricted N-linker was favorable for cytotoxic activity. Consequently, all series 6 compounds contain a 6-methoxy-1,2,3,4-tetrahydroquinoline moiety connected to an aromatic (naphthalene) or heteroaromatic (quinoline or quinazoline) bicyclic ring system, in which the resonance system of the A-ring is extended over an additional fused aromatic ring. As shown in Table 2, except for 6a with a naphthyl A-ring system, compounds with either a quinoline (6b) or quinazoline (6c-6e) moiety exhibited extremely high cytotoxicity in the HTCL assays. Among them, 6d with a 2-chloroquinazoline moiety (A-ring) was the most potent with GI50 values ranging from 1.5 to 1.7 nM. Thus, 6d was more potent than paclitaxel in the same assays, especially against the drug-resistant KBvin cell line (GI50 0.0017 μM versus 1.21 μM, respectively). Compounds 6b (2-methylquinoline), 6c (2-methylquinazoline), and 6e (2-methylaminoquinazoline) also showed high cytotoxicity with a GI50 range of 11-91 nM, more potent by at least ten-fold than 1a and series 3-5 compounds. Similarly to 6d, analogs 6b, 6c, and 6e were also much more potent than paclitaxel against KBvin cell growth, indicating that this compound type has great potential to overcome resistance to paclitaxel.
Based on these results in cellular assays, active compounds 4f, 4i, 5f, and 6b-e (GI50 < 1 μM) were evaluated in tubulin inhibition assays, in parallel with CA-4, a drug candidate in clinical trials, as a reference. As shown in Table 3, while N-methyl linked compounds 4f and 4i (open N-linker) showed good inhibitory potency against tubulin assembly (IC50 1.6–2.2 μM), the N-phenyl-6-methoxy-1,2,3,4-tetrahydroquinoline compound 5f (ring-restricted N-linker) exhibited improved potency in both tubulin assembly (IC50 1.0 μM) and colchicine binding (75%) assays. Thus, the presence of the ring-restricted N-linker probably increases compound affinity for tubulin. Furthermore, 6b-e with both a ring-restricted N-linker and a fused bicyclic aromatic ring system (A-ring) also displayed high potency in the tubulin assembly (IC50 0.92–1.00 μM) and inhibition of colchicine binding (87–99% at 5 μM and 61–95% at 1 μM) assays, comparable with and greater than those of 5f, respectively. Consistent with the cellular assays, compound 6d was the most potent compound with an IC50 value of 0.93 μM against tubulin assembly and inhibitory rates of 99% and 95% at 5 μM and 1 μM, respectively, for competitively inhibiting colchicine binding to tubulin. These data are comparable or better than those for the reference compound CA-4 (IC50 0.96 μM, 98% and 90% respectively). Thus, N-aryl-6-methoxy-1,2,3,4-tetrahydroquinoline compounds, e.g., series 5-6, likely present a novel class of highly potent tubulin polymerization inhibitors targeted at the colchicine binding site. Meanwhile, we postulated that a fused bicyclic aromatic A-ring might interact with the colchicine site in some way, possibly resulting in increased affinity for tubulin. On the other hand, the active 6 series compounds might also interact with other unidentified target(s), because 6b-e displayed much higher potency than 5f in the cellular cytotoxicity assays, but similar inhibitory activity against tubulin polymerization.
Table 3.
| Compound | Inhibition of tubulin assembly IC50 (μM) ± SD |
Inhibition of colchicine binding (%) inhibition ± SD |
|
|---|---|---|---|
| at 5 μM | at 1 μM | ||
| 4f | 2.2 ± 0.2 | 71 ± 0.3 | NDc |
| 4i | 1.6 ± 0.04 | 69 ± 3.0 | ND |
| 5f | 1.0 ± 0.1 | 75 ± 1.0 | ND |
| 6b | 1.0 ± 0.1 | 87 ± 2.0 | 61 ± 2.0 |
| 6c | 1.0 ± 0.1 | 98 ± 0.2 | 89 ± 2.0 |
| 6d | 0.93 ± 0.04 | 99 ± 0.6 | 95 ± 0.7 |
| 6e | 0.92 ± 0.06 | 95 ± 0.2 | 76 ± 0.1 |
|
| |||
| CA4 d | 0.96 ± 0.07 | 98 ± 0.6 | 90 ± 0.2 |
The tubulin assembly assay measured the extent of assembly of 10 μM tubulin after 20 min at 30 °C.
Tubulin: 1 μM. [3H]colchicine: 5 μM. Inhibitor: 5 μM or 1 μM (compounds inhibiting colchicine binding by more than 80% at 5 μM were further tested). Incubation was performed for 10 min at 37 °C.
Not determined.
reference compound is a drug candidate in phase II/III clinical trials.
3.2 Molecular Modeling
To better understand interactions between the newly synthesized active inhibitors and tubulin, we investigated potential binding modes of active compounds 4i, 5f, and 6d at the colchicine site in the tubulin dimer by using the CDOCKER program in the Discovery Studio 3.0 software with the tubulin crystal structure (PDB: 1SA0) [17,18], as in our previous study. In the binding models shown in Figure 3A, both 4i (purple) and 5f (green) displayed low energy binding conformations (-30.58 and -33.31 kcal/mol, respectively) that superimposed well with each other and with DAMA-colchicine (cyan), the ligand used in the tubulin crystal structure. As seen in Figure 3A, a hydrogen bond is present between Val181 in the α-T5 loop of tubulin and the amide carbonyl group on the A ring of 4i and 5f. This hydrogen bond is also observed between Val181 and the carbonyl group on the C-ring of DAMA-colchicine. In addition, the N-cyclopropyl of the amide group in both 4i and 5f overlaps with the methoxy group on the seven-membered ring (C-ring) of DAMA-colchicine and stretches into a small lipophilic pocket around the amino acids in α-T5, β-H8, β-S8 and β-S9 of tubulin. In addition, the 4-methoxyphenyl moiety (B-ring) in 4i and 5f, regardless of whether the N-linker is open or ring-restricted, overlaps with the trimethoxyphenyl portion of DAMA-colchicine. The 4-OCH3 group on the B-ring of 4i and 5f forms a hydrogen bond with the Cys241 side chain in β-H7 of tubulin, and a similar interaction is also observed with the 2′-OCH3 on the A-ring of DAMA-colchicine. Cys241 is a key amino acid that interacts with most tubulin inhibitors bound at the colchicine site. In the crystal structure, the C5 and C6 atoms of the B-ring of DAMA-colchicine are involved in hydrophobic interactions with the side chains of Alaβ250 and Leuβ255 in tubulin's β-H8 region. Although the N-CH3 linker in 4i is not in close proximity to Alaβ250 and Leuβ255 (> 4Å), the added carbons in the six-membered tetrahydropyridine cyclic linker in 5f are closer to the hydrophobic side chains of Alaβ250 and Leuβ255 (< 4Å). The additional van der Waal's force between the cyclic linker and the binding site would likely strengthen affinity of 5f for tubulin. Therefore, our current modeling results might explain, at least partially, the higher potency of 5f than 4f and 4i in the tubulin assays (Table 3).
Fig. 3.
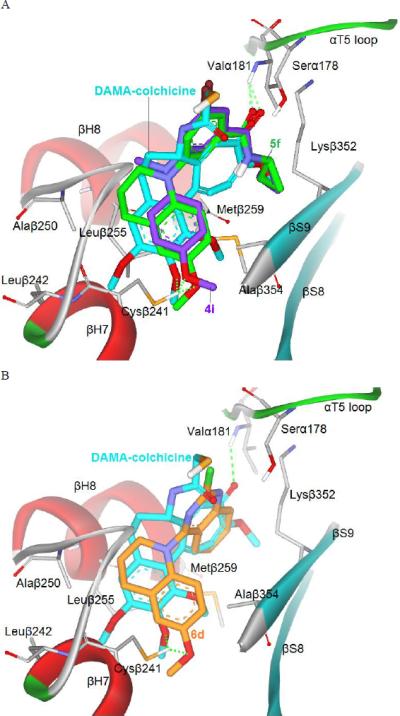
(A) Predicted modes for 4i (purple stick) and 5f (green) binding with tubulin (PDB code: 1SA0), and overlapping with DAMA-colchicine (cyan, the native ligand of 1SA0); (B) Superimposition of docked compound 6d (orange) with DAMA-colchicine (cyan). Surrounding amino acid side chains are shown in gray stick format and labeled. Hydrogen bonds are shown by green dashed lines, and the distance between ligands and protein is less than 3 Å.
Consistent with our earlier hypothesis [6,7], the steric hindrance of the N-CH3 linker [19] results in a binding torsional angle of 74.89 ° between the two aromatic rings in 4i, allowing the N-alkyl amide group on the A-ring to interact with the binding site as described above. When the N-linker is connected with additional carbons into a six-membered ring, a similar torsional angle (70.52 °) was found, maintaining a favorable binding conformation for 5f and all major interactions with the binding site as mentioned above for 4i. Figure 3B shows that 6d (binding energy: -34.39 kcal/mol) has a similar binding orientation as those of 4i and 5f, as well as good superimposition with DAMA-colchicine. The major interactions of the B-ring and the cyclic linker with tubulin are maintained. The torsional angle (69.07°) between the two rings in 6d is similar to that of 5f, resulting in a superimposition between the quinazoline ring of 6d and the aromatic C-ring of DAMA-colchicine. Although we postulated that the fused aromatic ring introduced in 6d would play a role similar to that of the C-ring of colchicine within the binding site on tubulin, no interaction was obvious in the computational model. Because of the torsional angle between the two aryl rings, the 2-chloro group on the quinazoline in 6d [as well as the m-bromo group on the phenyl (A-ring) in 4i and 5f (Figure 3A)] was orientated similarly to the acetyl group on the B-ring in DAMA-colchicine. Thus, the X substituent on the quinazoline (or other aromatic ring system) might interact with tubulin in some way and be modifiable.
3.3 Drug-like property evaluations
Next, we evaluated aqueous solubility and logP parameters of 4f, 4i, 5f, 6b, 6c, 6d and 6e as described previously [20]. As shown in Table 4, compounds 6b, 6c, and 6e showed much better aqueous solubility (3.21–7.67 μg/mL) than the other four compounds (□ 1.0 μg/mL). Consistently, log P values of 6b, 6c, and 6e (1.07–3.98) were also lower (4f, 4i, 5f, 6d >4). Subsequently, metabolic stability of 6b-6e was further evaluated by an in vitro human liver microsome incubation assay with propranolol (moderate metabolism in vivo, t1/2 3–5 h) and terfenadine (fast metabolism in vivo, t1/2 < 3 h) as positive controls. Compounds 6c (t1/2 25.19 min) and 6e (t1/2 25.97 min) were more stable than terfenadine (t1/2 21.14), while 6b and 6d were even less stable with short metabolic half-lives of 7.89 min and 10.59 min, respectively, indicating quick metabolism. In general, a potent compound with low metabolic stability is not usually recognized as a promising drug candidate. However, compounds targeting the colchicine site of tubulin are proposed as VDAs to treat cancers. For this new type of anticancer drug candidate, rapid metabolism might be desirable to decrease drug toxicity in the body, as exemplified by ZD6126 (Fig. 1), which was developed based on this hypothesis [21].
Table 4.
Physicochemical parameters of selected compounds
| Compound | at pH 7.4 |
Human liver microsome t1/2 min |
|
|---|---|---|---|
| Water Solubility (μg/mL) |
Log P | ||
| 4f | 0.81 ± 0.01 | 4.02 ± 0.11 | ND c |
| 4i | 0.16 ± 0.03 | 4.33 ± 0.12 | ND |
| 5f | 0.26 ± 0.05 | 4.75 ± 0.09 | ND |
| 6b | 3.21 ± 0.60 | 3.98 ± 0.02 | 7.89 |
| 6c | 7.67 ± 0.40 | 3.65 ± 0.03 | 25.19 |
| 6d | 0.45 ± 0.06 | 4.13 ± 0.05 | 10.59 |
| 6e | 7.21 ± 0.06 | 1.07 ± 0.21 | 25.97 |
|
| |||
| Propranolola | 40.82 | ||
|
| |||
| Terfenadineb | 21.14 | ||
Data presented as mean from three separate experiments with or without ± standard deviation (SD).
Propranolol has moderate metabolic stability with t1/2 of 3–5 h in vivo;
Terfenadine has fast metabolic stability with t1/2 of < 3 h in vivo.
Not determined.
4. Conclusion
Modifications of prior lead 1a resulted in the synthesis and biological evaluation of 24 new compounds (series 3-6). During this process, active compound 6d showed extremely high cytotoxicity against the screening HTCL panel with a GI50 value range of 1.5–1.7 nM, more potent than paclitaxel in the same assays, especially against resistant KBvin. Compound 6d also displayed significant inhibitory activity in tubulin assembly (IC50 0.93 μM) and colchicine binding (inhibition 99% at 5 μM and 95% at 1 μM) assays, with greater potency than CA-4. Similarly, analogues 6b, 6c, and 6e also showed high potency in the cellular (GI50 11–91 nM) and tubulin (IC50 0.92–1.0 μM) assays. Therefore, N-aryl-6-methoxy-1,2,3,4-tetrahydroquinoline derivatives were identified as a novel class of tubulin polymerization inhibitors targeted at the colchicine site. Current SAR studies lead to the following conclusions: (1) a ring-restricted N-linker is beneficial and provides a feasible torsional angle (about 70-75°) between the two aryl ring systems (A and B rings) for enhancing molecular affinity for tubulin; (2) the 6-methoxy-1,2,3,4-tetrahydroquinoline moiety can serve as a pharmacophore and the methoxy group on the B-ring is a necessary binding point to tubulin; (3) isosteric replacement of the A-ring is feasible and a fused aromatic ring, such as quinoline and quinazoline, improved molecular potency; (4) various m-substituents on the A-ring are present in active compounds, and either halogen or N-alkyl amide/amino might be beneficial. Molecular modeling results illustrated some possible interactions of the active compounds with the colchicine site of tubulin and supported our earlier hypothesis that a feasible torsional angle between the two aryl rings might be important for high potency. An evaluation of essential drug-like properties, i.e. water solubility, log P, and metabolic stability in vitro, also provided useful information. These results will help us to develop drug candidates from this novel class of tubulin inhibitors targeted at the colchicine site in further studies.
5. Experimental Section
5.1. Chemistry
Proton and carbon nuclear magnetic resonance (1H and 13C NMR) spectra were measured on a JNM-ECA-400 (400 MHz) spectrometer using tetramethylsilane (TMS) as internal standard. The solvent used was CDCl3 unless otherwise indicated. Mass spectra (MS) were measured on an API-150 mass spectrometer with an electrospray ionization source from ABI, Inc. Melting points were measured by a SGW X-4 Micro-Melting point detector without correction. The mw reactions were performed on a mw reactor from Biotage, Inc. Medium- pressure column chromatography was performed using a CombiFlash® Companion system from ISCO, Inc. Thin-layer chromatography (TLC) was performed on silica gel GF254 plates. Silica gel GF254 and H (200–300 mesh) from Qingdao Haiyang Chemical Company were used for TLC and column chromatography, respectively. All commercial chemical reagents were purchased from Beijing Chemical Works or Sigma-Aldrich, Inc. Reagents NADPH, MgCl2, KH2PO4, K2HPO4, and reference compounds propranolol and terfenadine were purchased from Sigma-Aldrich. HPLC grade acetonitrile for LC–MS analysis was purchased from VWR. Pooled human liver microsomes (lot no. 28831) were purchased from BD Biosciences (Woburn, MA). Purities of target compounds were determined by using an Agilent HPLC-1200 with UV detector and an Agilent Eclipse XDB-C18 column (150 mm × 4.6 mm, 5 μm), flow rate 0.8 mL/min, UV detection at 254 nm, and injection volume of 15 μL. Mobile elution was conducted with a mixture of solvents A and B [Condition 1: acetonitrile (ACN)/water 60~80/40~20; Condition 2: MeOH/water 70~90/30~10]. For 6b, 6c and 6e, solvent B contained 0.025 mM ammonium acetate.
5.1.1. Methyl 4-chloro-2-(4-methoxyphenyl)aminobenzoate (2a)
A mixture of 2,4-dichloro-benzoic acid (1.91 g, 10 mmol), 4-methoxyaniline (1.29 g, 10.5 mmol), copper powder (58 mg, 9% mmol), cuprous oxide (58 mg, 4% mmol), anhydrous potassium carbonate (1.38 g, 10 mmol) and 2-ethoxyethanol (7.5 mL) was refluxed under N2 protection for 24 h with stirring. The mixture was poured into water and active carbon added. After filtration through Celite, the filtrate was adjusted to pH 3-4 with aq HCl. The collected gray solid was dried to obtain 1.85 g of 4-chloro-2-(4-methoxyphenyl)aminobenzoic acid (0.83 g, 3.0 mmol), which was methylated directly with DMF-DMA (0.79 mL, 6.0 mmol) in toluene (12 mL) under reflux for 2 h. After removal of solvent in vacuo, the residue was purified by flash column chromatography (gradient elution: EtOAc/petroleum ether, 0-30%) to obtain 0.74 g of 2a in a 57% yield over two steps. It was used next without further purification.
5.1.2. Methyl 3-chloro-5-(4-methoxyphenylamino)benzoate (2b)
A mixture of methyl 3-chloro-5-iodobenzoate (296 mg, 1.0 mmol), 4-methoxyaniline (148 mg, 1.2 mmol), Cs2CO3 (455 mg, 1.4 mmol), X-Phos (17 mg, 0.04 mmol), Pd(OAc)2 (11 mg, 0.05 mmol), Celite (228 mg), and t-BuOH (0.5 mL) in toluene (3 mL) was heated at 120 °C for 1 h under mw irradiation with stirring. EtOAc (10 mL) was added to the mixture, and, after brief stirring, insoluble solid was removed by filtration. After removal of organic solvent in vacuo, the residue was purified by flash column chromatography (gradient elution: EtOAc/petroleum ether, 0-40%) to obtain 2b as a yellow solid, 216 mg, 74% yield, which was used directly in the next step without further purification.
5.1.3. Methyl 3-trifluoromethyl-5-(4-methoxyphenylamino)benzoate (2c)
A mixture of methyl 3-trifluoromethyl-5-bromobenzoate (285 mg, 1.0 mmol), 4-methoxyaniline (147 mg, 1.2 mmol), Cs2CO3 (455 mg, 1.4mmol), X-Phos (17 mg, 0.04 mmol), Pd(OAc)2 (11 mg, 0.05 mmol), Celite (228 mg), and t-BuOH (0.5 mL) in toluene (3.0 mL) was heated at 150 °C for 30 min under mw irradiation. After work-up as for 2b, 240 mg of 2c was obtained in 74% yield, yellow solid, which was directly used for the next step without further purification.
5.1.3. Methyl 3-bromo-5-(4-methoxyphenylamino)benzoate (2d)
A mixture of methyl 3,5-dibromobenzoate (294 mg, 1.0 mmol), 4-methoxyaniline (146 mg, 1.2 mmol), Cs2CO3 (455 mg, 1.4mmol), BINAP (31 mg, 0.05 mmol), and Pd(OAc)2 (11 mg, 0.05 mmol) in toluene (5- 10 mL) was refluxed for 12 h under nitrogen protection. After the mixture was cooled to rt, EtOAc (10 mL) was added. After stirring, the insoluble material was removed by filtration. The solvent was removed in vacuo, and the residue was purified by flash column chromatography (gradient elution: EtOAc/petroleum ether, 0-40%) to produce 215 mg of 2d in 64% yield as a brown oil.
5.1.4. General procedure for methylation of aniline NH with methyl iodide to prepare 3a and 4a-c
To a solution of a diarylamine (2) and MeI (molar ratio: 1/2-3) in anhydrous DMF (ca. 3 mL) was slowly added NaH (2-3 equiv, 60% oil suspension) at 0 °C with stirring over about 1 h. When the reaction was completed as monitored by TLC, the mixture was poured into ice-water and extracted with EtOAc three times. The combined organic phase was washed with water and brine successively and dried over anhydrous Na2SO4 overnight. After removal of solvent in vacuo, the crude product was purified by flash column chromatography (gradient elution: EtOAc/petroleum ether, 0-50%) to give the corresponding methylated tertiary amine compounds.
5.1.4.1. Methyl 4-chloro-2-(N-(4-methoxyphenyl)-N-methylamino)benzoate (3a)
Starting with 2a (670 mg, 2.3 mmol), methyl iodide (0.28 mL, 4.6 mmol) and NaH (184 mg, 4.6 mmol) to produce 662 mg of 3a in 82% yield, yellow solid, mp 52-53 °C; 1H NMR δ ppm 3.27 (3H, s, NCH3), 3.51 (3H, s, OCH3), 3.76 (3H, s, OCH3), 6.80 (4H, m, ArH-2′,3′,5′,6′), 7.02 (1H, dd, J = 8.4 and 2.0 Hz, ArH-5), 7.15 (1H, d, J = 2.0 Hz, ArH-3), 7.55 (1H, d, J = 8.0, ArH-6); MS m/z (%) 306 (M + 1, 100), 308 (M + 3, 26); HPLC purity 98.3%.
5.1.4.2. Methyl 3-chloro-5-(N- (4-methoxyphenyl)-N-methylamino)benzoate (4a)
Starting with 2b (322 mg, 1.10 mmol), methyl iodide (0.14 mL, 2.2 mmol) and NaH (88 mg, 2.2 mmol) to produce 328 mg of 4a in 98% yield, yellow solid, mp 58~60 °C; 1H NMR δ ppm 3.27 (3H, s, NCH3), 3.84 (3H, s, OCH3), 3.87 (3H, s, OCH3), 7.04 (1H, t, J = 2.0 Hz, ArH-2), 6.94 (2H, d, J = 8.8 Hz, ArH-2′,6′), 7.10 (2H, d, J = 8.8 Hz, ArH-3′,5′), 7.26 (1H, m, ArH-4), 7.35 (1H, t, J = 2.0 Hz, ArH-6). MS m/z (%) 306 (M + 1,39), 308 (M + 3, 13), 291 (100); HPLC purity 99.3%.
5.1.4.3. Methyl 3-trifluoromethyl-5-(N-(4-methoxyphenyl)-N-methylamino)benzoate (4b)
Starting with 2c (188 mg, 0.58 mmol), methyl iodide (0.07 mL, 1.2 mmol), and NaH (48 mg, 1.2 mmol) to produce 176 mg of 4c in 89% yield, yellow oil; 1H NMR δ ppm 3.32 (3H, s, NCH3), 3.85 (3H, s, OCH3), 3.90 (3H, s, OCH3), 6.95 (2H, d, J = 8.8 Hz, ArH-2′,6′), 7.04 (1H, s, ArH-2), 7.12 (2H, d, J = 8.8 Hz, ArH-3′,5′), 7.51 (1H, s, ArH-4), 7.61 (1H, s, ArH-6). MS m/z (%) 340 (M + 1, 55), 325 (M − 14, 100); HPLC purity 98.7%.
5.1.4.4. Methyl 3-bromo-(N-(4-methoxyphenyl)-N-methylamino)benzoate (4c)
Starting with 2d (128 mg, 0.38 mmol), methyl iodide (0.07 mL, 1.1 mmol) and sodium hydride (44 mg, 1.1 mmol) to produce 115 mg of 4c in 90% yield, yellow oil; 1H NMR δ ppm 3.27 (3H, s, NCH3), 3.84 (3H, s, OCH3), 3.87 (3H, s, OCH3), 6.94 (3H, m, ArH-2,2′,6′), 7.10 (2H, d, J = 8.8 Hz, ArH-3′,5′), 7.29 (1H, m, ArH-4), 7.50 (1H, s, ArH-6). MS m/z (%) 350 (M + 1, 34), 352 (M + 3, 29), 337 (M − 14, 100); HPLC purity 95.0%.
5.1.5. 5-Chloro-2-hydroxymethyl-N-(4-methoxy)phenyl-N-methylaniline (3b)
A solution of 3a (427 mg, 1.40 mmol) in THF (4 mL) was added dropwise to LiAlH4 (108 mg, 2.89 mmol, excess) in anhydrous THF (5 mL) at 0 °C with stirring, which was continued at the same temperature for another 1 h. After the reaction was complete, as monitored by TLC, to the mixture was added 0.11 mL of water, 0.33 mL of 15% aq NaOH, and 0.33 mL of water, successively, with stirring for another 10 min at rt. Then the mixture was filtered through Celite, solvent was removed in vacuo, and the residue was purified by flash column chromatography (gradient elution: EtOAc/petroleum ether 0-40%) to produce 377 mg of 3b in 97% yield, brown oil; 1H NMR δ ppm 3.19 (3H, s, NCH3), 3.76 (3H, s, OCH3), 4.48 (2H, s, OCH2), 6.68 (2H, d, J = 9.2 Hz, ArH-2′,6′), 6.79 (2H, d, J = 9.2 Hz, ArH-3′,5′), 7.12 (1H, d, J = 2.0 Hz, ArH-3), 7.19 (1H, dd, J = 8.0 & 2.0Hz, ArH-5), 7.38 (1H, d, J = 8.0, ArH-6). MS m/z (%) 278 (M + 1, 100), 280 (M + 3, 36).
5.1.6. 5-Chloro-2-(methoxymethyl)-N-(4-methoxy)phenyl-N-methylaniline (3c)
Prepared in the same manner as for 3a, starting with 3b (227 mg, 0.82 mmol), methyl iodide (0.10 mL, 1.6 mmol), and NaH (66 mg, 1.6 mmol) to produce 228 mg of 3c in 95% yield, yellow oil; 1H NMR δ ppm 3.17 (3H, s, NCH3), 3.32 (3H, s, OCH3), 3.76 (3H, s, OCH3), 4.26 (2H, s, OCH2), 6.60 (2H, d, J = 9.2 Hz, ArH-2′,6′), 6.79 (2H, d, J = 9.2 Hz, ArH-3′,5′), 7.11 (1H, d, J = 2.0 Hz, ArH-3), 7.20 (1H, dd, J = 8.4 & 2.0Hz, ArH-5), 7.45 (1H, d, J = 8.4, ArH-6). MS m/z (%) 292 (M + 1, 22), 294 (M+ 3, 7), 228 (M-63, 100).
5.1.7. General procedure for preparing N-methylamides 3d, 4d-f from esters under mw irradiation
A solution of ester compound (0.17 mmol ~ 0.40 mmol) in 3 mL 30% methylamine in MeOH was heated to 100-120 °C under mw irradiation for 1-2 h. Then the mixture was poured into ice-water, neutralized with aq HCl (2 N), and extracted with EtOAc three times. The combined organic phase was washed with water and brine, successively, and dried over anhydrous Na2SO4 overnight. After removal of solvent in vacuo, the crude product was purified by flash column chromatography (gradient elution: EtOAc/petroleum ether, 0-70%) to give the corresponding target compound.
5.1.7.1. N-methyl-4-chloro-2-(N-(4-methoxy)phenyl-N-methylamino)benzamide (3d)
Starting with 3a (115 mg, 0.38 mmol) at 120 °C for 2 h to produce 92 mg of 3d, 79% yield, yellow oil; 1H NMR δ ppm 2.89 (3H, d, J = 5.2 Hz, NCH3), 3.12 (3H, s, NCH3), 3.78 (3H, s, OCH3), 6.82 (4H, m, ArH on the B-ring), 7.02 (1H, d, J = 2.0 Hz, ArH-3), 7.26 (1H, m, ArH-5), 8.14 (1H, d, J = 8.0, ArH-6). MS m/z (%) 305 (M + 1, 12) 307 (M + 3, 4), 274 (M − 31, 100).
5.1.7.2. 3-Chloro-5-(N-(4-methoxyphenyl)-N-methylamino)-N-methylbenzamide (4d)
Starting with 4a (61 mg, 0.20 mmol) at 100 °C for 1 h to produce 52 mg of 4d, 85% yield, white solid, mp 144~145 °C; 1H NMR δ ppm 2.96 (3H, d, J = 4.8 Hz, NCH3), 3.26 (3H, s, NCH3), 3.83 (3H, s, OCH3), 5.96 (1H, s, CONH), 6.73 (1H, s, ArH-2), 6.93 (2H, d, J = 8.8 Hz, ArH-2′,6′), 6.96 (1H, s, ArH-4), 6.99 (1H, s, ArH-6), 7.10 (2H, d, J = 8.8 Hz, ArH-3′,5′). MS m/z (%) 305 (M + 1, 100), 307 (M + 3, 31); HPLC purity 99.3%.
5.1.7.3. 5-(N-(4-Methoxyphenyl)-N-methylamino)-3-trifluoromethyl-N-methylbenzamide (4e)
Starting with 4b (102 mg, 0.30 mmol) at 100 °C for 1 h to produce 90 mg of 4e in 89% yield, yellow solid, mp 115~116 °C; 1H NMR δ ppm 2.99 (3H, d, J = 4.8 Hz, NCH3), 3.31 (3H, s, NCH3), 3.84 (3H, s, OCH3), 6.05 (1H, s, CONH), 6.94 (2H, d, J = 8.8 Hz, ArH-2',6'), 6.97 (1H, s, ArH-2), 7.11 (2H, d, J = 8.8 Hz, ArH-3',5′), 7.20 (1H, s, ArH-4), 7.26 (1H, s, ArH-6). MS m/z (%) 339 (M + 1, 100); HPLC purity 96.3%.
5.1.7.4. 3-Bromo-5-(N-(4-methoxyphenyl)-N-methylamino)-N-methylbenzamide (4f)
Starting with 4c (100 mg, 0.29 mmol) at 100 °C for 1 h to produce 82 mg of 4f in 82% yield, white solid, mp 155~156 °C; 1H NMR δ ppm 2.96 (3H, d, J = 4.4 Hz, NCH3), 3.26 (3H, s, NCH3), 3.83 (3H, s, OCH3), 5.98 (1H, s, CONH), 6.89 (1H, s, ArH-2), 6.93 (2H, d, J = 8.8 Hz, ArH-2′,6′), 7.03 (1H, s, ArH-4), 7.08 (1H, s, ArH-6), 7.10 (2H, d, J = 8.8 Hz, ArH-3′,5′). MS m/z (%) 349 (M + 1, 100), 351 (M + 3, 95); HPLC purity 97.9%.
5.1.8. 3-Bromo-5-(N-(4-methoxyphenyl)-N-methylamino)-N,N-dimethylbenzamide (4g)
Prepared in the same manner as 3c. Starting with 4f (70 mg, 0.20 mmol), methyl iodide (0.02 mL, 0.32 mmol) and NaH (16 mg, 0.40 mmol) to produce 65 mg of 4g, 90% yield, brown solid, mp 80~81 °C; 1H NMR δ ppm 2.94 (3H, s, NCH3), 3.05 (3H, s, NCH3), 3.23 (3H, s, NCH3), 3.83 (3H, s, OCH3), 6.61 (1H, dd, J = 1.2 Hz and 1.2 Hz, ArH-4) 6.82 (1H, m, ArH-2), 6.83 (1H, m, ArH-6), 6.92 (2H, d, J = 8.8 Hz, ArH-2′,6′), 7.10 (2H, d, J = 8.8 Hz, ArH-3′,5′). MS m/z (%) 363 (M + 1, 100), 365 (M + 3, 95); HPLC purity 98.4%.
5.1.9. 3-Bromo-5-(N-(4-methoxyphenyl)-N-methylamino)benzoic acid (4h)
To a solution of 4c (76 mg, 0.20 mmol) in THF/MeOH (2/2 mL) was added aq NaOH (3 N, 2 mL), and the mixture was heated at reflux for 5 h. After removal of solvent in vacuo, the mixture was poured into ice-water, and the insoluble solid (pH 12) was removed by filtration. The filtrate was acidified to pH 2, until there was no further precipitation. The solid was collected by filtration, washed with water until neutral, and dried to obtain 178 mg of 4h in 90% yield, yellow solid, mp 242~244 °C; 1H NMR (DMSO-d6) δ ppm 3.17 (3H, s, NCH3), 3.77 (3H, s, OCH3), 7.46 (1H, m, ArH-4), 6.97 (2H, d, J = 8.8 Hz, ArH-2′,6′), 7.11 (2H, d, J = 8.8 Hz, ArH-3′,5′), 7.18 (1H, d, J = 1.2 Hz, ArH-6), 7.31 (1H, s, ArH-2). MS m/z (%) 336 (M + 1, 100), 338 (M + 3, 97).
5.1.10. 3-Bromo-5-(N-(4-methoxyphenyl)-N-methylamino)-N-cyclopropylbenzamide (4i)
A mixture of 4h (105 mg, 0.31 mmol), EDCI (90 mg, 0.47 mmol), and HOBt (64 mg, 0.47 mmol) in CH2Cl2 (5 mL) was stirred at rt for 30 min, and then cyclopropylamine (27 mg, 0.47 mmol) was added dropwise at 0 °C with stirring over 15 min, followed by warming to rt for an additional 24 h. The mixture was added to water (20 mL) and extracted with CH2Cl2 three times. The combined organic phase was washed with water and brine, successively, and dried over anhydrous Na2SO4 overnight. After removal of solvent in vacuo, crude product was purified by flash column chromatography (gradient elution: EtOAc/petroleum ether, 0-50%) to give 95 mg of pure 4i in 82% yield, white solid, mp 184-186 °C; 1H NMR δ ppm 0.59 (2H, m, CH2), 0.85 (2H, m, CH2), 2.84 (1H, m, CH), 3.26 (3H, s, NCH3), 3.83 (3H, s, OCH3), 6.09 (1H, br, NH), 6.87 (1H, dd, J = 1.6 & 2.0 Hz, ArH-2), 6.93 (2H, d, J = 8.8 Hz, ArH-2′,6′), 7.05 (2H, m, ArH-4,6), 7.09 (2H, d, J = 8.8 Hz, ArH-3′,5′). MS m/z (%) 375 (M + 1, 69), 377 (M + 3, 79), 320 (M - 54, 100); HPLC purity 99.1%.
5.1.11. 3-Bromo-5-(N-(4-methoxyphenyl)-N-methylamino)-N-cyclopentylbenzamide (4j)
As for 4i. Starting with 4h (168 mg, 0.50 mmol), EDCI (144 mg, 0.75 mmol), HOBt (101 mg, 0.75 mmol), and cyclopentylamine (64 mg, 0.75 mmol) to produce 161 mg of 4j in 80% yield, white solid, mp 173-174 °C; 1H NMR δ ppm1.46 (2H, m, CH2), 1.69 (4H, m, 2×CH2), 2.08 (2H, m, CH2), 3.26 (3H, s, NCH3), 3.83 (3H, s, OCH3), 4.33(1H, m, CH), 5.90 (1H, br, NH), 6.86 (1H, dd, J = 2.0 and 2.0 Hz, ArH-2), 6.93 (2H, d, J = 10.0 Hz, ArH-2′,6′), 7.04 (1H, s, ArH-4), 7.10 (3H, m, ArH-3′,5′,6). MS m/z (%) 403 (M + 1, 100), 405 (M + 3, 91).
5.1.12. N-(3-Chloro-5-carbomethoxy)phenyl-6-methoxy-1,2,3,4-tetrahydroquinoline (5a)
Prepared in the same manner as 2b. Starting with methyl 3-chloro-5-iodobenzoate (297 mg, 1.0 mmol), 6-methoxy-1,2,3,4-tetrahydroquinoline (7) (163 mg, 1.0 mmol) at 120 °C for 30 min to produce 255 mg of 5a in 77% yield, yellow oil; 1H NMR δ ppm 1.96 (2H, m, 3′-CH2), 2.76 (2H, t, J = 6.4 Hz, 4′-CH2), 3.61 (2H, t, J = 6.4 Hz, 2′-CH2), 3.78 (3H, s, OCH3), 3.89 (3H, s, OCH3), 6.64 (1H, d, J = 3.2 Hz, ArH-5′), 6.67 (1H, dd, J = 8.8 & 3.2 Hz, ArH-7′), 6.96 (1H, d, J = 8.8 Hz, ArH-8′), 7.28 (1H, m, ArH-6), 7.51 (1H, m, ArH-4), 7.66 (1H, m, ArH-2). MS m/z (%) 332 (M + 1, 100), 334 (M + 3, 30); HPLC purity 97.2%.
5.1.13. N-(3-Bromo-5-carbomethoxy)phenyl-6-methoxy-1,2,3,4-tetrahydroquinoline (5b)
Prepared in the same manner as 2d. Starting with methyl 3,5-dibromobenzoate (294 mg, 1.0 mmol), 7 (179 mg, 1.1 mmol) for 12 h to produce 255 mg of 5b in 68% yield, yellow oil; 1H NMR δ ppm 1.96 (2H, m, 3′-CH2), 2.76 (2H, t, J = 6.4 Hz, 4′-CH2), 3.61 (2H, t, J = 6.4 Hz, 2′-CH2), 3.78 (3H, s, OCH3), 3.89 (3H, s, OCH3), 6.65 (1H, dd, J = 9.2 and 2.8 Hz, ArH-7′), 6.68 (1H, d, J = 2.8 Hz, ArH-5′), 6.95 (1H, d, J = 9.2 Hz, ArH-8′), 7.43 (1H, t, J = 2.0 Hz, ArH-4), 7.66 (1H, t, J = 2.0 Hz, ArH-6), 7.66 (1H, m, ArH-2). MS m/z (%) 376 (M + 1, 100), 378 (M + 3, 91).
5.1.14. N-(3-Chloro-5-(N-methyl) carbamoyl)phenyl-6-methoxy-1,2,3,4-tetrahydroquinoline (5c)
Prepared in the same manner as 3d. Starting with 5a (100 mg, 0.30 mmol) at 100 °C for 1 h to produce 91 mg of 5c, 92% yield, pale yellow solid, mp 179~180 °C; 1H NMR δ ppm 1.96 (2H, m, 3′-CH2), 2.75 (2H, t, J = 6.0 Hz, 4′-CH2), 2.98 (3H, d, J = 4.8 Hz, NCH3), 3.61 (2H, t, J = 6.0 Hz, 2′-CH2), 3.78 (3H, s, OCH3), 6.05 (1H, bs, NH), 6.68 (2H, m, ArH-5′,7′), 6.97 (1H, d, J = 8.4 Hz, ArH-8′), 7.17 (1H, d, J = 1.6 Hz, ArH-6), 7.20 (1H, d, J = 1.6 Hz, ArH-4), 7.40 (1H, s, ArH-2). MS m/z (%) 331 (M + 1, 100), 333 (M + 3, 32); HPLC purity 99.0%.
5.1.15. N-(3-Bromo-5-(N-methyl)carbamoyl)phenyl-6-methoxy-1,2,3,4-tetrahydroquinoline (5d)
Prepared in the same manner as 3d. Starting with 5b (64 mg, 0.17 mmol) at 100 °C for 1 h to produce 57 mg of 5d, 92% yield, white solid, mp 161~162 °C; 1H NMR δ ppm 1.96 (2H, m, 3′-CH2), 2.75 (2H, t, J = 6.0 Hz, 4′-CH2), 2.99 (3H, d, J = 4.8 Hz, NCH3), 3.60 (2H, t, J = 6.0 Hz, 2′-CH2), 3.78 (3H, s, OCH3), 6.02 (1H, bs, NH), 6.68 (2H, m, ArH-5′,7′), 6.96 (1H, d, J = 8.4 Hz, ArH-8′), 7.32 (1H, s, ArH-4), 7.36 (1H, s, ArH-6), 7.44 (1H, s, ArH-2). MS m/z (%) 375 (M + 1, 100), 377 (M + 3, 97); HPLC purity 98.2%.
5.1.16. N-(3-Bromo-5-carboxy)phenyl-6-methoxy-1,2,3,4-tetrahydroquinoline 5e
Prepared in the same manner as (4h). Starting with 5b (226 mg, 0.60 mmol) to produce 178 mg of 5e in 82% yield, yellow solid, mp 176~178 °C; 1H NMR δ ppm 1.97 (2H, m, 3′-CH2), 2.76 (2H, t, J = 6.0 Hz, 4′-CH2), 3.61 (2H, t, J = 6.0 Hz, 2′-CH2), 3.79 (3H, s, OCH3), 6.68 (2H, m, ArH-5′,7′), 6.98 (2H, d, J = 8.8 Hz, ArH-8′), 7.48 (1H, t, J = 2.0 Hz, ArH-6), 7.70 (1H, dd, J = 2.0 and 1.6 Hz, ArH-4), 7.48 (1H, dd, J = 2.0 and 1.6 Hz, ArH-2). MS m/z (%) 362 (M + 1, 100), 364 (M + 3, 72).
5.1.17. N-(3-Bromo-5-(N-cyclopropyl)carbamoyl)phenyl-6-methoxy-1,2,3,4-tetrahydroquinoline 5f
Prepared in the same manner as 4i. Starting with 5e (109 mg, 0.30 mmol), EDCI (115 mg, 0.60 mmol), HOBt (81 mg, 0.60 mmol), and cyclopropylamine (56 mg, 1.0 mmol) to produce 108 mg of 5f in 90% yield, white solid, mp 163-164 °C; 1H NMR δ ppm 0.61 (2H, m, CH2), 0.86 (2H, m, CH2), 1.95 (2H, m, 3′-CH2), 2.75 (2H, t, J = 6.4 Hz, 4′-CH2), 2.86 (1H, m, CH), 3.60 (2H, t, J = 6.0 Hz, 2′-CH2), 3.78 (3H, s, OCH3), 6.12 (1H, br, NH), 6.66 (2H, m, ArH-5′,7′), 6.95 (1H, d, J = 8.4 Hz, ArH-8′), 7.26 (1H, t, J = 2.0 Hz, ArH-4), 7.35 (1H, d, J = 2.0 Hz, ArH-2), 7.43 (1H, t, J = 2.0 Hz, ArH-6); 13C NMR δ ppm 6.91 (CH2), 23.38 (CH), 23.39 (CH2), 27.71 (CH2), 49.78 (CH2), 55.71 (OCH3), 112.59 (CH), 114.35 (CH), 117.61 (CH), 120.62 (CH), 121.48 (CH), 123.02 (C), 125.54 (CH), 130 40 (C), 135.66 (C), 137.15 (C), 150.75 (C), 154.51 (C), 167.95 (C), 195.50 (CO). MS m/z (%) 401 (M + 1, 100), 403 (M + 3, 90); HPLC purity 98.4%.
5.1.18. 6-Methoxy-N-(naphthalen-1-yl)-1,2,3,4-tetrahydroquinoline 6a
Prepared in the same manner as (2d). Starting with 1-bromonaphthalene (206 mg, 1.0 mmol) and 7 (179 mg, 1.1 mmol) for 12 h to produce 196 mg of 6a in 68% yield, pale yellow solid, mp 93-95 °C; 1H NMR δ ppm 2.15 (2H, br, 3-CH2), 2.98 (2H, br, 4-CH2), 3.63 (2H, t, J = 5.6 Hz, 2-CH2), 3.72 (3H, s, 6-OCH3), 6.06 (1H, d, J = 8.8 Hz, ArH-8), 6.41 (1H, dd, J = 8.8 & 2.8 Hz, ArH-7), 6.68 (1H, d, J = 2.8 Hz, ArH-5), 6.97 (1H, d, J = 7.2 Hz, ArH-2′), 7.46 (3H, m, ArH-3′,6′,7′), 7.74 (1H, d, J = 8.0 Hz, ArH-4′), 7.89 (1H, d, J = 8.0 Hz, ArH-5′), 8.02 (1H, d, J = 8.0 Hz, ArH-8′). MS m/z (%) 290 (M + 1, 100), 274 (M-15, 100); HPLC purity 98.2%.
5.1.19. 6-Methoxy-2′-methyl-3,4-dihydro-2H-1,4′-biquinoline 6b
Prepared in the same manner as (2d). Starting with 4-chloro-2-methylquinoline (178 mg, 1.0 mmol) and 7 (180 mg, 1.1 mmol) for 12 h to produce 228 mg of 6b in75% yield, yellow solid, mp 120~121 °C; 1H NMR δ ppm 2.03 (2H, m, 3-CH2), 2.65 (3H, s, 2′-CH3), 2.94 (2H, t, J = 6.4 Hz, 4-CH2), 3.71 (2H, t, J = 5.6 Hz, 2-CH2), 3.77 (3H, s, 6-OCH3), 6.46 (1H, d, J = 9.2 Hz, ArH-8), 6.53 (1H, dd, J = 9.2 & 2.8 Hz, ArH-7), 6.71 (1H, d, J = 2.8 Hz, ArH-5), 6.97 (1H, s, ArH-3′), 7.38 (1H, m, ArH-6′), 7.64 (1H, m, ArH-7′), 7.90 (1H, d, J = 8.4 Hz, ArH-8′), 8.01 (1H, d, J = 8.4 Hz, ArH-5′) ); 13C NMR δ ppm 22.41 (CH2), 25.59 (CH3), 27.67 (CH2), 51.39 (CH2), 55.68 (OCH3), 112.78 (CH), 114.34 (CH), 116.44 (CH), 120.25 (CH), 123.58 (C), 124.31 (CH), 124.96 (CH), 127.55 (C), 129.27 (CH), 129.50 (CH), 138.14 (C), 149.92 (C), 153.93 (C), 154.75 (C), 159.78 (C). MS m/z (%) 305 (M + 1, 100); HPLC purity 99.3%.
5.1.20. 6-Methoxy-N-(2′-methylquinazol-4′-yl)-1,2,3,4-tetrahydroquinoline (6c)
A mixture of 4-chloro-2-methylquinazoline (89 mg, 0.5 mmol), 7 (98 mg, 0.5 mmol), and NaHCO3 (126 mg, 1.5 mmol) in absolute anhydrous EtOH (5 mL) was refluxed for 3 h. The mixture was poured into ice-water, acidified to pH 3 with aq HCl (2 N), and extracted with EtOAc three times. The combined organic phases were washed with water and brine, successively, and dried over anhydrous Na2SO4 overnight. After removal of solvent in vacuo, the crude product was purified by flash column chromatography (gradient elution: EtOAc/petroleum ether, 0-50%) to obtain 121 mg of pure 6c in79% yield, yellow solid, mp 134~136 °C; 1H NMR δ ppm 2.11 (2H, m, 3′-CH2), 2.73 (3H, s, CH3), 2.88 (2H, t, J = 6.8 Hz, 4′-CH2), 3.79 (3H, s, OCH3), 4.05 (2H, t, J = 6.8 Hz, 2′-CH2), 6.53 (1H, dd, J = 8.8 & 2.8 Hz, ArH-7′), 6.63 (1H, d, J = 8.8 Hz, ArH-8′), 6.78 (1H, d, J = 2.8 Hz, ArH-5′), 7.12 (1H, m, ArH-6), 7.32 (1H, dd, J = 8.4 &1.2 Hz, ArH-5), 7.62 (1H, m, ArH-7), 7.79 (1H, d, J = 8.0 Hz, ArH-8) ; 13C NMR δ ppm 24.42 (CH2), 26.65 (CH3), 27.39 (CH2), 47.49 (CH2), 55.62 (OCH3), 112.00 (CH), 113.70 (CH), 115.68 (C), 122.28 (CH), 124.40 (CH), 126.29 (CH), 128.00 (CH), 132.35 (CH), 132.45 (C), 136.22 (C), 152.43 (C), 156.07 (C), 161.95 (C), 163.99 (C). MS m/z (%) 306 (M + 1, 100); HPLC purity 98.8%.
5.1.21. N-(2′-Chloroquinazol-4′-yl)-6-methoxy-1,2,3,4-tetrahydroquinoline (6d)
Similar procedure to that of 6c. Starting with 2,4-dichloroquinazoline (200 mg, 1.0 mmol), 7 (163 mg, 1.0 mmol), and NaHCO3 (252 mg, 3.0 mmol) in absolute anhydrous EtOH (8 mL) for 3 h to produce 282 mg of 6d in 87% yield, yellow solid, mp 136~138 °C; 1H NMR δ ppm 2.12 (2H, m, 3′-CH2), 2.86 (2H, t, J = 6.8 Hz, 4′-CH2), 3.81 (3H, s, OCH3), 4.07 (2H, t, J = 6.8 Hz, 2′-CH2), 6.55 (1H, dd, J = 8.8 &2.8 Hz, ArH-7′), 6.70 (1H, d, J = 8.8 Hz, ArH-8′), 6.81 (1H, d, J = 2.8 Hz, ArH-5′), 7.13 (1H, m, ArH-6), 7.32 (1H, dd, J = 8.8 &1.2 Hz, ArH-5), 7.63 (1H, m, ArH-7), 7.78 (1H, dd, J = 8.8 Hz and 1.2 Hz, ArH-8) ; 13C NMR δ ppm 23.67 (CH2), 26.84 (CH2), 48.86 (CH2), 55.78 (OCH3), 112.82 (CH), 113.14 (C), 114.06 (CH), 123.21 (CH), 123.99 (CH), 126.68 (CH×2), 132.44 (C), 135.44 (CH), 135.86 (C), 145.79 (C), 152.50 (C), 159.10 (C), 160.89 (C). MS m/z (%) 326 (M + 1, 100), 328 (M + 3, 31); HPLC purity 98.4%.
5.1.22. 6-Methoxy-N-(2′-(N-methyl)aminoquinazol-4′-yl)-1,2,3,4-tetrahydroquinoline (6e)
Prepared in the same manner as 3d. Staring with 6d (65 mg, 0.20 mmol) in 3 mL of methylamine (30%) was heated to 100 ºC for 1 h to produce 42 mg of 6e in 66% yield, yellow solid, mp 139~140 °C; 1H NMR δ ppm 2.07 (2H, m, 3′-CH2), 2.85 (2H, t, J = 6.8 Hz, 4′-CH2), 3.10 (3H, d, J = 5.2 Hz, NCH3), 3.79 (3H, s, OCH3), 3.94 (2H, t, J = 6.8 Hz, 2′-CH2), 6.53 (1H, dd, J = 9.2 & 2.8 Hz, ArH-7′), 6.67 (1H, d, J = 9.2 Hz, ArH-8′), 6.76 (1H, d, J = 2.8 Hz, ArH-5′), 6.83 (1H, m, ArH-6), 7.29 (1H, d, J = 8.0 Hz, ArH-5), 7.46 (1H, m, ArH-7), 7.52 (1H, d, J = 8.0 Hz, ArH-8). MS m/z (%) 321 (M + 1, 100); HPLC purity 98.6%.
5.2. Antiproliferative Activity Assay
Target compounds were assayed by the SRB method for cytotoxic activity using a HTCL assay according to procedures described previously [22-24]. The panel of cell lines included human lung carcinoma (A-549), epidermoid carcinoma of the nasopharynx (KB), P-gp-expressing epidermoid carcinoma of the nasopharynx (KBvin), and prostate cancer (DU145). The cytotoxic effects of each compound were expressed as GI50 values, which represent the molar drug concentrations required to cause 50% tumor cell growth inhibition.
5.3. Tubulin Assays
Tubulin assembly was measured by turbidimetry at 350 nm as described previously [25]. Assay mixtures contained 1.0 mg/mL (10 μM) tubulin and varying compound concentrations were pre-incubated for 15 min at 30 °C without guanosine 5′-triphosphate (GTP). The samples were placed on ice, and 0.4 mM GTP was added. Reaction mixtures were transferred to 0 °C cuvettes, and turbidity development was followed for 20 min at 30 °C following a rapid temperature jump. Compound concentrations that inhibited increase in turbidity by 50% relative to a control sample were determined.
Inhibition of the binding of [3H]colchicine to tubulin was measured as described previously [26]. Incubation of 1.0 μM tubulin with 5.0 μM [3H]colchicine and 5.0 μM or 1.0 μM inhibitor was for 10 min at 37 °C, when about 40-60% of maximum colchicine binding occurs in control samples.
5.4. Aqueous Solubility
Determination
Solubility was measured at pH 7.4 by using an HPLC−UV method. Test compounds were initially dissolved in DMSO at a concentration of 1.0 mg/mL. Ten microliters of this stock solution were added to pH 7.4 phosphate buffer (1.0 mL) with the final DMSO concentration being 1%. The mixture was stirred for 4 h at rt and then centrifuged at 3000 rpm for 10 min. The saturated supernatants were transferred to other vials for analysis by HPLC−UV. Each sample was performed in triplicate. For quantification, a model 1200 HPLC−UV (Agilent) system was used with an Agilent Eclipse XDB-C18 column (150 mm × 4.6 mm, 5 μm) and elution was with 50%-80% ACN in water. The flow rate was 0.8 mL/min, and injection volume was 20 μL. Aqueous concentration was determined by comparison of the peak area of the saturated solution with a standard curve plotted peak area versus known concentrations, which were prepared by solutions of test compound in ACN at 50,12.5, 3.13, 0.78, and 0. 20 μg/mL.
5.5. Log P Measurement
One to two mg of test compound was dissolved in 1.0−2.0 mL of n-octane to obtain a 1.0 mg/mL solution. Next, the same volume of water as n-octane was added to each vial. The mixture was stirred at rt for 24 h, and then was left without stirring overnight. The aqueous and organic phases of each mixture were transferred to separate vials for HPLC analysis. The instrument and conditions were the same as those for water solubility determinations. The log P was calculated by the peak area ratio in n-octane and in water.
5.6. Microsomal Stability Assay
Stock solutions of test compounds (1 mg/mL) were prepared by dissolving the pure compound in DMSO, and the solutions were stored at 4 °C. Before assay, the stock solution was diluted with ACN to 0.1 mM. For measurement of metabolic stability, all compounds were brought to a final concentration of 1 μM with 0.1 M potassium phosphate buffer at pH 7.4, which contained 0.1 mg/mL human liver microsomes and 5 mM MgCl2. The incubation volumes were 300 μL, and reaction temperature was 37 °C. Reactions were started by adding 60 μL of NADPH (final concentration, 1.0 mM) and quenched by adding 600 μL of ice-cold ACN to stop the reaction at 5, 15, 30, and 60 min time points. Samples at the 0 min time point were prepared by adding 600 μL of ice-cold ACN first, followed by 60 μL of NADPH. All samples were prepared in duplicate. After quenching, all samples were centrifuged at 12000 rpm for 5 min at 0 °C. The supernatant was collected, and 20 μL of the supernatant was directly injected into a Shimadzu LC−MS 2010 system with an electrospray ionization source for further analysis. The following controls were also used: (1) positive control incubation containing liver microsomes, NADPH, reference compound propranolol or terfenadine; (2) negative control incubation omitting NADPH; (3) baseline control containing only liver microsomes and NADPH. The peak heights of test compounds at different time points were converted to percentage of compound remaining, and the peak height values at initial time (0 min) served as 100% values. The slope of the linear regression from log percentage remaining versus incubation time relationships (−k) was used to calculate the in vitro half-life (t1/2) by the formula of in vitro t1/2 = 0.693/k, regarded as first-order kinetics. Conversion to in vitro CLint (in units of mL/min/mg of protein) was calculated by the formula CLint = [0.693/(in vitro t1/2)][(mL incubation)/(mg of microsomes)]. The HPLC−MS analysis was carried out on a Shimadzu LC−MS 2010 with an electrospray ionization source. An Alltima C18 column (5 μm, 150 mm × 2.1 mm) was used for HPLC with a gradient elution at a flow rate of 0.2 mL/min. The elution condition was ACN (B) in water (A) at 30% for 0−2 min, 85% for 2−6 min, 100% for 6−9 min and 30% for 9−12 min. The MS conditions were optimized to a detector voltage of +1.7 kV, with acquisition mode selected ion monitoring of the appropriate molecular weights of the test compounds. The curved desolvation line temperature was 250 °C, heat block temperature was 200 °C, and neutralizing gas flow was 1.5 L/min. Samples were injected by an autosampler. Electrospray ionization was operated in positive and negative modes.
5.7. Molecular Modeling Studies
All molecular modeling studies were performed with Discovery Studio 3.0 (Accelrys, San Diego, USA). The crystal structures of tubulin in complex with DAMA-colchicine (PDB: 1SA0) and with TN16 (PDB: 3HKD) were downloaded from the RCSB Protein Data Bank (http://www.rcsb.org/pdb) for possible use in the modeling study. We selected the structure 1SA0 as our modeling system. CDOCKER was used to evaluate and predict in silico binding free energy of the inhibitors and automated docking. The protein protocol was prepared by several operations, including standardization of atom names, insertion of missing atoms in residues and removal of alternate conformations, insertion of missing loop regions based on SEQRES data, optimization of short and medium size loop regions with Looper Algorithm, minimization of remaining loop regions, calculation of pK, and protonation of the structure. The receptor model was typed with the CHARMm force field. A binding sphere with radius of 8.5 Å was defined through the original ligand (DAMA-colchicine) as the binding site for the study. The docking protocol employed total ligand flexibility, and the final ligand conformations were determined by the simulated annealing molecular dynamics search method set to a variable number of trial runs. The docked ligands (4i, 5f, and 6d) were further refined using in situ ligand minimization with the Smart Minimizer algorithm by standard parameters. The ligand and its surrounding residues within the above defined sphere were allowed to move freely during the minimization, while the outer atoms were frozen. The implicit solvent model of Generalized Born with Molecular Volume (GBMV) was also used to calculate the binding energies.
Supplementary Material
HIGHLIGHTS.
New N-aryl-6-methoxy-1,2,3,4-tetrahydroquinolines inhibited tubulin polymerization. Compound 6d significantly inhibited colchicine binding (99%, 5 μM; 95%, 1 μM). Compound 6d was more potent than anticancer drug paclitaxel (GI50 1.5–1.7 nM). A fused aromatic ring, such as quinoline and quinazoline, improved molecular potency A feasible torsional angle between the two aryl rings is important for high potency.
Acknowledgements
This investigation was supported by Grants 81120108022 and 30930106 from the Natural Science Foundation of China (NSFC) awarded to Dr. Lan Xie and NIH Grant CA17625-32 from the National Cancer Institute awarded to Dr. K. H. Lee. This study was also supported in part by the Taiwan Department of Health, China Medical University Hospital Cancer Research Center of Excellence (DOH100-TD-C-111-005).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer. 2004;4:253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 2.Dumontet C, Jordan MA. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010;9:790–803. doi: 10.1038/nrd3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sengupta S, Thomas SA. Drug target interaction of tubulin-binding drugs in cancer therapy. Expert Rev. Anticancer Ther. 2006;6:1433–1447. doi: 10.1586/14737140.6.10.1433. [DOI] [PubMed] [Google Scholar]
- 4.Siemann DW. The unique characteristics of tumor vasculature and preclinical evidence for its selective disruption by tumor-vascular disrupting agents. Cancer Treat. Rev. 2011;37:63–74. doi: 10.1016/j.ctrv.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mason RP, Zhao D, Liu L, Trawick L, Pinney KG. A perspective on vascular disrupting agents that interact with tubulin: preclinical tumor imaging and biological assessment. Integr. Biol. 2011;3:375–387. doi: 10.1039/c0ib00135j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang XF, Tian XT, Ohkoshi E, Qin BJ, Liu YN, Wu PC, Hung HY, Hour MJ, Qian K, Huang R, Bastow KF, Janzen WP, Jin J, Morris-Natschke SL, Lee KH, Xie L. Design and synthesis of diarylamines and diarylethers as cytotoxic antitumor agents. Bioorg. Med. Chem. Lett. 2012;22:6224–6228. doi: 10.1016/j.bmcl.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 7.Wang XF, Ohkoshi E, Wang SB, Hamel E, Bastow KF, Morris-Natschke SL, Lee KH, Xie L. Synthesis and biological evaluation of N-alkyl-N-(4-methoxyphenyl)pyridin-2- amines as a new class of tubulin polymerization inhibitors. Bioorg. Med. Chem. 2013;21:632–642. doi: 10.1016/j.bmc.2012.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blagg J. Structure-activity relationships for in vitro and in vivo toxicity. In: Anthony W, editor. Annu. Rep. Med. Chem. Vol. 41. Academic Press; 2006. pp. 353–368. [Google Scholar]
- 9.Kerns EH, Di L. In drug-like properties: concepts, structure design and methods. Academic Press; San Diego: 2008. Metabolic stability; p. 151. [Google Scholar]
- 10.Mei X, August AT, Wolf C. Regioselective copper-catalyzed amination of chlorobenzoic acids: synthesis and solid-state structures of N-aryl anthranilic acid derivatives. J. Org. Chem. 2006;71:142–149. doi: 10.1021/jo0518809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jensen TA, Liang X, Tanner D, Skjaerbaek N. Rapid and efficient microwave-assisted synthesis of aryl aminobenzophenones using Pd-catalyzed amination. J. Org. Chem. 2004;69:4936–4947. doi: 10.1021/jo049572i. [DOI] [PubMed] [Google Scholar]
- 12.Onnis V, Cocco MT, Lilliu V, Congiu C. Synthesis and evaluation of antitumor activity of ester and amide derivatives of 2-arylamino-6-trifluoromethyl-3-pyridinecarboxylic acids. Bioorg. Med. Chem. 2008;16:2367–2378. doi: 10.1016/j.bmc.2007.11.069. [DOI] [PubMed] [Google Scholar]
- 13.Nose A, Kudo T. Reduction of heterocyclic compounds. II. Reducation of heterocyclic compounds with sodium borohydride-transition metal salt systems. Chem. Pharm. Bull. 1984;32:2421–2425. [Google Scholar]
- 14.Perez-Sayans M, Somoza-Martin JM, Barros-Angueira F, Diz PG, Rey JM, Garcia-Garcia A. Multidrug resistance in oral squamous cell carcinoma: The role of vacuolar ATPases. Cancer Lett. 2010;295:135–143. doi: 10.1016/j.canlet.2010.03.019. [DOI] [PubMed] [Google Scholar]
- 15.Hung HY, Ohkoshi E, Goto M, Bastow KF, Nakagawa-Goto K, Lee KH. Antitumor agents. 293. Nontoxic dimethyl-4,4’-dimethoxy-5,6,5’,6’-dimethylenedioxylbiphenyl-2,2’- dicarboxylate (DDB) analogues chemosensitize multidrug-resistant cancer cells to clinical anticancer drugs. J. Med. Chem. 2012;55:5413–5424. doi: 10.1021/jm300378k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rubinstein LV, Shoemaker RH, Paull RM, Tosini S, Skehan P, Scudiero DA, Mpnks A, Boyd MR. Comparison of in vitro anticancer-drug-screening data generated with a tetrazolium assay versus a protein assay against a diverse panel of human tumor cell lines. J. Natl. Cancer Inst. 1990;82:1113–1117. doi: 10.1093/jnci/82.13.1113. [DOI] [PubMed] [Google Scholar]
- 17.Ravelli RBG, Gigant B, Curmi PA, Jourdain I, Lachkar S, Sobel A, Knossow M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature. 2004;428:198–202. doi: 10.1038/nature02393. [DOI] [PubMed] [Google Scholar]
- 18.Dorleans A, Gigant B, Ravelli RBG, Mailliet P, Mikol V, Knossow M. Variations in the colchicine-binding domain provide insight into the structural switch of tubulin. PNAS. 2009;106:13775–13779. doi: 10.1073/pnas.0904223106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gangjee A, Zhao Y, Lin L, Raghavan S, Roberts EG, Risinger AL, Hamel E, Mooberry SL. Synthesis and discovery of water-soluble microtubule targeting agents that binding to the colchicine site on tubulin and circumvent Pgp medicated resistance. J. Med. Chem. 2010;53:8116–8128. doi: 10.1021/jm101010n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun LQ, Zhu L, Qian K, Qin B, Huang L, Chen CH, Lee KH, Xie L. Design, synthesis, and preclinical evaluations of novel 4-substituted 1,5-diarylanilines as potent HIV-1 non-nucleoside reverse transcriptase inhibitor (NNRTI) drug candidates. J. Med. Chem. 2012;55:7219–7229. doi: 10.1021/jm3007678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cai SX. Small molecule vascular disrupting agents: potential new drugs for cancer treatment. Anti-Cancer Drugs. 2007;2:79–101. doi: 10.2174/157489207779561462. [DOI] [PubMed] [Google Scholar]
- 22.Boyd MR. Status of the NCI preclinical antitumor drug discovery screen. In: Devita VT, Hellman S, Rosenberg SA, editors. Cancer: Principles and Practice of Oncology Updates. J.d B. Lippincott; Philadelphia: 1989. pp. 1–12. [Google Scholar]
- 23.Monks A, Scudiero D, Skehan P, Shoemaker R, Paull K, Vistica D, Hose C, Langley J, Cronise P, Vaigro-Woiff A, Gray-Goodrich M, Campbell H, Mayo J, Boyd M. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J. Natl. Cancer Inst. 1991;83:757–766. doi: 10.1093/jnci/83.11.757. [DOI] [PubMed] [Google Scholar]
- 24.Houghton P, Fang R, Techatanawat I, Steventon G, Hylands PJ, Lee CC. The sulphorhodamine (SRB) assay and other approaches to testing plant extracts and derived compounds for activities related to reputed anticancer activity. Methods. 2007;42:377–387. doi: 10.1016/j.ymeth.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 25.Hamel E. Evaluation of antimitotic agents by quantitative comparisons of their effects on the polymerization of purified tubulin. Cell Biochem. Biophys. 2003;38:1–22. doi: 10.1385/CBB:38:1:1. [DOI] [PubMed] [Google Scholar]
- 26.Lin CM, Ho HH, Pettit GR, Hamel E. Antimitotic natural products combretastatin A-4 and combretastatin A-2: studies on the mechanism of their inhibition of the binding of colchicine to tubulin. Biochemistry. 1989;28:6984–6991. doi: 10.1021/bi00443a031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.