

Abstract
Background
We report our findings in two siblings with late-onset cone–rod dystrophy (CRD) with no visible macular degeneration.
Cases and methods
Case 1 was an 82-year-old man who first noticed a decrease in vision and color blindness in his early seventies. His mother and younger sister also had visual disturbances. His decimal visual acuity was 0.3 in the right eye and 0.2 in the left eye. Ophthalmoscopy showed normal fundi, and fluorescein angiography was also normal in both eyes. The photopic single flash and flicker eletroretinograms (ERGs) were severely attenuated and the scotopic ERGs were slightly reduced in both eyes. Case 2 was the 80-year-old younger sister of Case 1. She first noticed a decline in vision and photophobia in both eyes in her early seventies. Her decimal visual acuity was 0.4 in the right eye and 0.2 in the left eye. Ophthalmoscopy showed mottling of the retinal pigment epithelium in the midperiphery with no visible macular degeneration. The photopic single flash and flicker ERGs were severely attenuated, and the scotopic ERGs were slightly reduced in both eyes.
Conclusion
These siblings are the oldest reported cases of CRD with no visible macular degeneration. Thus, CRD should be considered in patients with reduced visual acuity, color blindness, and photophobia even if they are older than 70 years.
Keywords: cone-rod dystrophy, peripheral cone dystrophy, occult macular dystrophy, late onset, macular degeneration, negative ERG
Introduction
Cone–rod dystrophy (CRD) is an inherited retinal dystrophy that is characterized by reduced visual acuity, color blindness, and photophobia.1–3 The age of onset generally ranges from the teens to the thirties,2,3 and most patients with CRD have atrophic macular degeneration with a bull’s eye lesion, or midperipheral degeneration in the late stages of the disease.2,3 Patients with CRD can also have a central scotoma, and constriction of the peripheral visual fields at the end stages of the disease.2,3 The cone-driven electroretinograms (ERGs) are attenuated even in the early stages, and this reduction is essential for making a diagnosis of CRD.2,3
Cases of atypical CRD, such as CRD with normal fundi,4–7 occult macular dystrophy (OMD; Miyake disease),8–11 peripheral cone dystrophy,12–15 fundus albipunctatus associated with cone dystrophy,16,17 and cone dystrophy with supernormal rod ERGs18,19 have also been reported.
We present the clinical features of two siblings with CRD who had only mild fundus abnormalities. Their mother was also known to have visual difficulties, but was already dead at the time the two siblings were examined.
Cases and methods
Our two cases were siblings who underwent ophthalmoscopy, visual acuity measurements, fundus fluorescein angiography (FA), Goldmann kinetic perimetry, Farnsworth D-15 test, Goldmann–Weekers dark adaptometry, full-field ERGs, multifocal ERGs (mfERGs), and optical coherence tomography (OCT). OCT images recorded from 24 age-matched normal subjects served as controls.
The ERGs and mfERGs were recorded according to the protocol recommended by the International Society for Clinical Electrophysiology of Vision. The mfERGs were recorded with the VERIS™ recording system (Electro-Diagnostic Imaging, Inc, Redwood, CA, USA). A Cirrus™ high-definition spectral-domain OCT instrument (Carl Zeiss Meditec AG, Jena, Germany) was used to obtain the OCT images.
Case reports
Case 1 was an 82-year-old man who first noticed a decrease in his vision and color vision in both eyes in his early seventies. His decimal best-corrected visual acuity (BCVA) at our initial examination was 0.3 oculus dexter (OD) with +3.25 diopters (D) and 0.2 oculus sinister (OS) with +3.25 D. His pupillary light reflexes and intraocular pressures were normal in both eyes. There was no history of the use of retinotoxic drugs. His cataracts were removed in both eyes at age 75; however, his vision was not improved.
Ophthalmoscopy showed that the fundus was normal in both eyes, and FA showed small hyperfluorescent spots at the parafoveal region of the left eye (Figure 1). Goldmann kinetic perimetry showed a central scotoma in both eyes (Figure 2), and Farnsworth D-15 test showed a tritan axis error. Goldmann–Weekers dark adaptometry revealed a slight elevation of the threshold in both eyes (Figure 3). The full-field scotopic ERGs elicited by both low- and high-intensity stimuli were slightly reduced. The scotopic b-wave elicited by a high-intensity stimulus was smaller than the a-wave, resulting in a negative-type ERG (Figure 4). On the other hand, photopic single-flash and 30 Hz flicker ERGs were severely attenuated in both eyes (Figure 4). The mfERGs were nonrecordable in the central area, but small responses were recorded in the midperiphery (Figure 5). The photoreceptor inner segment/outer segment junction line was indistinct in the OCT images (Figure 6). The thickness of the outer nuclear layer was 76 μm OD and 65 μm OS (normal mean 172 ± 17 μm) at the fovea, 65 μm OD and 43 μm OS (normal mean 128 ± 19 μm) at 0.5 mm superior to the fovea, and 65 μm OD and 54 μm OS (normal mean 135 ± 23 μm) at 0.5 mm inferior to the fovea. At 2 mm superior to the fovea, the thickness of the outer nuclear layer was 65 μm OD and 43 μm OS (normal mean 97 ± 13 μm), and at 2 mm inferior to the fovea it was 43 μm OD and 54 μm OS (normal mean 88 ± 18 μm). Thus, the outer nuclear layer was thin, especially in the parafoveal region in both eyes (Figure 6). The thickness of the middle and inner layers of the retina were within normal limits.
Figure 1.

Fundus photographs (Fundi) and FA.
Abbreviations: OD, oculus dexter; OS, oculus sinister; FA, fluorescein fundus angiograms.
Figure 2.

Results of Goldmann kinetic perimetry.
Abbreviations: L, left; R, right.
Figure 3.
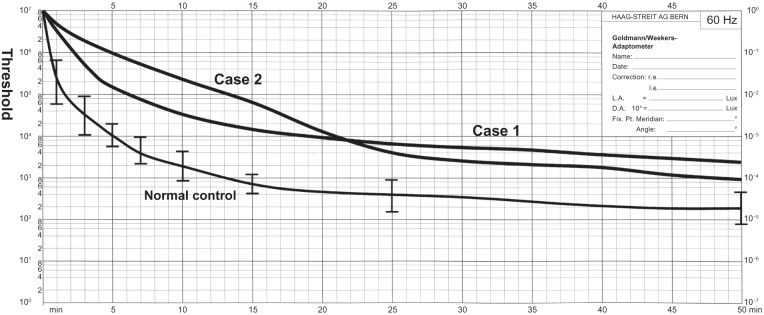
Results of Goldmann–Weekers dark adaptometry.
Note: The thin line indicates the average ± standard deviation isopter of normal controls.
Figure 4.

International Society for Clinical Electrophysiology of Vision-standard ERGs.
Note: The responses from both eyes are superimposed.
Abbreviation: ERG, electroretinography.
Figure 5.

Multifocal ERGs.
Note: Responses were contaminated with much noise which was caused by photophobia.
Abbreviations: OS, oculus sinister; OD, oculus dexter; ERG, electroretinography.
Figure 6.

Results of optical coherence tomography.
Note: The photoreceptor inner segment/outer segment junction line is indistinct in the macular area.
Abbreviations: OD, oculus dexter; OS, oculus sinister; CRD, cone–rod dystrophy; MD, macular degeneration.
Case 2 was the younger sister of case 1. She was 80 years old and had first noticed a decrease in her vision and photophobia in both eyes in her early seventies. Her cataract was removed in both eyes at age 76; however, her vision was not improved.
Our examination showed that her decimal BCVA was 0.4 OD with +0.25 D and 0.2 OS with +0.5 D. Her pupillary light reflexes and intraocular pressures were normal in both eyes. There was no history of retinotoxic drug use.
Her fundus was normal except for a slight mottling of the retinal pigment epithelium in the midperiphery. No macular degeneration was seen in either eye (Figure 1). FA was not performed because of her allergy to fluorescein sodium. Goldmann kinetic perimetry revealed a central scotoma in the left eye and a mild constriction of the visual fields in both eyes (Figure 2). Farnsworth D-15 test showed tritan axis errors in both eyes. Goldmann–Weekers dark adaptometry showed a slight elevation of the light threshold in both eyes (Figure 3). The full-field scotopic ERGs elicited by low-intensity stimuli were slightly reduced, and the scotopic high-intensity ERGs were normal, except the oscillatory potentials were reduced. The photopic single-flash and 30 Hz flicker ERGs were nonrecordable in both eyes (Figure 4). The mfERGs were nonrecordable in the right eye and reduced in the central and midperipheral areas of the left eye (Figure 5). OCT showed similar findings to her elder brother in the macular area (Figure 6). The photoreceptor inner segment/outer segment junction line was indistinct. The thickness of the outer nuclear layer was 130 μm OD and 129 μm OS (normal mean 172 ± 17 μm) at the fovea, 43 μm OD and 65 μm OS (normal mean 128 ± 19 μm) at 0.5 mm superior to the fovea, and 32 μm OD and 22 μm OS (normal mean 135 ± 23 μm) at 0.5 mm inferior to the fovea. At 2 mm superior to the fovea, the thickness of the outer nuclear layer was 65 μm OD and 76 μm OS (normal mean 97 ± 13 μm), and at 2 mm inferior to the fovea it was 65 μm OD and 43 μm OS (normal mean 88 ± 18 μm). Thus, the outer nuclear layer was thin, especially in the parafoveal region of both eyes (Figure 6).
Discussion
These two high-aged siblings, both in their eighties, had severely attenuated cone ERGs and mfERGs, and there was no evidence of visible macular abnormality. Case 1 had small hyperfluorescent spots in the fluorescein angiograms, and case 2 had mottling in the midperipheral fundus. Goldmann kinetic perimetry showed a central scotoma and reduced sensitivity in the area surrounding the scotoma in both patients. The mfERGs were nonrecordable in the foveal area, and only very small responses were recorded in the midperiphery; these findings are consistent with results of the Goldmann kinetic perimetry.
We reviewed 497 CRD cases in 181 papers. Among these, 86 eyes in 43 patients in 20 papers were reported to have either normal fundus or only subtle fundus abnormalities (Figure 7).4–7,20–35
Figure 7.

Age and visual acuity in cases with CRD in the past 181 papers published between 1963 and 2012.
Abbreviation: CRD, cone–rod dystrophy.
The vision of these cases is summarized in Figure 7, and the findings suggested that the vision in CRD cases with normal fundi is better than that in CRD cases with retinal/macular degeneration. Thirty-eight of 86 eyes (44%) with CRD and normal fundi had visual acuity better than 0.5, whereas 169 eyes of 908 eyes (19%) with retinal/macular degeneration had visual acuity better than 0.5.
Sixty-two of 86 eyes (72%) of the CRD cases with normal or subtle fundus abnormalities4–7,20–35 had a family member with visible retinal degeneration, except the cases noted in studies by Ohba,4 Rowe et al,5 Miyake,6 and Hayashi et al.7 Ohba4 reported on four CRD cases with normal fundi at ages 22 years, 23 years, 41 years, and 45 years. The patients’ decimal BCVAs ranged from 0.1 to 0.07 in both eyes, and all were sporadic cases.4 Rowe et al5 reported four cases that were all sporadic and their ages were 55 years, 56 years, 60 years, and 68 years. Their decimal BCVAs ranged from 0.5 to 0.1 in both eyes.5 Miyake6 reported three cases of CRD with normal fundi from the same family whose ages were 32 years, 35 years, and 66 years. Their BCVAs were not reported. A sporadic, 53-year-old CRD case with normal fundi reported by Hayashi et al7 had BCVAs of 0.1 OD and 0.7 OS.
CRD cases with normal fundi and no family history of retinal degeneration have a late onset, with an average age of 46 years and an average BCVA of 0.24. In comparison, the CRD cases with retinal or macular degeneration had an average age at onset of 23 years and the average BCVA was 0.09. The two siblings in our study had a very late onset with a positive family history and normal fundi. Their BCVAs after cataract removal ranged from 0.4 to 0.2 in both eyes, which are beyond the normal range reported,36 and are comparable to the CRD cases with normal fundi and no family history of retinal degeneration.4–7
OMD is a kind of cone dystrophy with a normal fundus appearance.8–11 Recently, a point mutation was found in the RP1L1 gene in patients with autosomal-dominant OMD.10 The ERG findings on our siblings are different from these cases of OMD because of the almost nonrecordable full-field cone response in our cases, which is due to diffuse cone dysfunction in these cases.
Peripheral cone dystrophy (or peripheral cone disease) is another kind of cone dystrophy that is characterized by cone dysfunction in the midperiphery to the periphery.12–15 All of the past cases were reported as peripheral cone dystrophy and all had normal fundi. The gene mutation causing this cone dystrophy has not been identified. The results of mfERGs in our two cases were different from past cases of peripheral cone dystrophy because the mfERGs from the macula were nonrecordable. However, the two siblings in our study were probably at an advanced stage of `the peripheral cone dystrophy because their outer nuclear layer at the fovea was well preserved compared with that of patients with cone dystrophy and a bull’s eye lesion (Figure 6).
Several gene mutations have been identified in cases of CRD. Mutations of the CRX gene,26,27,32,37–40 the GUCY2D gene,29,33,35,41 the GUCA1A gene,42 and peripherin/RDS gene43 have been found in cases of autosomal-dominant CRD. Mutations of the ABCA4 gene,44CNGB3 gene,28 and KCNV2 gene,19,30,31,34 have been reported for the autosomal-recessive cases of CRD. Mutations of the CACNA1F gene45 and RPGR gene46 have been detected in X-linked recessive cases of CRD.
Mutations of the CRX gene,26,27,32 as well as the GUCY2D,29,33,35KCNV2,30,31,34 and CNGB328 genes might be the cause of CRD in eyes with normal or mild fundus abnormalities. Cases with mutations in the CRX gene have diverse phenotypes ranging from Leber’s congenital amaurosis39,47 to CRD with normal fundi.26,27,32 Sohocki et al39 suggested that these varied phenotypes in patients with the CRX-gene mutations were due to deletion or point mutations in the gene. Deletion mutations in this gene would result in late-onset and mild CRD.39 Our cases are similar to CRD cases with mutations in the CRX gene.
Swain et al38 and Itabashi et al40 reported CRD cases caused by a CRX gene mutation that had negative ERGs. Case 1 in our study had a reduced b-wave in the high-intensity ERGs that resembled the ERGs reported by Swain et al38 and Itabashi et al.40
Mutations in other genes such as the GUCY2D gene,26,27,32KCNV2 gene30,31,34 and CNGB3 gene28 have also been reported to cause CRD with normal fundi; however, these patients were relatively young and some had supernormal rod responses, unlike our cases.
We investigated the gene in the siblings presented in our study using next-generation sequencing. However, detection of the causative mutation in these siblings was difficult because most of their family members, including their parents, were already deceased.
In summary, we report our findings in two siblings with late-onset CRD. Ophthalmoscopy showed that the macula was essentially normal in both cases. The scotopic ERGs were slightly reduced, but the photopic ERGs were nonrecordable. We recommended that older patients, >.45 years of age, who had good vision earlier in their lives but had developed reduced vision, color vision abnormalities, and photophobia be examined by electroretinography to rule out CRD.
Acknowledgments
This research was supported in part by research grants from the Ministry of Health, Labor and Welfare, Japan, and Japan Society for the Promotion of Science, Japan.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Steinmetz RD, Ogle KN, Rucker CW. Some physiologic considerations of hereditary macular degeneration. Am J Ophthalmol. 1956;42(4 Pt 2):304–319. doi: 10.1016/0002-9394(56)90382-8. [DOI] [PubMed] [Google Scholar]
- 2.Krill AE, Deutman AF, Fishman M. The cone degenerations. Doc Ophthalmol. 1973;35(1):1–80. doi: 10.1007/BF00234530. [DOI] [PubMed] [Google Scholar]
- 3.Thiadens AA, Phan TM, Zekveld-Vroon RC, et al. Writing Committee for the Cone Disorders Study Group Consortium Clinical course, genetic etiology, and visual outcome in cone and cone-rod dystrophy. Ophthalmology. 2012;119(4):819–826. doi: 10.1016/j.ophtha.2011.10.011. [DOI] [PubMed] [Google Scholar]
- 4.Ohba N. Progressive cone dystrophy; four cases of unusual form. Jpn J Ophthalmol. 1974;18(1):50–69. [Google Scholar]
- 5.Rowe SE, Trobe JD, Sieving PA. Idiopathic photoreceptor dysfunction causes unexplained visual acuity loss in later adulthood. Ophthalmology. 1990;97(12):1632–1637. doi: 10.1016/s0161-6420(90)32366-7. [DOI] [PubMed] [Google Scholar]
- 6.Miyake Y. Phenotypes of cone dysfunction syndrome. Folia Ophthalmol Jpn. 2000;51(8):725–733. Japanese. [Google Scholar]
- 7.Hayashi K, Yamada H, Wakakura M. A case of late-onset cone-rod dystrophy (CRD) which lacked ophthalmoscopic findings and diagnosed by electrophysiological examination. Neuro-Ophthalmol Japan. 2005;22(2):246–252. Japanese. [Google Scholar]
- 8.Miyake Y, Ichikawa K, Shiose Y, Kawase Y. Hereditary macular dystrophy without visible fundus abnormality. Am J Ophthalmol. 1989;108(3):292–299. doi: 10.1016/0002-9394(89)90120-7. [DOI] [PubMed] [Google Scholar]
- 9.Miyake Y, Horiguchi M, Tomita N, et al. Occult macular dystrophy. Am J Ophthalmol. 1996;122(5):644–653. doi: 10.1016/s0002-9394(14)70482-9. [DOI] [PubMed] [Google Scholar]
- 10.Akahori M, Tsunoda K, Miyake Y, et al. Dominant mutations in RP1L1 are responsible for occult macular dystrophy. Am J Hum Genet. 2010;87(3):424–429. doi: 10.1016/j.ajhg.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsunoda K, Usui T, Hatase T, et al. Clinical characteristics of occult macular dystrophy in family with mutation of RP1L1 gene. Retina. 2012;32(6):1135–1147. doi: 10.1097/IAE.0b013e318232c32e. [DOI] [PubMed] [Google Scholar]
- 12.Pinckers A, Deutman AF. Peripheral cone disease. Ophthalmologica. 1977;174(3):145–150. doi: 10.1159/000308592. [DOI] [PubMed] [Google Scholar]
- 13.Kondo M, Miyake Y, Kondo N, Ueno S, Takakuwa H, Terasaki H. Peripheral cone dystrophy: a variant of cone dystrophy with predominant dysfunction in the peripheral cone system. Ophthalmology. 2004;111(4):732–739. doi: 10.1016/j.ophtha.2003.07.005. [DOI] [PubMed] [Google Scholar]
- 14.Okuno T, Oku H, Kurimoto T, Oono S, Ikeda T. Peripheral cone dystrophy in an elderly man. Clin Experiment Ophthalmol. 2008;36(9):897–899. doi: 10.1111/j.1442-9071.2008.01905.x. [DOI] [PubMed] [Google Scholar]
- 15.Baek J, Lee HK, Kim US. Spectral domain optical coherence tomography findings in bilateral peripheral cone dystrophy. Doc Ophthalmol. 2013;126(3):247–251. doi: 10.1007/s10633-013-9377-y. [DOI] [PubMed] [Google Scholar]
- 16.Miyake Y, Shiroyama N, Sugita S, Horiguchi M, Yagasaki K. Fundus albipunctatus associated with cone dystrophy. Br J Ophthalmol. 1992;76(6):375–379. doi: 10.1136/bjo.76.6.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakamura M, Hotta Y, Tanikawa A, Terasaki H, Miyake Y. A high association with cone dystrophy in Fundus albipunctatus caused by mutations of the RDH5 gene. Invest Ophthalmol Vis Sci. 2000;41(12):3925–3932. [PubMed] [Google Scholar]
- 18.Gouras P, Eggers HM, MacKay CJ. Cone dystrophy, nyctalopia, and supernormal rod responses. A new retinal degeneration. Arch Ophthalmol. 1983;101(5):718–724. doi: 10.1001/archopht.1983.01040010718003. [DOI] [PubMed] [Google Scholar]
- 19.Wu H, Cowing JA, Michaelides M, et al. Mutations in the gene KCNV2 encoding a voltage-gated potassium channel subunit cause “cone dystrophy with supernormal rod electroretinogram” in humans. Am J Hum Genet. 2006;79(3):574–579. doi: 10.1086/507568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goodman G, Ripps H, Siegel IM. Cone dysfunction syndromes. Arch Ophthalmol. 1963;70:214–231. doi: 10.1001/archopht.1963.00960050216013. [DOI] [PubMed] [Google Scholar]
- 21.Berson EL, Gouras P, Gunkel RD. Progressive cone degeneration, dominantly inherited. Arch Ophthalmol. 1968;80(1):77–83. doi: 10.1001/archopht.1968.00980050079011. [DOI] [PubMed] [Google Scholar]
- 22.François J, de Rouck A, Verriest G, de Laey JJ, Cambie E. Progressive generalized cone dysfunction. Ophthalmologica. 1974;169(4):255–284. doi: 10.1159/000307127. [DOI] [PubMed] [Google Scholar]
- 23.Ripps H, Noble KG, Greenstein VC, Siegel IM, Carr RE. Progressive cone dystrophy. Ophthalmology. 1987;94(11):1401–1409. doi: 10.1016/s0161-6420(87)33272-5. [DOI] [PubMed] [Google Scholar]
- 24.van Schooneveld MJ, Went LN, Oosterhuis JA. Dominant cone dystrophy starting with blue cone involvement. Br J Ophthalmol. 1991;75(6):332–336. doi: 10.1136/bjo.75.6.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iijima H, Yamaguchi S, Kogure S, Hosaka O, Shibutani T. Electroretinogram in cone dystrophy. Jpn J Ophthalmol. 1991;35(4):453–466. [PubMed] [Google Scholar]
- 26.Jacobson SG, Cideciyan AV, Huang Y, et al. Retinal degenerations with truncation mutations in the cone-rod homeobox (CRX) gene. Invest Ophthalmol Vis Sci. 1998;39(12):2417–2426. [PubMed] [Google Scholar]
- 27.Tzekov RT, Sohocki MM, Daiger SP, Birch DG. Visual phenotype in patients with Arg41Gln and ala196+1 bp mutations in the CRX gene. Ophthalmic Genet. 2000;21(2):89–99. [PubMed] [Google Scholar]
- 28.Michaelides M, Aligianis IA, Ainsworth JR, et al. Progressive cone dystrophy associated with mutation in CNGB3. Invest Ophthalmol Vis Sci. 2004;45(6):1975–1982. doi: 10.1167/iovs.03-0898. [DOI] [PubMed] [Google Scholar]
- 29.Smith M, Whittock N, Searle A, Croft M, Brewer C, Cole M. Phenotype of autosomal dominant cone-rod dystrophy due to the R838C mutation of the GUCY2D gene encoding retinal guanylate cyclase-1. Eye (Lond) 2007;21(9):1220–1225. doi: 10.1038/sj.eye.6702612. [DOI] [PubMed] [Google Scholar]
- 30.Wissinger B, Dangel S, Jägle H, et al. Cone dystrophy with supernormal rod response is strictly associated with mutations in KCNV2. Invest Ophthalmol Vis Sci. 2008;49(2):751–757. doi: 10.1167/iovs.07-0471. [DOI] [PubMed] [Google Scholar]
- 31.Ben Salah S, Kamei S, Sénéćhal A, et al. Novel KCNV2 mutations in cone dystrophy with supernormal rod electroretinogram. Am J Ophthalmol. 2008;145(6):1099–1106. doi: 10.1016/j.ajo.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 32.Kitiratschky VB, Nagy D, Zabel T, et al. Cone and cone-rod dystrophy segregating in the same pedigree due to the same novel CRX gene mutation. Br J Ophthalmol. 2008;92(8):1086–1091. doi: 10.1136/bjo.2007.133231. [DOI] [PubMed] [Google Scholar]
- 33.Kitiratschky VB, Wilke R, Renner AB, et al. Mutation analysis identifies GUCY2D as the major gene responsible for autosomal dominant progressive cone degeneration. Invest Ophthalmol Vis Sci. 2008;49(11):5015–5023. doi: 10.1167/iovs.08-1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robson AG, Webster AR, Michaelides M, et al. “Cone dystrophy with supernormal rod electroretinogram”: a comprehensive genotype/phenotype study including fundus autofluorescence and extensive electrophysiology. Retina (Philadelphia, Pa) 2010;30(1):51–62. doi: 10.1097/IAE.0b013e3181bfe24e. [DOI] [PubMed] [Google Scholar]
- 35.Garcia-Hoyos M, Auz-Alexandre CL, Almoguera B, et al. Mutation analysis at codon 838 of the guanylate cyclase 2D gene in Spanish families with autosomal dominant cone, cone-rod, and macular dystrophies. Mol Vis. 2011;17:1103–1109. [PMC free article] [PubMed] [Google Scholar]
- 36.Sjöstrand J, Laatikainen L, Hirvelä H, Popovic Z, Jonsson R. The decline in visual acuity in elderly people with healthy eyes or eyes with early age-related maculopathy in two Scandinavian population samples. Acta Ophthalmol. 2011;89(2):116–123. doi: 10.1111/j.1755-3768.2009.01653.x. [DOI] [PubMed] [Google Scholar]
- 37.Freund CL, Gregory-Evans CY, Furukawa T, et al. Cone-rod dystrophy due to mutations in a novel photoreceptor-specific homeobox gene (CRX) essential for maintenance of the photoreceptor. Cell. 1997;91(4):543–553. doi: 10.1016/s0092-8674(00)80440-7. [DOI] [PubMed] [Google Scholar]
- 38.Swain PK, Chen S, Wang QL, et al. Mutations in the cone-rod homeobox gene are associated with the cone-rod dystrophy photoreceptor degeneration. Neuron. 1997;19(6):1329–1336. doi: 10.1016/s0896-6273(00)80423-7. [DOI] [PubMed] [Google Scholar]
- 39.Sohocki MM, Sullivan LS, Mintz-Hittner HA, et al. A range of clinical phenotypes associated with mutations in CRX, a photoreceptor transcription-factor gene. Am J Hum Genet. 1998;63(5):1307–1315. doi: 10.1086/302101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Itabashi T, Wada Y, Sato H, Kawamura M, Shiono T, Tamai M. Novel 615delC mutation in the CRX gene in a Japanese family with cone-rod dystrophy. Am J Ophthalmol. 2004;138(5):876–877. doi: 10.1016/j.ajo.2004.05.067. [DOI] [PubMed] [Google Scholar]
- 41.Kelsell RE, Gregory-Evans K, Payne AM, et al. Mutations in the retinal guanylate cyclase (RETGC-1) gene in dominant cone-rod dystrophy. Hum Mol Genet. 1998;7(7):1179–1184. doi: 10.1093/hmg/7.7.1179. [DOI] [PubMed] [Google Scholar]
- 42.Payne AM, Downes SM, Bessant DA, et al. A mutation in guanylate cyclase activator 1A (GUCA1A) in an autosomal dominant cone dystrophy pedigree mapping to a new locus on chromosome 6p21.1. Hum Mol Genet. 1998;7(2):273–277. doi: 10.1093/hmg/7.2.273. [DOI] [PubMed] [Google Scholar]
- 43.Nakazawa M, Kikawa E, Chida Y, Wada Y, Shiono T, Tamai M. Autosomal dominant cone-rod dystrophy associated with mutations in codon 244 (Asn244His) and codon 184 (Tyr184Ser) of the peripherin/RDS gene. Arch Ophthalmol. 1996;114(1):72–78. doi: 10.1001/archopht.1996.01100130068011. [DOI] [PubMed] [Google Scholar]
- 44.Cremers FP, van de Pol DJ, van Driel M, et al. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt’s disease gene ABCR. Hum Mol Genet. 1998;7(3):355–362. doi: 10.1093/hmg/7.3.355. [DOI] [PubMed] [Google Scholar]
- 45.Jalkanen R, Mäntyjärvi M, Tobias R, et al. X linked cone-rod dystrophy, CORDX3, is caused by a mutation in the CACNA1F gene. J Med Genet. 2006;43(8):699–704. doi: 10.1136/jmg.2006.040741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Demirci FY, Rigatti BW, Wen G, et al. X-linked cone-rod dystrophy (locus COD1): identification of mutations in RPGR exon ORF15. Am J Hum Genet. 2002;70(4):1049–1053. doi: 10.1086/339620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Freund CL, Wang QL, Chen S, et al. De novo mutations in the CRX homeobox gene associated with Leber congenital amaurosis. Nat Genet. 1998;18(4):311–312. doi: 10.1038/ng0498-311. [DOI] [PubMed] [Google Scholar]