

SUMMARY
Fusobacterium nucleatum (Fn) has been associated with colorectal cancer (CRC), but causality and underlying mechanisms remain to be established. We demonstrate that Fn adheres to, invades and induces oncogenic and inflammatory responses to stimulate growth of CRC cells through its unique FadA adhesin. FadA binds to E-cadherin, activates β-catenin signaling, and differentially regulates the inflammatory and oncogenic responses. The FadA-binding site on E-cadherin is mapped to an 11 amino acid region. A synthetic peptide derived from this region of E-cadherin abolishes FadA-induced CRC cell growth, and oncogenic and inflammatory responses. FadA levels in the colon tissue from patients with adenomas and adenocarcinomas is >10–100 times higher compared to normal individuals. The increased FadA expression in CRC correlates with increased expression of oncogenic and inflammatory genes. This study unveils a mechanism by which Fn can drive CRC and identifies FadA as a potential diagnostic and therapeutic target for CRC.
INTRODUCTION
The human intestinal microbiome contains >1000 different species totaling 1014 microorganisms and plays an extremely important role in the maintenance of the normal physiology of the gut, including energetic metabolism, proliferation and survival of epithelial cells, and protection against pathogens (Tremaroli and Backhed, 2012). The microbiota exerts both beneficial and detrimental effects on host contributing to healthy or disease (Sekirov et al., 2010). Recently, two research teams simultaneously reported overabundance of a specific microorganism, Fusobacterium nucleatum (Fn), in colorectal carcinoma tissues (Castellarin et al., 2012; Kostic et al., 2012). However, it was unknown if Fn was a cause or a consequence of CRC.
Fn is an opportunistic commensal anaerobe in the oral cavity, implicated in various forms of periodontal diseases. Outside the oral cavity, it is one of the most prevalent species in extra-oral infections (Han, 2011). It is highly prevalent in intrauterine infections associated with pregnancy complications such as preterm birth, stillbirth, and neonatal sepsis (Han et al., 2009; 2010; Wang XW et al., 2012). Fn adheres to and invades endothelial and epithelial cells, a likely mechanism utilized for its systemic dissemination (Han et al., 2000; 2004). The attachment and invasion take place via adhesin FadA, a virulence factor identified from Fn (Han et al., 2005; Xu et al., 2007). Immunofluorescent staining of non-permeabilized Fn using anti-FadA monoclonal antibodies showed that FadA was expressed on the bacterial surface (Ikegami et al., 2009).
FadA is highly conserved among Fn (Han et al., 2005). It exists in two forms, the non-secreted intact pre-FadA consisting of 129 amino-acid (aa) residues, and the secreted mature FadA (mFadA) consisting of 111 aa without the 18-aa signal sequence (Han et al., 2005). The mFadA monomer is predominantly alpha-helical and forms a “hairpin” like structure (Nithianantham et al., 2009). The monomers are linked together in a head-to-tail pattern to form uniformly long and thin filaments, with no binding activity (Temoin et al., 2012). When mFadA is mixed with pre-FadA, they form activity complexes, FadAc, consisting of heterogeneous filaments, presumably due to varying degrees of filament bundling (Xu et al., 2007; Temoin et al., 2012). We have recently demonstrated that the vascular endothelial (VE) cadherin (CDH5), a member of the cadherin superfamily, is a receptor for FadA, required for Fn to attach and invade endothelial cells (Fardini et al., 2011). The receptor for FadA on epithelial cells was not identified.
In the present study, we demonstrate that FadA binds to E-cadherin on CRC and non-CRC cells, mediating Fn attachment of and invasion into the cells. FadA modulates E-cadherin and activates β-catenin signaling, leading to increased expression of transcription factors, oncogenes, Wnt genes, and inflammatory genes, as well as growth stimulation of CRC cells. Further, we show that while FadA binding to CRC cell is sufficient to turn on the Wnt and oncogenes, its internalization mediated by clathrin is needed to activate the inflammatory genes. This study reveals a mechanism by which Fn contributes to CRC and identifies FadA as a potential diagnostic and therapeutic target for CRC.
RESULTS
Fn stimulates human CRC cell proliferation
Wild-type Fn 12230 significantly stimulated proliferation of human colon cancer cells HCT116, DLD1, SW480, and HT29, but only weakly stimulated RKO. It did not stimulate the non-CRC cells HEK293. Compared to the untreated cells or those incubated with E. coli DH5α, the growth stimulation increased by approximately 100% for HCT116, DLD1, SW480, and HT29, but only 18% for RKO, after 72 hours (Fig. 1a). The fadA-deletion mutant US1 weakly stimulated the growth of all cancer cell lines. The fadA-complementing clone, USF81, restored proliferation of HCT116, DLD1, SW480, and HT29 to the wild-type level. Furthermore, HCT116 growth was enhanced by purified FadAc in a dose-dependent manner, with the maximum stimulation observed at 1 mg/ml, while mFadA exhibited no stimulatory effect (Fig. 1b). Neither FadAc or mFadA stimulated growth of RKO. These results indicate that stimulation of CRC cell by Fn is FadA-dependent.
Figure 1. Fn and FadA stimulate proliferation of human colon cancer cells via E-cadherin.

A. Wild type Fn (Fn) and the fadA-complementing USF81 (fadA+) stimulated proliferation of human CRC cells HCT116, DLD1, SW480, and HT29, compared to untreated cells or those incubated with E. coli. US1 (fadA−) only weakly stimulated their growth. Fn, USF81 and US1 all weakly stimulated the growth of CRC cells RKO, but not the non-CRC cells HEK293. B. Purified FadAc stimulated HCT116 cell growth in a dose-dependent manner, while mFadA did not. Neither FadAc nor mFadA stimulated RKO cell growth. C. Suppression of Fn-stimulated cell growth by inhibiting E-cadherin. Fn-stimulated HCT116 growth was inhibited by siRNA specific for CDH1, GST-EC5 fusion protein, and the inhibitory peptide (IP), but not by non-specific siRNA, GST, or the control peptide (CP). D. Fn stimulated the growth of RKO cells transfected with CDH1, but not mock-transfected RKO. Growth stimulation of CDH1-transfected RKO cells was suppressed by GST-EC5 and the inhibitory peptide, but not by GST or the control peptide. The results are presented as mean±SD. ***p<0.001. See also Figure S1.
FadA binds to E-cadherin on CRC cells
To investigate the mechanism by which FadA stimulates CRC cell growth, we set out to identify CRC cell receptors for FadA. It was previously shown that FadA binds to VE-cadherin on endothelial cells (Fardini et al., 2011). Cadherins are a large family of calcium-dependent cell adhesion glycoproteins, each composed of five extracellular repeat domains (EC1-EC5), a transmembrane domain, and a highly conserved cytoplasmic tail that binds other cytoplasmic components including β-catenin (Gumbiner, 2005) (Fig. 2a). Given the 33.5% similarity between VE- and E-cadherins, we speculated if FadA also bound to E-cadherin. E-cadherin is present on epithelial cells, including the non-cancerous HEK293, as well as the CRC cells, except RKO (Fig. 2b). FadA binding to E-cadherin was directly tested by co-immunoprecipitation. FadAc co-precipitated with E-cadherin, while mFadA did not (Fig. 2c). Using the GST pull-down assay, we determined that FadAc bound specifically to EC5 of E-cadherin, but not to EC1-4, the transmembrane, or the cytoplasmic domains (Fig. 2d). Deletions of various regions in EC5 showed that region 3 was responsible for FadA binding (Fig. S2).
Figure 2. E-cadherin is FadA receptor.
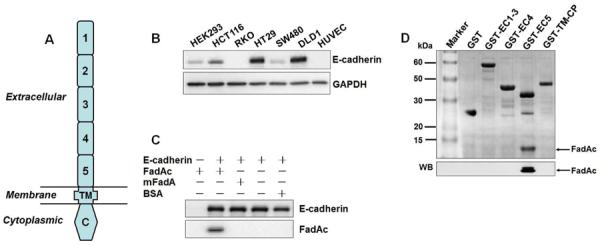
A. Schematic representation of the E-cadherin (CDH1) structure. E-cadherin has five extracellular cadherin (ECs) repeats, numbered EC1–5 starting from N-terminal. TM, transmembrane domain; C, cytoplasmic domain. B. E-cadherin is expressed in epithelial cell HEK293 and most CRC cells. E-cadherin in HEK293 and human CRC cell lines HCT116, RKO, HT29, SW480 and DLD1 was examined by Western blot. Human umbilical vein endothelial cells (HUVEC) were included as a negative control. The endogenous GAPDH was used as a loading control. C. E-cadherin co-immunoprecipitates with FadAc. HEK293 cell lysate expressing E-cadherin was mixed with E. coli lysates expressing FadAc, or mFadA, or BSA, followed by incubation with mouse anti-CDH1 monoclonal antibodies (mAb) and captured with agarose A/G beads. E-cadherin and FadA in the bead elutes were detected by Western blot. D. FadA binds to EC5. Purified GST or GST-fusion proteins carrying EC1–3, EC4, or EC5 were incubated with E. coli lysates expressing FadAc, followed by capture with GST resin. The eluted components were subjected to SDS-PAGE, followed by Coommassie blue staining (top panel) and Western blot (WB) using anti-FadA mAb 5G11-3G8 (bottom panel). See also Figure S2.
FadA promotes Fn attachment and invasion of E-cadherin-expressing cells
Attachment and invasion are hallmarks of Fn. Thus, we tested the role of FadA binding to E-cadherin in these processes. Fn attachment and invasion of non-CRC cells HEK293 was inhibited by mouse monoclonal antibody HECD-1 raised against the extracellular domain of E-cadherin (Fig. S3a). Deletion of fadA (US1) severely impaired the ability of Fn to bind and invade HEK293, whereas the fadA-complemented clone, USF81, restored the activities. Down-regulation of E-cadherin expression by siRNA in HEK293 significantly inhibited attachment and invasion by wild-type Fn and USF81 (Fig. S3b). These results indicate that Fn attachment and invasion of HEK293 requires FadA and E-cadherin.
Similar observations were made with CRC cells HCT116 expressing E-cadherin. US1 (fadA−) was defective in attachment and invasion of HCT116, compared to wild-type Fn or USF81 (fadA+) (Fig. 3a). Inhibition E-cadherin expression by siRNA reduced attachment and invasion (Fig. 3a). In contrast, no difference was observed among Fn 12230, US1 and USF81 in their weak binding and invasion of the non-E-cadherin-expressing RKO cells (Fig. 3b). Transfection of RKO with the full-length E-cadherin led to increased binding and invasion by Fn 12230 and USF81 to levels comparable to those observed in HCT116 (Fig. 3b). These results indicate that FadA mediates Fn attachment and invasion of CRC cells via E-cadherin.
Figure 3. Fn adheres to and invades E-cadherin-expressing CRC cells.
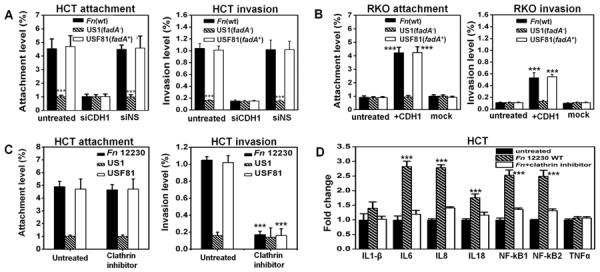
A. Fn adheres to and invades the E-cadherin-expressing HCT116 via FadA and E-cadherin. The fadA-deletion mutant US1 (fadA−) was defective for attachment and invasion, compared to wild type Fn and the fadA-complementing clone USF81 (fadA+). Transfection with siRNA to inhibit E-cadherin expression (siCDH1) reduced attachment and invasion, while the non-specific siRNA (siNS) did not. B. Wild-type Fn, US1, and USF81, were defective for attachment and invasion of the non-E-cadherin-expressing RKO cells. Transfection of full-length CDH1 into RKO enhanced attachment and invasion by wild type Fn and USF81 (fadA+), but not by US1 (fadA−). C. The clathrin inhibitor, Pitstop2, inhibits Fn and USF81 (fadA+) invasion of HCT116, without affecting their attachment. D. Wild type Fn stimulates expression of NF-kappaB and pro-inflammatory cytokines IL-6, 8, and 18 in HCT116, which was inhibited by the clathrin inhibitor. Expression levels in untreated HCT116 were designated as “1”. For A, B, and D, the attachment and invasion levels were expressed as percent bacteria recovered from the host cells relative to the initial inoculum. For wild type Fn, these levels reflect recovering approximately 9000 CFU per well (in a 96-well plate) from the attachment assay and approximately 2000 CFU from the invasion assay. The invasion level of E. coli DH5a into HCT116 was <0.01%, i.e. < 20 CFU recovered per well (data not shown). For C, the original attachment (4.4±0.8%) and invasion (1.3±0.1%) levels without inhibition were designated as “100%”, and the relative inhibition values were shown. The results are presented as the mean±SD. ***p<0.001. See also Figure S3.
E-cadherin can be internalized via clathrin (Bryant and Stow, 2004). Pitstop2, a clathrin inhibitor (von Kleist et al., 2011), prevented Fn invasion of HCT116, without affecting attachment (Fig. 3c). Fn stimulated expression of the inflammatory genes including NF-kappaB and cytokines IL-6, 8, and 18 from HCT116 (Fig. 3d). Such stimulation was abolished in the presence of the clathrin inhibitor, indicating that invasion was required for the stimulation of inflammation (Fig. 3d).
Identification of inhibitory peptides to prevent Fn attachment and invasion
Since FadA bound to the EC5 domain of E-cadherin, we tested the role of EC5 in Fn attachment and invasion. Fn attachment and invasion of HCT116 was inhibited by purified GST-EC5 fusion protein in a dose-dependent manner, with maximum inhibition observed at 0.1 μM (Fig. 4a & 4b). Synthetic peptides derived from different regions of EC5 were then tested for their ability to inhibit Fn attachment and invasion (Fig. 4a & 4c). Peptide 3, corresponding to region 3, exhibited similar inhibitory effect as EC5 (compare Fig 4b & 4c). To determine the minimal sequences required for inhibition, sequential deletions of peptide 3 were generated. The results showed that the 11-aa peptide (ASANWTIQYND) was the minimum required, and was designated as the “inhibitory peptide” (IP) (Fig. 4d).
Figure 4. Identification of synthetic peptides to inhibit Fn attachment and invasion of HCT116 cells.
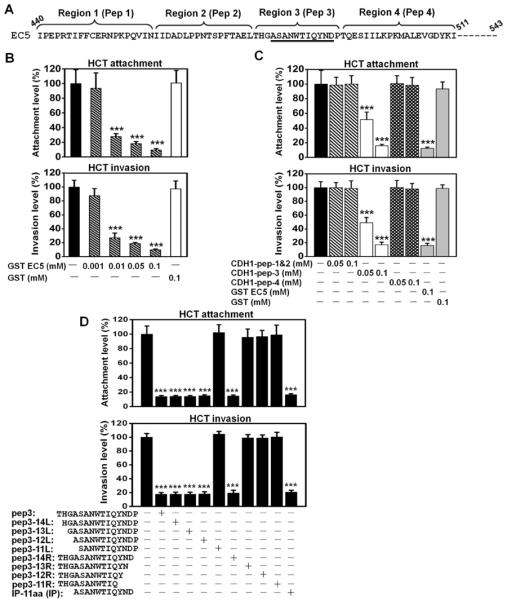
A. Partial amino-acid sequence of the EC5 domain. The regions and the corresponding peptides (pep) are shown above the sequence. The sequences corresponding to the inhibitory peptide (IP, see below) are underlined. Peptide 4 was the control peptide (CP) in all studies. B. Purified GST-EC5 fusion protein inhibits wild type Fn attachment and invasion of HCT116 in a dose-dependent manner. C. Fn attachment and invasion of HCT116 cells were inhibited by a synthetic peptide corresponding to region 3 (pep 3) on the EC5 domain, not by peptides corresponding to regions1&2, or 4. D. The inhibitory effects of synthetic oligopeptides carrying sequential deletions from the N- and C-termini of region 3 on Fn attachment and invasion. Deletion of 3 residues from N-terminal and 1 residue form C-terminal did not affect the inhibitory function. An 11-aa peptide (ASANWTIQYND) was found as the minimum sequence required for inhibition of Fn attachment and invasion. All values were expressed as relative to those without inhibition, which were designated as “100%”. The actual attachment and invasion levels were 6.3±1.4% and 1.6±0.1%, respectively, for B, and 5.9±0.7% and 1.4±0.1%, respectively, for C. The results are presented as mean±SD. ***p<0.001.
FadA promotes CRC cell proliferation via E-cadherin
Stimulation of HCT116 growth by Fn was inhibited by the CDH1-specific siRNA, EC5, and the inhibitory peptide (Fig. 1c). In the non-E-cadherin-expressing RKO cells, transfection of the full-length E-cadherin resulted in growth stimulation by Fn (Fig. 1d). As in HCT116, such stimulation was diminished by EC5 and the inhibitory peptide (Fig. 1d). Similar observations were made using purified FadAc instead of Fn (Fig. S1). These results elucidate the critical role of E-cadherin in promoting Fn-driven CRC cell growth.
FadAc activates E-cadherin-mediated cellular signaling
To investigate the downstream events subsequent to FadA binding to E-cadherin, HCT116 were fractionated following incubation with purified FadA. FadAc, but not mFadA, bound to the HCT116 membranes within five minutes of incubation, leading to E-cadherin phosphorylation on the membrane and internalization (Fig. 5a). This was accompanied by decreased phosphorylation of β-catenin, β-catenin accumulation in the cytoplasma and translocation into the nucleus, resulting in activation of β-catenin-regulated transcription (CRT), as evidenced by increased expression of transcription factors lymphoid enhancer factor (LEF)/T-cell factor (TCF), NF kappaB, and oncogenes Myc and Cyclin D1 (Fig. 5a). It was previously shown that protein tyrosine kinase plays a crucial role in E-cadherin endocytosis and recycling (Le et al., 2002). Interestingly, the protein tyrosine kinase (PTK) inhibitor, Genistein, not only prevented E-cadherin phosphorylation and internalization, but also abolished FadA binding to the membranes and its internalization, as well as the above-described CRT activation (Fig. 5a). These results suggest that phosphorylation of E-cadherin or other cellular components are required for activation of CRT. FadA may bind to phosphorylated E-cadherin, or it may bind to non-phosphorylated E-cadherin, which is then phosphorylated leading to positive feedback. The central role of β-catenin in regulating the cellular responses was confirmed using HCT116 β-catenin−/− cells, which did not affect FadA binding to E-cadherin, phosphorylation of E-cadherin, or the internalization, but prevented all gene activations tested in the nuclei (Fig 5a).
Figure 5. FadAc activates E-cadherin-mediated cellular signaling.
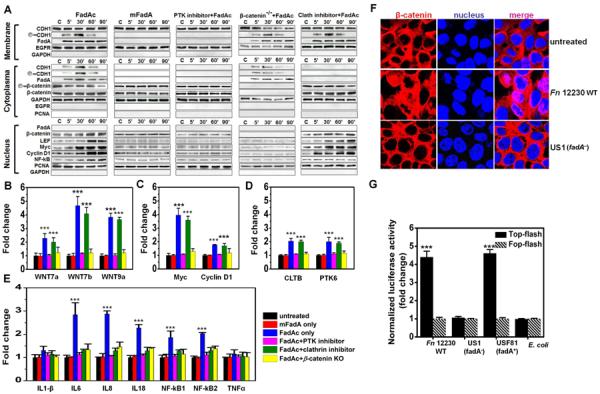
A. FadAc, but not mFadA, binds to the membranes of HCT116, accompanied by phosphorylation of E-cadherin on the membrane; internalization of E-cadherin and FadA, reduced phosphorylation of β-catenin, and accumulation of β-catenin in the cytoplasma; and translocation of β-catenin and activation of transcription factors lymphoid enhancer factor (LEF)/T-cell factor (TCF), NF kappaB, and oncogenes Myc and Cyclin D1 in the nuclei; all as detected by Western blot. Protein tyrosine kinase (PTK) inhibitor, Genistein, inhibits all FadAc-activated functions. No gene activation was detected in the HCT116 β-catenin−/− cells, despite binding and internalization of FadA and E-cadherin. The clathrin inhibitor, Pitstop2, prevented E-cadherin and FadA internalization and activation of NF kappaB, but did not affect nuclei translocation of β-catenin or expression of LEF/TCF, Myc or Cyclin D1. The epidermal growth factor receptor (EGFR), glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and proliferating cell nuclear antigen (PCNA) were used as loading controls for membrane, cytoplasmic, and nucleus, respectively, and for examination for cross-contamination between the subcellular fractions. B–E. FadAc, but not mFadA or BSA, activates expression of Wnt signaling genes 7a, 7b, 9a (B), oncogenes Myc and Cyclin D1 (C), clathrin (cltb) and protein tyrosine kinase genes (ptk6) (D), and NF-kappaB and pro-inflammatory cytokines IL-6, 8, and 18 (E) in wild-type or β-catenin knockout HCT116 cells following 2 hrs incubation as determined by qPCR. The clathrin inhibitor inhibited expression of the inflammatory genes but not the Wnt or oncogenes, while inhibition of β-catenin by siRNA suppressed expression of all genes. Expression levels in untreated HCT116 were designated as “1”. F. Wild type Fn (Fn), but not US1 (fadA−), induces nuclei translocation of β-catenin following 2 hrs incubation as observed by confocal microscopy. β-catenin was stained with Alex 634 (red) and the nuclei with 4',6-diamidino-2-phenylindole (DAPI) (blue). The purple color in the merged images indicates translocation of β-catenin into the nucleus. G. Luciferase reporter gene expression following HCT116 transfection with TOPFlash (activated by β-catenin) or FOPFlash (insensitive to β-catenin activation). Fn was incubated with the transfected cells at a MOI of 1,000:1 for 2 hours, followed by measurement of the luciferase activity. Values obtained with FOPFlash were designated as “1” and those obtained with TOPFlash were expressed as fold changes. Data are presented as mean fold changes ±SD of two independent experiments, each in triplicate. ***p<0.001.
The clathrin inhibitor, although did not affect FadA binding to or E-cadherin phosphorylation on the membranes, inhibited FadA and E-cadherin internalization. A surprise consequence was the divergent responses observed in the nuclei. Translocation of β-catenin and expression of LEF/TCF, Wnt and oncogenes were unaffected. In contrast, no NF kappaB activation was observed (Fig. 5a). These results indicate that tumor growth and inflammatory responses, although both requiring β-catenin, are differentially regulated.
The Western blot analysis of protein levels were corroborated with real-time quantitative PCR (qPCR) analysis of the mRNA levels (Fig 5b–5e). While PTK inhibitor and CDH1-specific siRNA inhibited activation of tumor growth and inflammatory genes, the clathrin inhibitor only inhibited the inflammatory genes, but not the Wnt or oncogenes.
To further confirm the role of FadA in CRT activation, we performed confocal microscopy analysis and observed nuclei translocation of β-catenin in HCT116 in response to wild-type Fn, but not to US1 (fadA−) (Fig. 5f). In addition, wild-type Fn and USF81 (fadA+), but not US1 (fadA−), activated the luciferase reporter gene in TOPFlash carrying the β-catenin-response promoter, but not in FOPFlash carrying β-catenin-non-response promoter (Fig 5g).
FadA promotes E-cadherin-mediated CRC tumor growth and induction of pro-inflammatory cytokines in xenograft mice
To examine the effects of FadA and Fn on CRC cell growth in vivo, HCT116 were inoculated into nude mice, followed by treatment with either purified protein or bacteria. Tumor growth was increased by 20% after 3 weeks of treatment by FadAc, compared to those treated with mFadA or BSA (Fig. 6a and 6d). No increase was detected in the presence of 0.01 mmol inhibitory peptide, but not the control peptide, indicating the role of FadA binding to E-cadherin in tumor growth (Fig. 6b and 6d). FadAc had no stimulatory effect on the non-E-cadherin-expressing RKO (Fig. 6c).
Figure 6. FadA promotes E-cadherin-mediated CRC tumor growth and induction of pro-inflammatory cytokines in xenograft mice.
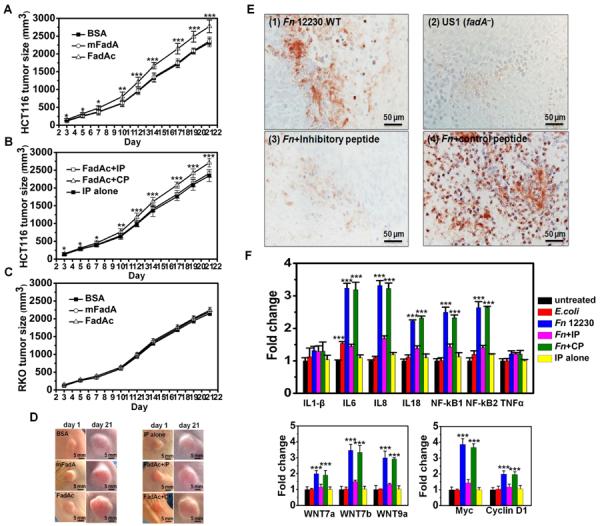
HCT116 or RKO were injected subcutaneously and bilaterally into female nude mice, which were then randomized (5 per group) to receive treatments. A–C. FadAc stimulates HCT116 but not RKO xenograft growth. Purified FadAc, mFadA, or BSA were injected into xenografts of HCT116 either alone (A), or along with the inhibitory (IP) or control (CP) peptides (B), or RKO alone (C) in nude mice. IP alone is injected into HCT116 as a negative control (B). D. Representative tumors from A and B are shown. The first day of protein injection was designated as “day 1”. All tumors look the same on day 1. Notice the size increase of the tumor treated with FadAc and FadAc+CP on day 21, compared to other tumors on the same day. E. Immunohistochemical staining of xenografts infected with wild-type Fn (Fn), alone and with the inhibitory or control peptides, and the fadA-deletion mutant US1 (fadA−) using rabbit anti-Fn polyclonal antibodies. For controls, xenografts infected with wild-type Fn was stained with pre-serum, and xenografts infected with E. coli DH5α were stained with anti-Fn antibodies (data not shown). F. Wild-type Fn induces expression of NF-kappaB and pro-inflammatory cytokines IL-6, 8, and 18; Wnt 7a, 7b, and 9a; and Myc and Cyclin D1 in HCT116 xenografts, as determined by qPCR. The inductions were inhibited by IP, but not by CP. E. coli only weakly induced IL-6. The results are presented as mean±SD. ***p<0.001. See also Figure S4.
When wild-type Fn was injected into HCT116 xenografts, abscess formation was observed within 3–5 days (data not shown). Immunohistochemical analysis using anti-Fn antibodies showed that wild-type Fn invaded into the tumor tissues while US1 (fadA−) did not (Fig. 6e). Nor did E. coli DH5α (data not shown). Fn invasion was prevented by the inhibitory peptide, but not by the control peptide (Fig. 6e). FadAc (Fig. S4) and wild-type Fn (Fig. 6f) stimulated the tumor growth genes and the inflammatory genes to same extent, which were inhibited by the inhibitory peptide, but not the control peptide, consistent with the observations in vitro.
Patients with CRC and precancerous adenomas have elevated FadA gene and expression levels compared to normal individuals
We examined FadA gene and expression levels in human colon specimens from the following 5 groups: (1) normal non-cancerous individuals (n=14), (2) normal tissues from patients with precancerous adenomas (n=16); (3) precancerous adenomas (n=16); (4) normal tissues from patients with adenocarcinomas (n=19); and (5) adenocarcinomas (n=19). A step-wise increase of FadA gene copies was observed from Group 1 to Group 2–4, and to Group 5, with >1 log difference between each step (Fig. 7a). The biggest difference was observed between the non-cancerous controls and CRC, with > 2 logs difference (Fig. 7a). The FadA mRNA levels in the colon tissues, when normalized to GAPDH, also showed a stepwise increase correlating with the FadA gene copy numbers (data not shown). When the FadA mRNA levels were normalized to Fn 16S RNA to reflect FadA expression in Fn, a significance increase was only observed in the carcinoma tissues (Group 5), indicating Fn exhibits increased virulence in CRC, compared to the normal and precancerous tissues (Fig. 7b). Consistent with the increase of FadA, expression of a representative Wnt gene, Wnt7b, and a representative inflammatory gene, NF kappa b2, were also significantly increased in CRC, corroborating with the results obtained in vitro and in xenograft mice.
Figure 7. Quantification of fadA gene copies and FadA, Wnt7b and NFkB2 expression in health, precancerous adenomas, and carcinomas.
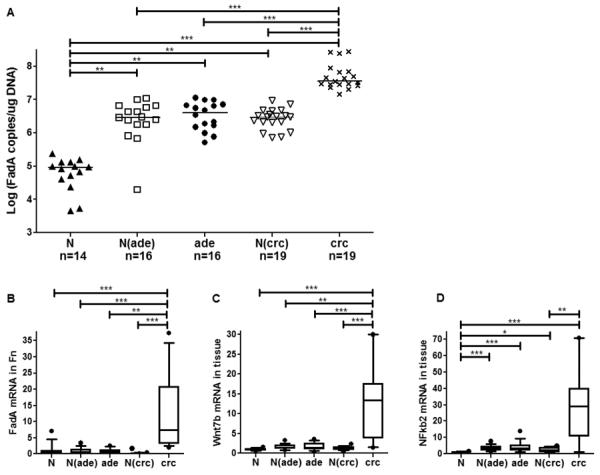
DNA and RNA were extracted from full-thickness colon specimens from the following 5 groups: (1) normal non-cancerous controls (N; n=14); (2) Normal tissues from patients with precancerous adenomas [N(ade); n=16]; (3) Precancerous adenomas (ade; n=16); (4) Normal tissues from patients with carcinomas [N(crc); n=19]; and (5) Carcinomas (crc; n=19). Gene copy numbers of fadA (A) were measured using DNA and determined using the standard curves. FadA mRNA levels in Fn were normalized to Fn 16 rRNA (B), and Wnt7b (C) and NFkb2 (D) mRNA levels were each normalized to the endogenous GAPDH. The average value of Group 1 (N) was designated “1”, and the fold changes of the other groups were determined by comparing to Group 1. The horizontal bars in a represent the median values. For B–D, the boxes show the 25/75 percentiles and the lines within the boxes the median values. Whiskers show the 10/90 percentiles. *p<0.05, **p<0.01 and *** p<0.001.
DISCUSSION
CRC is the second leading cause of cancer death in men and women combined in the U.S. (ACS, 2012). Worldwide, over one million new cases are diagnosed each year (Bartlett and Chu, 2012). Understanding the mechanisms of CRC and identifying risk factors are important to human health. Increasing evidence supports a relationship between infective agents and various human malignancies (Plottel and Blaser, 2011). Previous studies have provided mechanistic insights into the microbial involvement in the development of CRC (Arthur et al., 2012; Cuevas-Ramos et al., 2010; Lee et al., 2010; Wu et al., 2009). It was reported that Bacteroides fagilis enterotoxin BFT cleaves E-cadherin to activate the β-catenin signaling (Wu et al., 1998; 2003). Here, we report a mechanism by which Fn modulates E-cadherin/β-catenin signaling.
Fn significantly stimulates proliferation of E-cadherin-expressing CRC cells, but not those without E-cadherin. The stimulation requires FadA, an E-cadherin ligand. The fadA-deletion mutant US1 has weak stimulatory activity. These observations indicate that the interactions between FadA and E-cadherin play a primary role in promoting CRC tumor growth, while the FadA-independent pathway(s) play a minor role.
E-cadherin is a tumor suppressor, which functions through β-catenin (Bryant and Stow, 2004; Peifer and Polakis, 2000). It has been previously reported that E-cadherin is present in 100% of the normal mucosa of CRC patients and in 75% of cancer specimens (Mohri, 1997). Loss or heterogeneous expression of E-cadherin correlated with advanced stages of CRC and poor prognosis (Mohri, 1997; Dorudi et al., 1993). FadA binding to E-cadherin inhibits its tumor suppressor activity, resulting in increased CRT. This phenotype is consistent with the adenomatous polyposis coli (APC) mutation, the most common mutation in CRC (Kinzler and Vogelstein, 2002). The four human colon cancer cell lines stimulated by FadA, HCT116, DLD1, SW480, and HT29, carry either an APC mutation (DLD1, SW480, and HT29) (Yang et al., 2006) or a β-catenin mutation (HCT116) (Morin et al., 1997), thus have increased CRT background (Chan et al., 2002). FadA, both in purified form and in Fn, further enhanced CRT and stimulated their growth, attesting to its carcinogenic potency. Interestingly, FadA does not stimulate the growth of the non-cancerous HEK293, suggesting that it promotes oncogenesis after a mutation occurs.
FadA is the major component mediating Fn attachment and invasion (Xu et al., 2007). It binds to an 11-aa region in the EC5 domain on E-cadherin, and is internalized with E-cadherin. The 11-aa inhibitory peptide inhibits Fn from binding and invading, and abolishes all subsequent host responses, including tumor growth and inflammatory responses. Thus, this inhibitory peptide not only prevents Fn colonization but also inhibits Fn-mediated oncogenesis.
Inflammation plays a key role in CRC oncogenesis (Baron and Sandler, 2000; Ekbom et al., 1990;). In this issue of Cell Host and Microbe, Kostic et al showed that Fn generates a pro-inflammatory microenvironment that is conducive for colorectal neoplasia progression (Kostic et al., 2013). Both the observations herein and by Kostic et al support that inflammatory stimulation by Fn further reinforces the oncogenic potential of this microorganism. As shown in the xenografts, FadA alone was able to stimulate the inflammatory responses to the same extent as Fn, indicating FadA is the major stimulant of inflammation.
Our results reveal that tumor growth and inflammation are differentially regulated, although both require β-catenin, the master regulator. It is often difficult to distinguish the roles of adherence and invasion as these two processes are tightly correlated (Martin et al., 2004; Swidsinski et al., 1998), with the former being a prerequisite for the latter. Using the clathrin inhibitor, we are able to functionally separate Fn attachment from invasion and demonstrate that although invasion is not required for tumor growth, it is essential for the induction of inflammation, which is consistent with a previous report that the invasive potential of Fn correlates with the host IBD status (Strauss et al., 2011).
The mechanistic investigation is corroborated with studies in humans. Previous studies have shown that Fn is readily detected in the tissues, but not in the stools (Chen et al., 2012). This may be due to its adherent and invasive properties. Hence, we examined the presence of Fn in colon tissue specimens, rather than stools. The results unveil a stepwise increase of FadA gene copies from the baseline of normal individuals to adenomas and normal tissues adjacent to adenomas and adenocarcinomas, and to adenocarcinomas. The “normal” tissues from patients with adenomas and adenocarcinomas are thus “pseudo-normal”, compared to the non-cancerous controls. This is consistent with the findings by McCoy et al (McCoy et al., 2013). Given the fact that FadA mediates Fn binding to both CRC and non CRC cells, and the fact that the normal tissues also express E-cadherin, it is not surprising that Fn can colonize both tumor and non-tumor sites. Elevated Fn colonization in the normal tissues may predispose the host to the development of adenomas and/or adenocarcinoma, with carcinogenesis being accelerated when a mutation occurs. The fact that FadA expression levels in Fn, as well as the host inflammatory and oncogenic responses, are increased only in the cancerous tissues, but not in the precancerous state, further supports this notion.
The finding of elevated FadA levels in the precancerous state implies translational potential. Compared to the metagenomic approach in the previous studies (Castellarin et al., 2012; Kostic et al., 2012), qPCR is significantly less time consuming and more cost effective, suitable for clinical purposes. FadA is unique to Fn (Han et al., 2005), hence, is an ideal potential diagnostic marker to identify individuals at risk for developing adenomas and/or adenocarcinomas. Based on the results shown in Fig 7, it is possible that diagnostic criteria may be developed to define healthy, precancerous, and cancerous states according to FadA gene copy levels. One limitation of the current study is the small sample size. The potential use of FadA as a diagnostic marker needs to be evaluated in a much larger prospective cohort. Future studies are also warranted to examine the use of the inhibitory peptide and/or its derivatives to eradicate Fn, either as a preventive therapy for individuals with increased risk, or as a complementary therapy for those with CRC.
In conclusion, we have elucidated an oncogenic mechanism of CRC. Findings from this study will significantly impact our understanding, diagnosis, prevention and treatment of CRC.
EXPERIMENTAL PROCEDURES
Bacterial strains, cell cultures, construction of plasmids, protein purification and GST pull-down assays
Bacteria and human cell lines were cultured as previously described (Fardini et al., 2011; Han, 2006; Zhang et al., 2012). EC domains of E-cadherin were amplified by PCR from pcDNA3-E-cadherin (AddGene, MA) vector using primers listed in Table S1. Protein expression and purification, GST pull-down assays were all performed as previously described (Fardini et al., 2011; Xu et al., 2007).
Tissue culture attachment and invasion assays
These assays were performed as previously described (Han et al., 2000). Briefly, cells were seeded in 24-well or 96-well plates at 8×104 cells or 2.5×104 cells per well in the growth medium and grown to 100% confluent. Bacteria were added to the cells at a multiplicity of infection (MOI) of 50. Following 1 hr incubation at 37°C in 5% CO2, the monolayers were washed 3 times with D-PBS, pH 7.1, supplemented with Ca2+ and Mg2+. Cells were lyzed with water for 20 min at 37°C. Serial dilutions of the lysates were plated onto blood agar plates to enumerate the total cell-associated bacteria. For invasion assays, the bacteria were incubated with the monolayers at 37°C for 4 hrs, followed by washes with PBS. Fresh media containing 300 μg/ml gentamicin and 200 μg/ml metronidazole were added to the monolayers and incubated for an additional hour to kill extracellular bacteria. The cells were then washed and lyzed with water as described above. The levels of attachment and invasion were expressed as the percentage of bacteria recovered following cell lysis relative to the total number of bacteria initially added. Each experiment was performed in triplicates and repeated at least twice.
Antibodies, peptides, and Western blot analysis
The following antibodies were used: anti-FadA monoclonal antibody (mAb) 5G11-3G8 (Xu et al., 2007), mAb anti-CDH1 (Abcam or Cell Signaling Tech), polyclonal anti-phospho-CDH1 (ECM Biosciences), mAb anti-GAPDH (Invitrogen), mAb anti-β-catenin (R&D systems), polyclonal anti-phospho-β-catenin (Cell Signaling Tech), mAb anti-LEF/TCF1 (Invitrogen), mAb anti-NFkB (Invitrogen), mAb anti-Myc (Cell Signaling Tech), mAb anti-cyclin D1 (Cell Signaling Tech), mAb anti-EGFR (Cell Signaling Tech), mAb anti-GAPDH (Cell Signaling Tech), mAb anti-PCNA (Cell Signaling Tech), polyclonal goat-anti-mouse or goat-anti-rabbit secondary antibody conjugated with horseradish peroxidase (HRP) (Pierce Biotechnology). Peptides were synthesized by Neo Group, Inc (Cambridge, MA). Western blot analyses were performed as previously described (Fardini et al., 2011).
Co-immunoprecipitation (Co-IP) assay
500 μg of HEK293 cell lysate prepared with CO-IP Lysis/Wash Buffer (Pierce) were mixed with 100 μg of BSA or E. coli lysate expressing FadAc or mFadA, followed by the addition of mouse monoclonal anti-CDH1 antibodies. The protein complex was captured by agarose A/G beads (Santa Cruz) as previously described (Chan et al., 2002), followed by elution and Western blot analysis. An equal volume of the elutes were loaded onto SDS-PAGE.
DNA and siRNA transfection
CDH1 transfections were performed as previously described (Lebreton et al., 2011). siRNA assays were performed using the FlexiTube siRNA anti-CDH1 or anti-beta-catenin reagent and All Stars FlexiTube Control siRNA from Qiagen (Valencia, CA) according the manufacturer's instructions.
Cell proliferation assay
CRC cells were seeded in 24-well plates at 1×104 cells per well in the growth medium. Cells were untreated, or incubated with FadAc or peptides at indicated concentration, or with bacteria at a multiplicity of infection (MOI) of 1000:1. Cell numbers were counted at 24-hour intervals using a hemocytometer. Each experiment was performed in triplicates and repeated at least twice.
Preparation of subcellular fractions
Cells were incubated with 1 mg/ml of FadAc or mFadA followed by the extraction of subcellular fractions. When indicated, 50 μM of the corresponding protein tyrosine kinase inhibitor (Genistein) and clathrin inhibitor (Pitstop2) were pre-incubated with cells for 1 hour. Membrane, cytosolic and nuclei fractions were prepared using the Compartmental Protein Extraction kit (Millipore) according to the manufacturer's instructions.
Luciferase reporter assay
HCT116 cells were seeded in 96-well plates at 2.5×104 cells per well in the growth medium and grown to 100% confluent. Cells were transfected with 0.2 μg of TOPFlash, a luciferase reporter vector carrying TCF promoter upstream of the luciferase gene and can be activated by β-catenin, or FOPFlash (with mutations rendering insensitivity to β-catenin activation), using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. On the following day, cells were treated with bacteria at a multiplicity of infection (MOI) of 1000:1 for 2 hours. Reporter assays were performed using the luciferase reporter system (Promega, Madison, WI). The experiment was performed in triplicates and repeated twice.
Immunofluorescent and immunohistochemical (IHC) staining
Immunofluorescent staining of cells was performed as previously described. Mouse anti-β-catenin mAb and Alexa Fluor 634-conjugated goat anti-mouse polyclonal antibodies were used (Fardini et al., 2011). IHC analysis of xenograft tumors were performed as previously described, using rabbit anti-Fn polyclonal antibodies (Han et al., 2004). Pre-immune serum from the same rabbit was used as control.
Xenografts
The animal protocol was approved by the Case Western Reserve University Institutional Animal Care and Use Committee. An inoculum of 5×106 cells were injected s.c. and bilaterally into 4- to 6-week-old female nude mice (5 per group) as previously described (Zhang et al., 2012). The mice were randomized to receive one of the following: FadAc or mFadA (each at 80 μg), BSA (0.01 μmol, i.e. 660 μg), or peptide (0.01 μmol), or bacteria at 1×107 cfu, at each inoculation site. Xenograft volumes were calculated as previously described (Zhang et al., 2012).
Clinical specimens
This study was approved by the University Hospitals of Cleveland Institutional Review Board. A total of 19 cases diagnosed with colonic adenocarcinoma and 16 cases of adenomas were retrieved from files at the Department of Pathology, University Hospitals Case Medical Center, Cleveland, Ohio. H&E slides were reviewed to confirm the presence of adenocarcinoma (or adenoma). A representative block of colon adenocarcinoma (or adenoma) and a block of normal colon from the same patient were used. In addition, normal colon tissues were derived from 14 individuals undergoing resection for benign colon pathology or resection of adjacent organs. Exclusion criteria were history of gastrointestinal malignancies, presence of prominent inflammation or abscess, and history of inflammatory bowel disease. All cases were from within the last 12 months. Genomic DNA was extracted from formalin-fixed paraffin-embedded tissue samples as previously described (Tang et al., 2009). RNA was extracted using PureLink FFPE Total RNA Isolation Kit (Invitrogen) or RNeasy FFPE Kit (Qiagen).
Real-time Quantitative PCR (qPCR)
Total RNA was extracted from CRC cells, xenografts, or clinical specimens. cDNA synthesis and RT-PCR were performed as previously described, using primers listed in Table S1 (Lebreton et al., 2011). Data were analyzed by the ΔΔCt method (Livak and Schmittgen, 2001) and normalized to the GAPDH or Fn-specific 16S rRNA. To quantify the fadA gene copies, plasmid carrying fadA was serially diluted to 102–108 fadA copies/μl and used to generate standard curves for Ct values. The fadA gene copies in the clinical samples were calculated based on the standard curves. Each experiment was performed in triplicates and repeated at least three times.
Statistical analysis
The differences between groups were examined by two-tailed one-way analysis of variance (ANOVA) followed by Student-Newman-Keuls (SNK) test. For the clinical specimens the Kruskal–Wallis non-parametric test was performed followed by Conover test. p<0.05 were considered statistically significant.
Supplementary Material
F. nucleatum (Fn) adhesin FadA binds E-cadherin and promotes CRC cell proliferation
A peptide from the FadA-binding region of E-cadherin prevents Fn mediated carcinogenesis
FadA promotes inflammation and E-cadherin-mediated CRC tumor growth in xenograft mice
Tissue from human adenomas and adenocarcinomas have elevated FadA expression levels
ACKNOWLEDGMENTS
We thank Zhenghe Wang and Sanford Markowitz for stimulating discussions, Zhenghe Wang for providing human cancer cell lines HCT116, DLD1, SW480, HT29, and RKO, and Bert Vogelstein and Zhenghe Wang for providing the HCT116 β-catenin−/− cell line. This work was supported by the National Institute of Dental and Craniofacial Research grant RO1 DE14924 to Y.W.H.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS Y.W.H conceived, designed, and supervised the study. W.L. provided clinical specimens. X.W., M.R., Y.H. and G.C. performed the experiments. Y.W.H., X.W., M.R. and W. L. interpreted the data and wrote the manuscript.
The authors declare no conflict of interest.
REFERENCES
- ACS Cancer Facts & Figures 2012. American Cancer Society (ACS) 2012:1–66. [Google Scholar]
- Arthur JC, Perez-Chanona E, Muhlbauer M, Tomkovich S, Uronis JM, Fan TJ, Campbell BJ, Abujamel T, Dogan B, Rogers AB, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science (New York, NY. 2012;338:120–123. doi: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron JA, Sandler RS. Nonsteroidal anti-inflammatory drugs and cancer prevention. Annual review of medicine. 2000;51:511–523. doi: 10.1146/annurev.med.51.1.511. [DOI] [PubMed] [Google Scholar]
- Bartlett DL, Chu E. Can metastatic colorectal cancer be cured? Oncology. 2012;26:266–275. [PubMed] [Google Scholar]
- Bryant DM, Stow JL. The ins and outs of E-cadherin trafficking. Trends in cell biology. 2004;14:427–434. doi: 10.1016/j.tcb.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, Barnes R, Watson P, Allen-Vercoe E, Moore RA, et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012;22:299–306. doi: 10.1101/gr.126516.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan TA, Wang Z, Dang LH, Vogelstein B, Kinzler KW. Targeted inactivation of CTNNB1 reveals unexpected effects of beta-catenin mutation. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:8265–8270. doi: 10.1073/pnas.082240999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Liu F, Ling Z, Tong X, Xiang C. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PLoS One. 2012;7:e39743. doi: 10.1371/journal.pone.0039743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuevas-Ramos G, Petit CR, Marcq I, Boury M, Oswald E, Nougayrede JP. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:11537–11542. doi: 10.1073/pnas.1001261107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorudi S, Sheffield JP, Poulsom R, Northover JM, Hart IR. E-cadherin expression in colorectal cancer. An immunocytochemical and in situ hybridization study. The American journal of pathology. 1993;142:981–986. [PMC free article] [PubMed] [Google Scholar]
- Ekbom A, Helmick C, Zack M, Adami HO. Ulcerative colitis and colorectal cancer. A population-based study. The New England journal of medicine. 1990;323:1228–1233. doi: 10.1056/NEJM199011013231802. [DOI] [PubMed] [Google Scholar]
- Fardini Y, Wang X, Temoin S, Nithianantham S, Lee D, Shoham M, Han YW. Fusobacterium nucleatum adhesin FadA binds vascular endothelial cadherin and alters endothelial integrity. Mol Microbiol. 2011;82:1468–1480. doi: 10.1111/j.1365-2958.2011.07905.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbiner BM. Regulation of cadherin-mediated adhesion in morphogenesis. Nat Rev Mol Cell Biol. 2005;6:622–634. doi: 10.1038/nrm1699. [DOI] [PubMed] [Google Scholar]
- Han YW. Laboratory maintenance of fusobacteria. In: Coico R, Kowalik T, Quarles J, Stevenson B, Taylor R, editors. Current Protocols in Microbiology. John Wiley & Sons, Inc.; 2006. [DOI] [PubMed] [Google Scholar]
- Han YW. Fusobacterium nucleatum interaction with host cells. In: Kolenbrander PE, editor. Chapter 15 in Oral Microbial Communities: Genomic Inquiry and Interspecies Communication. ASM Press; 2011. [Google Scholar]
- Han YW, Fardini Y, Chen C, Iacampo KG, Peraino VA, Shamonki JM, Redline RW. Term stillbirth caused by oral Fusobacterium nucleatum. Obstetrics and gynecology. 2010;115:442–445. doi: 10.1097/AOG.0b013e3181cb9955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han YW, Ikegami A, Rajanna C, Kawsar HI, Zhou Y, Li M, Sojar HT, Genco RJ, Kuramitsu HK, Deng CX. Identification and characterization of a novel adhesin unique to oral fusobacteria. J Bacteriol. 2005;187:5330–5340. doi: 10.1128/JB.187.15.5330-5340.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han YW, Redline RW, Li M, Yin L, Hill GB, McCormick TS. Fusobacterium nucleatum induces premature and term stillbirths in pregnant mice: implication of oral bacteria in preterm birth. Infect Immun. 2004;72:2272–2279. doi: 10.1128/IAI.72.4.2272-2279.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han YW, Shen T, Chung P, Buhimschi IA, Buhimschi CS. Uncultivated bacteria as etiologic agents of intra-amniotic inflammation leading to preterm birth. J Clin Microbiol. 2009;47:38–47. doi: 10.1128/JCM.01206-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han YW, Shi W, Huang GT, Kinder Haake S, Park NH, Kuramitsu H, Genco RJ. Interactions between periodontal bacteria and human oral epithelial cells: Fusobacterium nucleatum adheres to and invades epithelial cells. Infect Immun. 2000;68:3140–3146. doi: 10.1128/iai.68.6.3140-3146.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinzler KW, Vogelstein B. In: The Genetic Basis of Human Cancer. Vogelstein B, Kinzler KW, editors. McGraw-Hill; New York: 2002. pp. 565–587. [Google Scholar]
- Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, Clancy TE, Chung DC, Lochhead P, Hold GL, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor immune microenvironment. Cell Host Microbe. 2013 doi: 10.1016/j.chom.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, Ojesina AI, Jung J, Bass AJ, Tabernero J, et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012;22:292–298. doi: 10.1101/gr.126573.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le TL, Joseph SR, Yap AS, Stow JL. Protein kinase C regulates endocytosis and recycling of E-cadherin. American journal of physiology. 2002;283:C489–499. doi: 10.1152/ajpcell.00566.2001. [DOI] [PubMed] [Google Scholar]
- Lebreton A, Lakisic G, Job V, Fritsch L, Tham TN, Camejo A, Mattei PJ, Regnault B, Nahori MA, Cabanes D, et al. A bacterial protein targets the BAHD1 chromatin complex to stimulate type III interferon response. Science (New York, NY. 2011;331:1319–1321. doi: 10.1126/science.1200120. [DOI] [PubMed] [Google Scholar]
- Lee SH, Hu LL, Gonzalez-Navajas J, Seo GS, Shen C, Brick J, Herdman S, Varki N, Corr M, Lee J, et al. ERK activation drives intestinal tumorigenesis in Apc(min/+) mice. Nature medicine. 2010;16:665–670. doi: 10.1038/nm.2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods (San Diego, Calif. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Martin HM, Campbell BJ, Hart CA, Mpofu C, Nayar M, Singh R, Englyst H, Williams HF, Rhodes JM. Enhanced Escherichia coli adherence and invasion in Crohn's disease and colon cancer. Gastroenterology. 2004;127:80–93. doi: 10.1053/j.gastro.2004.03.054. [DOI] [PubMed] [Google Scholar]
- McCoy AN, et al. Fusobacterium is associated with colorectal adenomas. PloS one. 2013;8:e53653. doi: 10.1371/journal.pone.0053653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohri Y. Prognostic significance of E-cadherin expression in human colorectal cancer tissue. Surgery today. 1997;27:606–612. doi: 10.1007/BF02388215. [DOI] [PubMed] [Google Scholar]
- Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science (New York, NY. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- Nithianantham S, Xu M, Yamada M, Ikegami A, Shoham M, Han YW. Crystal structure of FadA adhesin from Fusobacterium nucleatum reveals a novel oligomerization motif, the leucine chain. J Biol Chem. 2009;284:3865–3872. doi: 10.1074/jbc.M805503200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peifer M, Polakis P. Wnt signaling in oncogenesis and embryogenesis--a look outside the nucleus. Science (New York, NY. 2000;287:1606–1609. doi: 10.1126/science.287.5458.1606. [DOI] [PubMed] [Google Scholar]
- Plottel CS, Blaser MJ. Microbiome and malignancy. Cell host & microbe. 2011;10:324–335. doi: 10.1016/j.chom.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekirov I, Russell SL, Antunes LC, Finlay BB. Gut microbiota in health and disease. Physiological reviews. 2010;90:859–904. doi: 10.1152/physrev.00045.2009. [DOI] [PubMed] [Google Scholar]
- Strauss J, et al. Invasive potential of gut mucosa-derived Fusobacterium nucleatum positively correlates with IBD status of the host. Inflamm Bowel Dis. 2011;17:1971–8. doi: 10.1002/ibd.21606. [DOI] [PubMed] [Google Scholar]
- Swidsinski A, Khilkin M, Kerjaschki D, Schreiber S, Ortner M, Weber J, Lochs H. Association between intraepithelial Escherichia coli and colorectal cancer. Gastroenterology. 1998;115:281–286. doi: 10.1016/s0016-5085(98)70194-5. [DOI] [PubMed] [Google Scholar]
- Tang W, et al. Cold Spring Harbor protocols 2009, pdb prot5138. 2009. DNA extraction from formalin-fixed, paraffin-embedded tissue. [DOI] [PubMed] [Google Scholar]
- Temoin S, Wu KL, Wu V, Shoham M, Han YW. Signal peptide of FadA adhesin from Fusobacterium nucleatum plays a novel structural role by modulating the filament's length and width. FEBS Lett. 2012;586:1–6. doi: 10.1016/j.febslet.2011.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremaroli V, Backhed F. Functional interactions between the gut microbiota and host metabolism. Nature. 2012;489:242–249. doi: 10.1038/nature11552. [DOI] [PubMed] [Google Scholar]
- von Kleist L, Stahlschmidt W, Bulut H, Gromova K, Puchkov D, Robertson MJ, MacGregor KA, Tomilin N, Pechstein A, Chau N, et al. Role of the clathrin terminal domain in regulating coated pit dynamics revealed by small molecule inhibition. Cell. 2011;146:471–484. doi: 10.1016/j.cell.2011.06.025. [DOI] [PubMed] [Google Scholar]
- Wang XW, Buhimschi CS, Temoin S, Bhandar V, Han YW, IA B. Comparative microbial analysis of paired amniotic fluid and cord blood from pregnancies complicated by preterm and early-onset neonatal sepsis. PLoS One. 2013;8:56131. doi: 10.1371/journal.pone.0056131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Lim KC, Huang J, Saidi RF, Sears CL. Bacteroides fragilis enterotoxin cleaves the zonula adherens protein, E-cadherin. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:14979–14984. doi: 10.1073/pnas.95.25.14979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, Huso DL, Brancati FL, Wick E, McAllister F, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nature medicine. 2009;15:1016–1022. doi: 10.1038/nm.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Morin PJ, Maouyo D, Sears CL. Bacteroides fragilis enterotoxin induces c-Myc expression and cellular proliferation. Gastroenterology. 2003;124:392–400. doi: 10.1053/gast.2003.50047. [DOI] [PubMed] [Google Scholar]
- Xu M, Yamada M, Li M, Liu H, Chen SG, Han YW. FadA from Fusobacterium nucleatum utilizes both secreted and nonsecreted forms for functional oligomerization for attachment and invasion of host cells. J Biol Chem. 2007;282:25000–25009. doi: 10.1074/jbc.M611567200. [DOI] [PubMed] [Google Scholar]
- Yang J, Zhang W, Evans PM, Chen X, He X, Liu C. Adenomatous polyposis coli (APC) differentially regulates beta-catenin phosphorylation and ubiquitination in colon cancer cells. The Journal of biological chemistry. 2006;281:17751–17757. doi: 10.1074/jbc.M600831200. [DOI] [PubMed] [Google Scholar]
- Zhang P, Guo A, Possemato A, Wang C, Beard L, Carlin C, Markowitz SD, Polakiewicz RD, Wang Z. Identification and functional characterization of p130Cas as a substrate of protein tyrosine phosphatase nonreceptor 14. Oncogene. 2012 doi: 10.1038/onc.2012.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.