

Abstract
As part of a continuous effort to develop efficient countermeasures against sulfur mustard injuries, several unique NSAID prodrugs have been developed and screened for anti-inflammatory properties. Presented herein are three classes of prodrugs which dually target inflammation and cholinergic dysfunction. Compounds 1–28 contain common NSAIDs linked either to choline bioisosteres or to structural analogs of acetylcholinesterase (AChE) inhibitors. These agents have shown utility as anti-vesicants and anti-inflammatory agents when screened in a mouse ear vesicant model (MEVM) against both 2-chloroethyl ethyl sulfide (CEES), a blistering agent, and 12-O-tetradecanoylphorbol-13-acetate (TPA), a common topical irritant. Many of the prodrugs have activity against CEES, with 5, 18, 22 and 27 reducing inflammation by more than 75 % compared to a control. Compounds 12, 13, 15 and 22 show comparable activity against TPA. Promising activity in the MEVM is related to half-lives of NSAID-release in plasma, moderate to high lipophilicity, and some degree of inhibition of AChE, a potential contributor to sulfur mustard-mediated tissue damage.
Introduction
Sulfur mustard (2,2′-dichloroethyl sulfide, SM) is a powerful chemical vesicant first used in World War I. In the 1980s, SM use resulted in an estimated 100,000 casualties during the Iraq-Iran conflict (Kehe and Szinicz, 2005). Known for its onion/garlic smell, SM primarily affects the skin, eyes, mucous membranes, and airways. Severe blistering from SM often results in weeks or months of hospital treatment and chronic symptoms such as bronchitis, pulmonary fibrosis, eye lesions, and hypo- and hyperpigmentation can occur (Kehe and Szinicz, 2005).
Tissue damage and other pathological effects of mustard poisoning are often observed after a latency period of 2 to 24 hours, depending on the degree of exposure. One of the most widely accepted theories is that SM-mediated DNA-alkylation initiates toxicity (Papirmeister et al., 1985 and Berger et al., 1986). However, SM can also alkylate other nucleophile-containing macromolecules, including RNA, membrane lipids, and proteins (Papirmeister et al., 1985). These modifications can result in the release of proteases, which attack and degrade major structural proteins including collagen and proteoglycans. Activation of proteolytic enzymes has been linked to the vesication following SM-exposure (Kam et al., 1997 and Powers et al., 2000). Activation of proteases can also initiate and promote inflammation, a well characterized response to SM-induced vesication (Cowan et al., 2003 and Rikimaru et al., 1991).
Although questions remain as to the specific role of inflammation in mustard pathogenesis, anti-inflammatory agents such as non-steroidal anti-inflammatory drugs (NSAIDs) have been extensively investigated as potential mustard treatments. Topical and systemic administration of anti-inflammatory drugs such as indomethacin, olvanil, and hydrocortisone have shown a protective effect against sulfur mustard lesions when screened in a mouse ear vesicant model (MEVM) (Casillas et al., 2000 and Babin et al., 2000). Mixtures of the steroid Adexone and the topical NSAID formulation Voltaren (diclofenac) showed promising results by reducing SM-induced edema by 60–70 % in the MEVM and seemed to promote faster healing when administered post-exposure (Dachir et al., 2004).
Recent evidence suggests that non-neuronal cholinergic activity also plays a role in regulating skin function (Kurzen et al., 2007). The neurotransmitter acetylcholine is produced by keratinocytes and infiltrating leukocytes during inflammation and may regulate epidermal cell proliferation and differentiation (Grando et al., 1997). Up-regulation of choline and use of cholinergic agonists such as inhibitors of acetylcholinesterase (AChE), an enzyme that degrades acetylcholine, have been shown to suppress proinflammatory cytokine release (Saeed et al., 2002 and Pavlov et al., 2009). Steinritz and co-workers (2007) also observed elevated AChE levels during the apoptotic pathway of SM-exposed pulmonary cells. Although AChE levels have not been directly correlated with in vivo cutaneous SM injury, the enzyme plays a crucial and often overlooked role in systemic inflammation (Tracey, 2007). The anti-Alzheimer’s drug and potent AChE inhibitor (AChEI), galantamine, when administered peripherally, significantly reduces circulating levels of various pro-inflammatory cytokines (Pavlov et al., 2009). In this regard, Amitai et al. (2006) demonstrated that an NSAID linked via a hydrocarbon chain to an AChEI provides more potent anti-inflammatory and anti-vesicant effects against SM injury than commercial NSAID preparations. The advantage of using such a drug is to target multiple mechanisms of dermal SM lesions by regulating acetylcholine levels and gradually inhibiting cyclooxygenases (COX).
Our group has previously reported the synthesis and biochemical evaluation of two classes of anticholinergic NSAID prodrugs (Young et al., 2010 and Laskin et al., 2010). The prodrugs contain NSAIDs such as ibuprofen, naproxen, indomethacin, and diclofenac linked to choline mimics either directly or indirectly via a hydrophobic linker. An additional class is presented herein containing NSAIDs conjugated directly via ester linkages to known AChEIs such as galantamine. Partially modified neostigmine and pyridostigmine analogs were employed when a direct combination was not possible without structural modification. Considering the role of inflammation and enhanced AChE levels in SM pathogenesis, the use of these novel molecules as anti-vesicants and anti-inflammatory agents was explored by screening them in a mouse ear inflammation model against both 2-chloroethyl ethyl sulfide (CEES), a blistering agent often used as a model for SM skin injury (Tewari-Singh et al., 2009), and 12-O-tetradecanoylphorbol-13-acetate (TPA), a common topical irritant (Akihisa et al., 2010 and Yasukawa et al., 2010). The results reported herein include in vitro and in vivo data on our previously synthesized NSAID derivatives as well as on the aforementioned class of novel compounds.
Materials and Methods
Synthesis
Generic structures of bifunctionals 1–28 are shown in Figure 1. Class 1 and 2 compounds 1–19 were synthesized via previously reported methods (Young et al., 2010). Class 3 compounds 20–28 were easily accessible using the same coupling method as that described previously for Class 2 (Young et al., 2010). Direct coupling of the NSAID to an AChEI or a derivative thereof using 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC·HCl) and 4-dimethylaminopyridine (DMAP) was employed. For galantamine esters 21, 24, and 27, the free base alkaloid was coupled to each NSAID to form the final products in moderate to high yields (81–95 %). In the same manner, the pyridostigmine conjugates were formed by EDC·HCl/DMAP coupling of commercially available 3-hydroxypyridine to the desired NSAID followed by quaternization using excess methyl iodide. The neostigmine conjugates (22, 25, and 28) were also accessed by EDC·HCl/DMAP coupling of an NSAID to 3-(dimethylamino)phenol, which was synthesized using the method of Grove et al. (2002), followed by quaternization.
Figure 1.
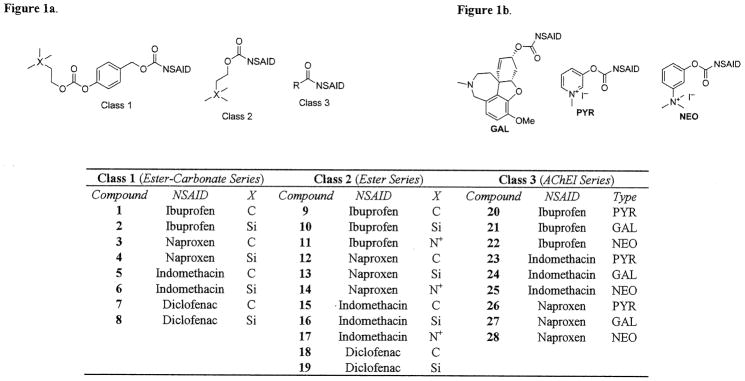
Figure 1a. Generic Structures of Anticholinergic NSAID Prodrugs.
Figure 1b. Structures of Types of Class 3 Prodrugs.
Detailed physical characterization and synthetic procedures for compounds 1 through 19 have been published previously (Young et al., 2010). Synthesis and physical characterization of novel compounds 20–28 are included under Supplementary data.
Assay of Acetylcholinesterase Inhibition
Acetylcholinesterase (Type V-S from electrophorus electricus), acetylthiocholine iodide (ATChI), 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) and tacrine hydrochloride were obtained from Sigma-Aldrich (Saint Louis, MO). Methanol was obtained from EMD Chemicals (Gibbstown, NJ). Cholinesterase inhibition was assayed spectrophotometrically at 412 nm according to Ellman’s method (Ellman, 1959). Assays were performed in polystyrene 96-well plates (Corning 96-well flat transparent) and a conventional micro-plate reader was employed for kinetic readings (Tecan’s Infinite 200 multimode). The following reagents were added to the wells: 200 μL of 0.5 mM DTNB in sodium phosphate buffer (100 mM, pH 8), 30 μL of inhibitor stock solution in methanol, 20 μL of 1.25 units/mL of AChE in sodium phosphate buffer (20 mM, pH 7), and 50 μL of 3 mM ATChI in buffer (100 mM, pH 8). Immediately after the substrate was added, the absorption signal was measured at 30 s intervals over 5 min at 25 °C. Percentage inhibition was calculated relative to a control (methanol). The background signal was measured in control wells containing every reagent except for the substrate. IC50 values were obtained from a minimum of eight concentrations in duplicate and by fitting the experimental data with a dose-response curve using Prism software (Version 5.00, GraphPad Software, San Diego, CA).
Plasma Hydrolysis
The rates of hydrolysis of prodrugs 20, 23–25, and 27 were determined at 37 °C in fresh human plasma (80 % plasma in PBS, pH 7.4). Human plasma was obtained from the pooled, heparinized blood of healthy donors and was stored at −80 °C prior to use. Prodrugs (20 μL of a 6 mM solution in DMSO) were added to pre-heated plasma (980 μL) and mixed gently (300 rpm) at 37 °C. Solutions of the same concentration of inhibitor in PBS:DMSO (98:2) were kept as a control for non-enzymatic hydrolysis. At suitable intervals, aliquots of 100 μL were withdrawn and 200 μL of cold precipitation buffer (90/10 acetonitrile/water with 0.1 % formic acid) were added to precipitate proteins. The resulting mixture was filtered through a Mini-Uniprep™ filter (Whatman, PVDF membrane, 0.45 μm) and the filtrate was analyzed by HPLC (vide infra). The remaining prodrug and NSAID released were monitored by single determination at 230, 277 or 289 nm. HPLC method - Agilent high resolution XDB-C18 column (1.8 μm, 4.6 × 50mm); mobile phase: water (A) and methanol (B), both containing 0.1 % formic acid; flow rate: 0.8 mL/min; gradient increase from A/B:85/15 to 15/85 over 8 min, 15/85 to 10/90 over 4 min, 10/90 to 5/95 over 4 min, 5/95 to 85/15 over 4 min, post-time 5 min; injection volume: 30 μL.
Animals
Animal studies were approved by the Rutgers University Institutional Animal Care and Use Committee and received human care in compliance with the institution’s guidelines, as outlined in the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences.
Mouse Ear Vesicant Model
Compounds were assessed as inhibitors of inflammation using the MEVM as previously described (Casillas et al., 2000), except that female CD-1 mice (4–6 weeks old) were used. Either CEES (65 μmoles) or TPA (1.5 nmol) was used to induce inflammation. To evaluate each compound, ears (3–4 mice per group) were treated with 20 μL of vehicle control (methylene chloride or acetone) or the test compound (1.5 μmol) in 20 μL of the appropriate vehicle. After 5 h, mice were euthanized and ear punches (6 mm in diameter) were taken and weighed. Once the raw data were obtained, masses of ear punches were averaged and the percent reduction of vesicant-induced edema and inflammation was calculated using a previously described method (Casillas et al., 2000). Data were analyzed using a one-way ANOVA to evaluate statistical significance (P < 0.05). One of the compounds with a high degree of activity against CEES (5) was then dosed at different levels (from 0.25 to 1.5 μmol/ear) to determine the effective dosage.
Results
Plasma Hydrolysis
In our previously described anticholinergic NSAID class (1–19), the parent NSAIDs were released at the same rate as the rate of hydrolysis of the prodrug (t1/2 of 5–458 min) (Young et al., 2010). Direct combination of the two therapeutic components via an ester linkage led to faster NSAID release in Class 2 agents (5–111 min) compared to Class 1. Both the neostigmine- and pyridostigmine-like conjugates released the parent NSAIDs with half-lives of less than 5 min. Galantamine esters 24 and 27 showed exceptionally long half-lives with no significant hydrolysis over 12 h (Table 1).
Table 1.
In vitro data for novel NSAID-AChEI Conjugates.*
| Compound # | IC50 (μM) | Plasma t1/2 (min−1) | Clog P** |
|---|---|---|---|
| 20 | 77.3 ± 9.43 | <5 | −0.09 |
| 21 | 40.4 ± 3.91 | nd | 5.87 |
| 22 | 5–15*** | nd | 1.37 |
| 23 | 51.8 ±16.9 | <5 | 0.41 |
| 24 | NA | >12 h | 6.38 |
| 25 | 10.6 ±2.2 | <5 | 1.87 |
| 26 | NA | nd | −0.95 |
| 27 | 35–40*** | >12 h | 5.01 |
| 28 | nd*** | nd | 0.50 |
| Galantamine HBr | 1.12±0.31 | −0.33 | |
| Pyridostigmine Mimic (Fig. 2) | NA | −3.49 | |
| Neostigmine Mimic (Fig. 2) | 4.75 ±0.41 | −2.76 |
NA – not active; nd – not determined.
Clog P calculated using ChemBioDraw Ultra 12.0.
Precise IC50 values could not be obtained due to limits in compound solubility.
Assay of Acetylcholinesterase Inhibition
Activities of Class 1 and 2 compounds against AChE have been presented previously and will be discussed only briefly (Young et al., 2010). The IC50 values of compounds 1–8 range from 0.51 to 2.29 μM, with 7 showing the highest degree of inhibition. Class 2 compounds (9–19) are at least five-fold less potent than their Class 1 counterparts, with IC50s ranging from 2.66 μM to 2 mM. Anticholinesterase activities of novel compounds 20–28 are presented in Table 1. As a control, the original or modified AChEIs were screened for activity and compared to the corresponding NSAID conjugates. For the pyridostigmine-like molecules (20, 23, and 26), 23 showed the most potent yet modest inhibitory activity, while all other agents, including the pyridostigmine mimic itself (Figure 2), showed little to no activity. Of the galantamine ester series, 21 and 27 were moderately active in the range of 35–40 μM, while indomethacin conjugate 24 showed no in vitro inhibition. Finally, within the neostigmine class, 22 and 25 showed good activity with IC50s ranging from 5–15 μM. In most cases, only a modest decrease in activity was observed as a result of linking the AChEIs or analogs thereof to NSAIDs.
Figure 2.
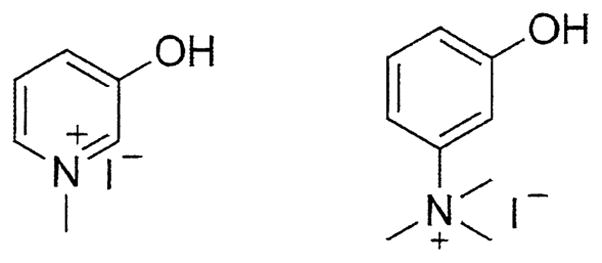
Structures of Pyridostigmine Mimic (left) and Neostigmine Mimic (right).
Mouse Ear Vesicant Model (CEES)
A MEVM was used as a screen to select effective anti-vesicants for further study. Listed in Table 2 are percentages of inhibition of edema and inflammation relative to a positive control (exposure to CEES only) for candidate compounds. Many of the novel agents reduced vesication between 60 and 80 %, values greater than those of the parent NSAIDs alone. It should be noted that ibuprofen, when screened against CEES, enhanced inflammation and edema by 15 %. Interestingly, most of the ibuprofen prodrugs in these sets were active in the MEVM from 20–76 %. Furthermore, diclofenac suppressed CEES-induced inflammation at a mere 17 % while prodrugs 7 and 18 showed activity to a much greater extent (90 and 113%, respectively).
Table 2.
% Reduction of Edema and Inflammation Following Exposure to CEES or TPA.*
| Class 1 (Bifunctionals) | Class 2 (Simple Esters) | Class 3 (AChEI Conjugates) | ||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Cmpd # | % Red. (CEES) | % Red. (TPA) | Cmpd # | % Red. (CEES) | % Red. (TPA) | Cmpd # | % Red. (CEES) | % Red. (TPA) |
| 1 | 20 | 41** | 9 | −35 | 6 | 20 | 60** | 80*** |
| 2 | 68 | 17 | 11 | 38 | 73*** | 22 | 76*** | 57** |
| 4 | 45 | 70** | 12 | NA | 94** | 24 | 75** | nd |
| 5 | 91** | 21** | 13 | 60** | 111*** | 27 | 86** | nd |
| 6 | 62 | 73*** | 15 | NA | 76** | |||
|
|
||||||||
| 7 | 90** | 24 | 16 | 38 | 47*** | NSAID Standards | ||
|
|
||||||||
| 8 | 24 | 31** | 18 | 113** | 29** | Diclofenac | 17 | 58*** |
| Indomethacin | 46** | 55** | ||||||
| Ibuprofen | −15 | −33 | ||||||
| Naproxen | NS | 104** | ||||||
NS – reduction not statistically significant; nd – not determined; NA – not active.
Value differs from positive control based on one-way ANOVA, with P < 0.05.
Value differs from positive control based on one-way ANOVA, with P < 0.005.
Significant levels of inflammation suppression were observed for several compound of Class 1, the prodrugs containing the hydrophobic linker. In general, the simple esters were found less active than their Class 1 counterparts except for the high level of activity observed with 18 (113 %). Prodrugs 5 and 7 exhibited the most potent effect against CEES in their class (91 and 90 %, respectively). Prodrugs 5 and 18 are also potent reversible inhibitors of AChE (< 3 μM). Very high levels of suppression were also observed with the combinations of NSAIDs with AChE inhibitors or mimic inhibitors. Galantamine derivatives 24 and 27 exhibited significant inhibition of vesication, with levels exceeding 70 %.
We have observed that prodrug lipophilicity also affects activity in the MEVM screen. Out of the three sets, the bifunctional ester-carbonates containing the alkyl-aryl linker are the most lipophilic by an average of one log unit compared to the simple esters in Class 2 (Young et al., 2010) and approximately five log units compared to the AChE inhibitor conjugates in Class 3 (Table 1). A general trend exists in that derivatives of the lipophilic NSAIDs (indomethacin, diclofenac, and ibuprofen) show a more pronounced effect against CEES than the more polar naproxen conjugates.
Mouse Ear Inflammation Model (TPA)
Presented in Table 2 are percent reductions of edema and inflammation relative to a positive control (exposure to TPA only). Several novel agents reduced TPA-induced inflammation by more than 70 %. Overall, compounds from Class 2 were more potent against TPA. Compounds 12, 13, and 20 had the highest degree of an anti-inflammatory effect.
As with the CEES data, a slight dependence on lipophilicity was observed in the TPA results. In this case, however, the slightly more hydrophilic esters of Classes 2 and 3 were more active against TPA than CEES. This trend is especially evident when comparing the activity of 11, 12, and 15 in the TPA versus CEES models. These agents were either inactive or had moderate activity against CEES, yet were especially effective against TPA (60–75 %).
Dose-Response of 5
To further investigate the dosage needed for optimal activity, lead 5 was tested against CEES and TPA at varying concentrations (Table 3). The minimum amount of drug needed for the desired anti-CEES effect is below the level used for screening (1.5 μmol/ear). Significant inflammation and edema suppression (up to 100 % compared to the positive control) were observed at doses between 0.5 and 0.75 μmol/ear. A similar range of doses applied after TPA-exposure did not result in a significant degree of inflammation suppression (24 % or less, Table 3).
Table 3.
Dose-response of indomethacin prodrug 5.
| Dose of 5 (μmol/ear) | % Reduction (CEES) | % Reduction (TPA) |
|---|---|---|
| 0.25 | 34* | 9* |
| 0.50 | 31* | 24** |
| 0.75 | 100** | nd |
| 1.0 | 96** | 12* |
| 1.5 | 91** | nd |
Reduction not statistically significant.
Value differs from positive control based on one-way ANOVA, with P < 0.05.
Discussion
The search for effective mustard therapeutics is moving towards the development of multifunctional drugs to target the various pathways of SM toxicity. Dachir et al. (2004) mechanically combined a steroid with an NSAID to optimize anti-inflammatory activity and deplete tissues of prostaglandin E. In this study, the combined drugs were more effective than either drug alone. Similarly, Wormser and co-workers (2000 and 2004) combined topical iodine with steroidal and nonsteroidal anti-inflammatory drugs to suppress SM-induced necrosis and hemorrhage while enhancing biomarkers of healing. Little work has been done to explore controlled-release SM therapeutics containing individual drugs such as anti-inflammatory agents and anticholinergics covalently linked in one molecule. An earlier study reported that conjugates formed by a covalent linkage of pyridostigmine with an NSAID via a decyl hydrocarbon spacer were as active as commercial NSAID preparations in ameliorating SM-induced toxicity in a mouse ear model (Amitai et al., 2006). The concept of linking an AChE inhibitor to common NSAIDs was innovative and effective; however, no in vitro or in vivo release of the individual pharmacologic components was shown. In the present work, integration of NSAIDs and anticholinergics into a novel scaffold to facilitate gradual drug-release has allowed us to target multiple mechanisms of SM-induced injury in a way that is unique to the field and will expand the current library of anti-mustard candidates.
A great deal of work has gone into the exploration of anticholinergics as potential anti-inflammatory agents. For example, the anticholinergic, galantamine, when administered peripherally, ameliorates systemic inflammation (Pavlov et al., 2009). Furthermore, maintaining cholinergic balance with these drugs has been reported to alleviate severe inflammation from sepsis, endotoxemia, and arthritis (Hofer et al., 2008, Pavlov et al., 2009, and van Maanen et al., 2009). Regarding the specific effects that SM has on AChE levels, enzyme synthesis has been reported to be increased in neuroblastoma cells following a SM treatment (Lanks et al., 1975). Steinritz and co-workers (2007) also observed elevated AChE levels during apoptosis of SM-exposed pulmonary epithelial cells. Our compounds were synthesized based on the emerging role of AChE in inflammation and the work of Amitai et al. (2006) demonstrating their potential as anti-mustard agents.
In our MEVM screen, high levels of activity against CEES were observed for several of our Class 1 and 2 bifunctionals containing lipophilic choline analogs linked to NSAIDs (Table 2). Several of the most promising prodrugs reduced inflammation and edema up to and greater than 90 % (compounds 5, 7, and 18). The anti-inflammatory effects observed were directly associated with potent inhibition of AChE, high lipophilicity, and long half-lives in plasma. We have shown previously that all compounds in these classes release their parent NSAIDs in plasma (Young et al., 2010). It is not yet established whether the therapeutic effect observed is from the intact compounds or from the in vivo release of both the NSAID and the anticholinergic.
Class 3 compounds structurally designed around potent AChE inhibitors like galantamine, neostigmine, and pyridostigmine also showed very promising results against CEES. Significant suppression of CEES-induced inflammation was observed, with levels exceeding 75 % reduction in some cases (compounds 22 and 27). Most of these compounds are potent anticholinergics and/or release an NSAID in plasma. The stability of Class 3 compounds in human plasma, however, could limit the in vivo application of these agents as prodrugs. In particular, the rapid release of the NSAID (t1/2 < 5 min) for some agents could be detrimental. Galantamine derivatives showed exceptionally long biological half-lives for enzymatic release of the NSAID and the AChEI. Further in vivo investigation is needed to determine the rate of release of the parent drugs following transdermal absorption.
To study the biological activity of the selected agents in-depth, the inhibitory activity against butyrylcholinesterase (BuChE) was also determined. BuChE is a less selective esterase that is highly homologous with AChE and is found principally in the circulation (Glick et al., 2003). Inhibitory activity against equine serum BuChE (eBuChE) was observed for our compounds with IC50s ranging from 3.8–55 μM and potencies lower than those observed for AChE (Young et al., unpublished work, 2010). Thus, our compounds are able to inhibit both AChE and BuChE, paralleling the known cholinesterase inhibitors, tacrine and galantamine (de los Ríos et al., 2010). Existing literature does not suggest that attenuation of BuChE levels will have an effect on SM dermal injury.
Several compounds of the set were also screened in the mouse ear model against the phorbol ester TPA, a common proinflammatory agent used in this case to assay the dual-action prodrugs for a wider range of anti-inflammatory properties (Akihisa et al., 2010 and Yasukawa et al., 2010). Many of our compounds in all three classes reduced TPA-induced inflammation and edema by at least 50–75 %. The potent anti-TPA effects of the simple esters suggest that activity is linked to both the rate of NSAID release and skin penetrability. It is important to note that both anti-CEES and anti-TPA activity likely depend upon both the parent NSAIDs and the prodrugs themselves as previous results have shown that NSAIDs have different rates of transdermal absorption (Singh et al., 1994 and Beetge et al., 2000).
In general, the simple ester series was found to be more active against TPA while Class 1 agents were more effective against CEES. TPA is a highly potent inflammatory agent which activates protein kinase C and induces the release of histamine, proteases, and proinflammatory mediators (Nakadate, 1989 and Goel et al., 2007). The rapid rate of NSAID release may explain the effectiveness that Class 2 agents have against TPA. On the other hand, the CEES or SM-induced blister is a slow-developing lesion resulting from DNA-alkylation, protease activation and an inflammatory response (Cowan et al., 2003). Considering the gradual development of SM lesions (Kehe and Szinicz, 2005), Class 1 agents seem more appropriate as SM therapeutics because of their slow-release of the parent NSAID and high lipophilicity allowing enhanced skin penetration.
Directly preventing SM-induced DNA modifications has been used as a strategy for decontaminating the skin following exposure to vesicants. Sulfur species such as sodium thiosulfate are commonly explored as decontaminats (Baskin et al., 2000). It is proposed that these SM scavengers intervene in macromolecular-alkylation either by interacting with nucleic acids or by directly reacting with the highly electrophilic sulfonium ion formed by SM (Baskin et al., 2000). Therapeutic intervention of DNA-alkylation has not been explored likely because SM penetrates the skin within 30–60 min after exposure (Wormser et al., 2002). Various groups have instead taken the approach of designing treatments which target the detrimental effects resulting from SM-induced DNA damage. For example, DNA damaging agents activate poly (ADP-ribose) polymerase (PARP) as well as proteases and inhibitors of these enzymes have been reported to reduce epidermal necrosis following SM exposure (Cowan et al., 2003). Antioxidants such as melatonin have also been explored as potential treatments because of their ability to reduce cellular damage from oxidative and nitrosative stress (Korkmaz et al., 2008, Laskin et al., 2010). Considering the structure of our novel bifunctional agents, which are not alkylating, antioxidant or intercalating agents, it is unlikely that they function by directly modulating SM-induced DNA modifications.
In summary, we have synthesized bifunctional compounds that combine an NSAID with an anticholinergic moiety. This combination results in a novel class of lipophilic NSAID prodrugs that possesses considerable anti-inflammatory activity against CEES and/or TPA in the MEVM. In most cases, the edema and inflammation suppression observed for these compounds is higher than that of the NSAIDs alone. Further studies are needed to more precisely understand their mechanism of action and to determine if they mitigate SM-induced skin injury.
Acknowledgments
This work was funded by the National Institutes of Health Center grant ES005022 and the CounterACT Program through the National Institute of Arthritis and Musculoskeletal and Skin Diseases (award #U54AR055073). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the federal government.
Footnotes
Supplementary data
Synthetic procedures and physical characterization of compounds 20–28 can be found under the Supplementary data for this journal.
References
- Akihisa T, Kojima N, Kikuchi T, Yasukawa K, Tokuda H, Masters ET, Manosroi A, Manosroi J. Anti-Inflammatory and Chemopreventive Effects of Triterpene Cinnamates and Acetates from Shea Fat. J Oleo Sci. 2010;59:273–280. doi: 10.5650/jos.59.273. [DOI] [PubMed] [Google Scholar]
- Amitai G, Adani R, Fishbein E, Meshulam H, Laish I, Dachir S. Bifunctional compounds eliciting anti-inflammatory and anti-cholinesterase activity as potential treatment of nerve and blister chemical agents poisoning. J Appl Toxicol. 2006;26:81–87. doi: 10.1002/jat.1111. [DOI] [PubMed] [Google Scholar]
- Babin M, Ricketts K, Skvorak J, Gazaway M, Mitcheltree L, Casillas R. Systemic administration of candidate antivesicants to protect against topically applied sulfur mustard in the mouse ear vesicant model (MEVM) J Appl Toxicol. 2000;20:S141–S144. doi: 10.1002/1099-1263(200012)20:1+<::aid-jat666>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Baskin SI, Prabhaharan V, Bowman JD, Novak MJ. In vitro effects of anionic sulfur compounds on the spectrophotometric properties of native DNA. J Appl Toxicol. 2000;20:S3–S5. doi: 10.1002/1099-1263(200012)20:1+<::aid-jat662>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Beetge E, du Plessis J, Müller DG, Goosen C, van Rensburg FJ. The influence of the physicochemical characteristics and pharmacokinetic properties of selected NSAID’s on their transdermal absorption. Int J Pharm. 2000;193:261–264. doi: 10.1016/s0378-5173(99)00340-3. [DOI] [PubMed] [Google Scholar]
- Berger S, Sudar D, Berger N. Metabolic consequences of DNA damage: DNA damage induces alterations in glucose metabolism by activation of poly(ADP-ribose) polymerase. Biochem Biophys Res Commun. 1986;134:227–232. doi: 10.1016/0006-291x(86)90551-6. [DOI] [PubMed] [Google Scholar]
- Casillas R, Kiser R, Truxall J, Singer A, Shumaker S, Niemuth N, Ricketts K, Mitcheltree L, Castrejon L, Blank J. Therapeutic approaches to dermatotoxicity by sulfur mustard I. Modulation of sulfur mustard-induced cutaneous injury in the mouse ear vesicant model. J Appl Toxicol. 2000;20:S145–S151. doi: 10.1002/1099-1263(200012)20:1+<::aid-jat665>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Cowan F, Broomfield C, Lenz D, Smith W. Putative role of proteolysis and inflammatory response in the toxicity of nerve and blister chemical warfare agents: implications for multi-threat medical countermeasures. J Appl Toxicol. 2003;23:177–186. doi: 10.1002/jat.901. [DOI] [PubMed] [Google Scholar]
- Dachir S, Fishbeine E, Meshulam Y, Sahar R, Chapman S, Amir A, Kadar T. Amelioration of sulfur mustard skin injury following a topical treatment with a mixture of a steroid and a NSAID. J Appl Toxicol. 2004;24:107–113. doi: 10.1002/jat.955. [DOI] [PubMed] [Google Scholar]
- de los Ríos C, Egea J, Marco-Contelles J, León R, Samadi A, Iriepa I, Moraleda I, Gálvez E, García AG, López MG, Villarroya M, Romero A. Synthesis, Inhibitory Activity of Cholinesterases, and Neuroprotective Profile of Novel 1,8-Naphthyridine Derivatives. J Med Chem. 2010;53:5129–5143. doi: 10.1021/jm901902w. [DOI] [PubMed] [Google Scholar]
- Ellman G. Tissue sulfhydryl groups. Arch Biochem Biophys. 1959;82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- Glick D, Ben Moyal L, Soreq H. Genetic variation in butyrylcholinesterase and the physiological consequences for acetylcholinesterase function. In: Giacobini E, editor. Butyrylcholinesterase: Function and Inhibition. Martin Dunitz; London: 2003. pp. 55–67. [Google Scholar]
- Goel G, Harinder PS, Makkar PS, Francis G, Becker K. Phorbol esters: structure, biological activity, and toxicity in animals. Int J Toxicol. 2007;26:279–288. doi: 10.1080/10915810701464641. [DOI] [PubMed] [Google Scholar]
- Grando S. Biological functions of keratinocyte cholinergic receptors. J Investig Dermatol Symp Proc. 1997;2:41–48. doi: 10.1038/jidsymp.1997.10. [DOI] [PubMed] [Google Scholar]
- Hofer S, Eisenbach C, Lukic I, Schneider L, Bode K, Brueckmann M, Mautner S, Wente M, Encke J, Werner J, Dalpke A, Stremmel W, Nawroth P, Martin E, Krammer P, Bierhaus A, Weigand M. Pharmacologic cholinesterase inhibition improves survival in experimental sepsis. Crit Care Med. 2008;36:404–408. doi: 10.1097/01.CCM.0B013E31816208B3. [DOI] [PubMed] [Google Scholar]
- Kam C, Selzler J, Bongiovanni R, Powers J. Enhanced Serine Protease Activities in the Sulfur Mustard-Exposed Homogenates of Hairless Guinea Pig Skin. Int J Toxicol. 1997;16:625–638. [Google Scholar]
- Kehe K, Szinicz L. Medical aspects of sulphur mustard poisoning. Toxicology. 2005;214:198–209. doi: 10.1016/j.tox.2005.06.014. [DOI] [PubMed] [Google Scholar]
- Korkmaz A, Kunak ZI, Paredes SD, Yaren H, Tan DX, Reiter RJ. The use of melatonin to combat mustard toxicity. Neuro Endocrinol Lett. 2008;29:614–619. [PubMed] [Google Scholar]
- Kurzen H, Wessler I, Kirkpatrick CJ, Kawashima K, Grando SA. The non-neuronal cholinergic system of human skin. Horm Metab Res. 2007;39:125–135. doi: 10.1055/s-2007-961816. [DOI] [PubMed] [Google Scholar]
- Lanks K, Turnbull J, Aloyo V, Dorwin A, Papirmeister B. Sulfur mustards induce neurite extension and acetylcholinesterase synthesis in cultured neuroblastoma cells. Experimental Cell Research. 1975;93:355–362. doi: 10.1016/0014-4827(75)90460-7. [DOI] [PubMed] [Google Scholar]
- Laskin JD, Fabio K, Lacey CJ, Young S, Mohanta P, Guillon C, Huang M-T, Heck DE, Heindel ND. Unique Dual-Action Therapeutics. 2010/051044. WO Patent. 2010:A1.
- Laskin JD, Black AT, Jan YH, Sinko PJ, Heindel ND, Sunil V, Heck DE, Laskin DL. Oxidants and antioxidants in sulfur mustard-induced injury. Ann N Y Acad Sci. 2010;1203:92–100. doi: 10.1111/j.1749-6632.2010.05605.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakadate T. The mechanism of skin tumor promotion caused by phorbol esters: possible involvement of arachidonic acid cascade/lipoxygenase, protein kinase C and calcium/calmodulin systems. Jpn J Pharmacol. 1989;49:1–9. doi: 10.1254/jjp.49.1. [DOI] [PubMed] [Google Scholar]
- Papirmeister B, Gross C, Meier H, Petrali J, Johnson J. Molecular basis for mustard-induced vesication. Fund Appl Toxicol. 1985;5:S134–S149. [PubMed] [Google Scholar]
- Pavlov V, Parrish W, Rosas-Ballina M, Ochani M, Puerta M, Ochani K, Chavan S, Al-Abed Y, Tracey K. Brain acetylcholinesterase activity controls systemic cytokine levels through the cholinergic anti-inflammatory pathway. Brain Behav Immun. 2009;23:41–45. doi: 10.1016/j.bbi.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers J, Kam C, Ricketts K, Casillas R. Cutaneous protease activity in the mouse ear vesicant model. J Appl Toxicol. 2000;20:S177–S182. doi: 10.1002/1099-1263(200012)20:1+<::aid-jat678>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Rikimaru T, Nakamura M, Yano T, Beck G, Habicht G, Rennie L, Widra M, Hirshman C, Boulay M, Spannhake E. Mediators, Initiating the Inflammatory Response, Released in Organ Culture by Full-Thickness Human Skin Explants Exposed to the Irritant, Sulfur Mustard. J Invest Dermatol. 1991;96:888–897. doi: 10.1111/1523-1747.ep12475292. [DOI] [PubMed] [Google Scholar]
- Saeed R, Varma S, Peng-Nemeroff P, Sherry B, Balakhaneh D, Huston J, Tracey K, Al-Abed Y, Metz C. Cholinergic stimulation blocks endothelial cell activation and leukocyte recruitment during inflammation. J Exp Med. 2002;201:1113–1123. doi: 10.1084/jem.20040463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove SJ, Kaur J, Muir A, Pow E, Tarver G, Zhang M. Oxyaniliniums as acetylcholinesterase inhibitors for the reversal of neuromuscular block. Bioorg and Med Chem Lett. 2002;12:193–196. doi: 10.1016/s0960-894x(01)00703-x. [DOI] [PubMed] [Google Scholar]
- Singh P, Roberts MS. Skin permeability and local tissue concentrations of nonsteroidal anti-inflammatory drugs after topical application. J Pharmacol Exp Ther. 1994;268:144–151. [PubMed] [Google Scholar]
- Steinritz D, Emmler J, Hintz M, Worek F, Kreppel H, Szinicz L, Kehe K. Apoptosis in sulfur mustard treated A549 cell cultures. Life Sci. 2007;80:2199–2201. doi: 10.1016/j.lfs.2006.11.052. [DOI] [PubMed] [Google Scholar]
- Tewari-Singh N, Rana S, Gu M, Pal A, Orlicky DJ, White CW, Agarwal R. Inflammatory biomarkers of sulfur mustard analog 2-chloroethyl ethyl sulfide-induced skin injury in SKH-1 hairless mice. Toxicol Sci. 2009;108:194–206. doi: 10.1093/toxsci/kfn261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracey K. Physiology and immunology of the cholinergic antiinflammatory pathway. J Clin Invest. 2007;117:289–296. doi: 10.1172/JCI30555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Maanen MA, Vervoordeldonk M, Tak P. The cholinergic anti-inflammatory pathway: towards innovative treatment of rheumatoid arthritis. Nat Rev Rheumatol. 2009;5:229–232. doi: 10.1038/nrrheum.2009.31. [DOI] [PubMed] [Google Scholar]
- Wormser U, Sintov A, Brodsky B, Nyska A. Topical iodine preparation as therapy against sulfur mustard-induced skin lesions. Toxicol Appl Pharmacol. 2000;169:33–39. doi: 10.1006/taap.2000.9056. [DOI] [PubMed] [Google Scholar]
- Wormser U, Brodsky B, Sintov A. Skin toxicokinetics of mustard gas in the guinea pig: effect of hypochlorite and safety aspects. Arch Toxicol. 2002;76:517–522. doi: 10.1007/s00204-002-0362-6. [DOI] [PubMed] [Google Scholar]
- Wormser U, Sintov A, Brodsky B, Casillas RP, Nyska A. Protective effect of topical iodine containing anti-inflammatory drugs against sulfur mustard-induced skin lesions. Arch Toxicol. 2004;78:156–166. doi: 10.1007/s00204-003-0523-2. [DOI] [PubMed] [Google Scholar]
- Yasukawa K, Matsubara H, Sano Y. Inhibitory effect of the flowers of artichoke (Cynaracardunculus) on TPA-induced inflammation and tumor promotion in two-stage carcinogenesis in mouse skin. J Nat Med. 2010;64:388–391. doi: 10.1007/s11418-010-0403-z. [DOI] [PubMed] [Google Scholar]
- Young S, Fabio K, Guillon C, Mohanta P, Halton TA, Heck DE, Flowers RA, II, Laskin JD, Heindel ND. Peripheral site acetylcholinesterase inhibitors targeting both inflammation and cholinergic dysfunction. Bioorg and Med Chem Lett. 2010;20:2987–2990. doi: 10.1016/j.bmcl.2010.02.102. [DOI] [PMC free article] [PubMed] [Google Scholar]