

Abstract
The isocitrate dehydrogenase (IDH) enzymes were initially identified as essential components of the Krebs cycle. IDH mutations were thought to be incompatible with cell survival. However, 90% of glioblastomas were recently shown to be associated with somatic mutations in these enzymes, indicating a possible role for IDH in promoting cellular survival in hypoxic environments. Our proteomic analysis of rats given 10 minutes of middle cerebral artery occlusion to induce transient ischemia demonstrates a significant decrease in IDH expression. We have recapitulated this decrease in an in vitro model using primary cortical neurons exposed to acute oxygen and glucose deprivation. Given the role of IDHs in energy metabolism and antioxidant production, we hypothesize that the IDHs may serve as first-line, rapid-response enzymes that regulate survival in environments of energetic or oxidative stress. In order to identify the specific events that regulate IDH enzymes, HT-22 neural cells were subjected to either a selective energetic challenge or a pure oxidative stress. In response to the non-lethal energetic challenge induced by substituting galactose for glucose, we observed increased IDH1, 2, and 3 expression and cessation of cellular proliferation. No change in expression of any IDH isoform was observed when neural cells were subjected to subtoxic oxidative stress via glutathione depletion. Taken together, these data imply that IDH expression rapidly responds to changes in energetic status, but not to oxidative stress. These data also suggest that IDH enzymes respond not only to allosteric modulation, but can also change patterns of expression in response to moderate stress in an effort to maximize ATP production and survival.
Keywords: Preconditioning, energetics, galactose challenge, glioblastoma, ischemia, isocitrate dehydrogenase, mitochondria, oxidative stress, oxygen glucose deprivation (OGD), stroke
Introduction
The components of the Krebs cycle and their modulation through changes in cell physiology have been well appreciated for over 60 years [1, 2]. Krebs cycle enzymes were thought to be essential to the survival of complex aerobic organisms such that mutations in these proteins were believed to be lethal. However, recent studies, as reviewed by Thompson (2009), link mutations in components of the Krebs cycle to human diseases. Gliomas, secondary glioblastomas, and acute myeloid leukemia (AML) have all been associated with heterozygous mutations in isocitrate dehydrogenase (IDH) [3], one of the key enzymes in the Krebs cycle. These disease-associated mutations result in the inability to convert isocitrate to alpha-ketoglutarate (α-KG). Consequently, there has been an increased focus on bioenergetics in pathological conditions, such as tumorigenesis, where hyper-proliferative cells undergo metabolic adaptation, priming them for growth in anaerobic conditions [4]. Moreover, in disease states, neurons can metabolically adapt to increase survival to a limited extent by switching to glycolytic pathways and consuming lactate similar to tumor cells [5], although the mechanisms that promote neuronal adaptation during hypoxia are not fully understood.
Hypoxia-induced injury in the brain during an ischemic attack is largely mediated by excess glutamate release and excitotoxic effects due to a significant influx of calcium through N-methyl-D-aspartate receptors, as well as increases in reactive oxygen species (ROS) [6]. While these changes can induce neuronal death, mitochondria can initiate neuroprotection through the sequestration of calcium and reduction of ROS via a mild uncoupling of the electron transport chain [6], ultimately eliciting changes in ATP production. Mitochondria, and thus regulators of the Krebs cycle, therefore must be able to both sense and rapidly react to stroke-like conditions in order to promote neuronal survival. While the benefits of utilizing available substrates to maximize ATP production during hypoxia and/or ischemia are well appreciated, the role of individual modulators of adaptation to injury are poorly understood. A recent report utilizing a proteomics screen found that several Krebs cycle enzymes and regulatory proteins are downregulated after neuronal preconditioning [7]. In keeping with these data, preliminary proteomic screens in our lab, identified a 30% reduction of IDH3 in the brains of animals exposed to transient ischemia by middle cerebral artery occlusion (MCAO), which elicits neuroprotection [8, 9]. This protective phenomenon, known as preconditioning (PC), results when a mild stress prepares the brain for and therefore protects it from the harmful effects of a subsequent, more severe stress. Hallmark features of PC include protein synthesis, KATP channel opening, and heat shock protein induction [10, 11].
Given the growing recognition that IDH regulation plays an essential role in both physiological and pathophysiological environments, we sought to analyze conditions associated with ischemia that regulate the expression of the IDH enzymes. Our goal was to determine the consequences of these alterations on the energetic and redox status of neural cells and their ability to survive subsequent injury. We hypothesize that IDH isoforms may serve as a rapid regulatory control point after mild injury, ensuring a prompt response that can alter neural fate.
Materials and Methods
User-friendly versions of all protocols and procedures can be found on our website at http://www.mc.vanderbilt.edu/root/vumc.php?site=mclaughlinlab&doc=17838.
Reagents
Hyclone defined fetal bovine serum was obtained from Fisher Scientific (SH3007003). Bovine serum albumin (BSA) and 1 M HEPES buffer were procured from Sigma (A1470 and H0887). Poly-L-ornithine hydrobromide (Sigma, P3655) was used to coat 12 mm or 25 mm glass coverslips (Carolina Biological Supply, 63-3029) for primary cultures. All remaining cell culture media and supplements were purchased from Invitrogen. Antibodies were purchased as follows: IDH3α (Aviva Systems Biology, ARP42237_T100), heat shock conjugate 70 (HSC70) (Assay Designs, SPA-816), hypoxia inducible factor-1α (HIF1α) (Novus Biologicals, NB100-110); IDH1 (sc-49996), IDH2 (sc-55668) and α-KG (sc-49589) antibodies were purchased from Santa Cruz Biotechnology, Inc. All other immunoblot supplies, unless noted, were purchased from Bio-Rad Laboratories. Secondary antibodies for immunocytochemistry were purchased through Jackson ImmunoResearch and included Cy3 conjugated anti-rabbit (711-165-152) and Cy2 conjugated anti-mouse (715-225-150). The antifade reagent, Prolong Gold (P36934) was purchased through Invitrogen. Western Lightning Chemiluminescence Reagent Plus was obtained from PerkinElmer Life Sciences (NEL104001EA), SuperSignal West Dura Extended Duration Substrate Chemiluminescence was procured through Thermo Scientific (34075), DC Protein Assay Kit II from Bio-Rad (500-0112), Hyblot CL Autoradiography Film (E3018) from Denville Scientific, Inc. and ViaLight HS ATP Kit from Lonza (LT07-211). All additional chemicals were purchased through Sigma.
Primary Neuronal Cultures
Cortical cultures were prepared from embryonic day 18 Sprague-Dawley rats as previously described [12, 13]. Briefly, cortices were digested in trypsin and dissociated. Resultant cell suspensions were adjusted to 335,000 cells/mL and plated 2 mL/well in 6-well tissue culture plates containing five 12 mm or one 25 mm poly-L-ornithine-coated glass coverslip(s). Cultures were maintained at 37°C, 5% CO2 in plating medium composed of a volume to volume mixture of 84% Dulbecco’s modified Eagle’s medium (DMEM), 8% Ham’s F12-nutrients, 8% fetal bovine serum, 24 U/mL penicillin, 24 μg/mL streptomycin, and 80 μM L-glutamine. Glial proliferation was inhibited after two days in culture with 1–2 μM cytosine arabinoside, after which cultures were maintained in Neurobasal medium containing 2% B27, 2x N2 and 4% NS21 supplements [14] with antibiotics. All experiments were performed between days in vitro 21–25 [15].
Oxygen Glucose Deprivation (OGD)
OGD was performed as previously described [13]. Briefly, growth media was replaced with deoxygenated, glucose-free Earle’s balanced salt solution (150 mM NaCl, 2.8 mM KCl, 1 mM CaCl2 and 10 mM HEPES; pH 7.3), bubbled with 10% H2/85% N2/5% CO2. Cultures were exposed to OGD in an anaerobic chamber (Billups-Rothberg) for a specified time at 37ºC. Upon OGD termination, cells were washed once with minimal essential medium (MEM)/BSA/HEPES (0.01% BSA and 25 mM HEPES) and allowed to recover in MEM/BSA/HEPES/2xN2 (0.01% BSA and 25 mM HEPES and 2x N2) for various time points. Following recovery, protein extracts were prepared for immunoblotting as previously described [10].
Primary Neuronal Preconditioning
Preconditioning was performed as previously described [16]. Briefly, mature neurons on glass coverslips were transferred to 35 mm Petri dishes containing glucose-free balanced salt solution (150 mM NaCl, 2.8 mM KCl, 1 mM CaCl2 and 10 mM HEPES; pH 7.3) that had been bubbled with an anaerobic gas mixture (10% H2/85% N2/5% CO2) for 5 min immediately prior to the addition of cells to remove dissolved oxygen. The neurons were exposed to OGD in an anaerobic chamber (Billups-Rothberg) for 5 min at 37ºC. Upon OGD termination coverslips were washed once with MEM/BSA/HEPES (0.01% BSA and 25 mM HEPES) and then transferred to 6-well plates containing fresh MEM/BSA/HEPES/2xN2 (0.01% BSA, 25 mM HEPES, and 2x N2) for 24 h. The following day, coverslips were again transferred to 35 mm Petri dishes and exposed to 5 min of OGD. Upon OGD termination, the coverslips were washed once in MEM/BSA/HEPES and placed back into the identical 6-well plates containing the MEM/BSA/HEPES/2xN2 from the initial OGD exposure and placed in the incubator for 24 h. The 5 min OGD preconditioning was repeated a final time on the third consecutive day, and following 24 h recovery, protein extracts were prepared for immunoblotting as previously described [10].
Every 24 h following 5 min OGD exposures, photomicrographs were taken and toxicity was determined using lactate dehydrogenase assays to confirm there was no neuronal cell death. In order to assess that the multi-day OGD resulted in PC, simultaneous experiments were performed in which neurons were exposed to the 3 day 5 min OGD PC paradigm followed 24 h later by a lethal 90 min OGD. Cellular death was then determined the following day using the lactate dehydrogenase toxicity assay.
Immunoblotting
All cell lysis and harvesting steps took place on ice. Cells were washed twice with ice-cold phosphate buffered saline (PBS) (4.3 mM Na2HPO4 ~7 H2O, 1.4 mM KH2PO4, 137 mM NaCl, 2.7 mM KCl; pH 7.4). Following the second wash, 250 – 500 μL of TNEB was added (50 mM Tris-Cl, pH 7.8, 2 mM EDTA, 100 mM NaCl, 1% NP-40, and a 1:1000 dilution of protease inhibitor) and cells were collected. Approximately 100–200 μL of the cell suspension was saved for protein determination and the remaining lysate was re-suspended in an equal volume of Laemmli buffer with β-mercaptoethanol (1:20). Protein samples were heated to 95°C for 10 min, and stored at −20°C. Protein concentrations were determined via the DC Protein Assay Kit II.
Equal protein concentrations were separated using 4–12% Bis-Tris gels followed by transfer onto Hybond PVDF membranes and then blocked in methanol for 5 min. After 15 min of drying, the membranes were incubated at 4°C overnight with their respective primary antibody prepared in 5% nonfat dry milk in a Tris-buffered saline solution containing 0.1% Tween 20 (TBS-Tween). Primary antibodies were used at the following concentrations: IDH1 (1:1000), IDH2 (1:500), IDH3α (1:800), α-KG (1:500), HIF1α (1:500), and HSC70 (1:1000). Following incubation in primary antibodies, membranes were washed three times with TBS-Tween, and incubated for 1 h at room temperature in a 1:5000 dilution of horseradish peroxidase–conjugated secondary antibodies dissolved in 5% nonfat dry milk in TBS-Tween. After three additional washes in TBS-Tween, protein bands were visualized using SuperSignal West Dura Extended Duration Substrate Chemiluminescence and exposed to Hyblot CL Autoradiography Film.
HT-22 Cell Line
HT-22 cells derived from the immortalized H4 hippocampal murine neuroblastoma cell line were received as a generous gift from Pamela Maher (Salk Institute). This neural cell line was chosen because HT-22 cells are a well-characterized model of neural oxidative injury [17, 18]. These cells undergo oxidative stress-induced toxicity upon exposure to glutamate, which blocks glutamate-cystine antiporters in the plasma membrane, resulting in glutathione (GSH) deficiency and oxidative cell death [18, 19]. Therefore, these cells model oxidative stress without activating exitotoxic cell death pathways. HT-22 cells were maintained in DMEM with glutamine, supplemented with 10% fetal bovine serum and penicillin/streptomycin (0.2%). Cells were grown in 75 cm2 cell culture flasks at 37°C with 5% CO2 and passed by trypsinization when confluency reached 50%–80% [20].
Glutamate Toxicity Assay
HT-22 neural cells were plated at a density of 100,000 cells/well in 6-well plates and grown at 37°C with 5% CO2 overnight. The following day, cells were switched into MEM/BSA/HEPES (0.01% BSA and 25 mM HEPES) culture medium containing 3 mM glutamate. As a positive control, 1 M sorbitol was prepared and added to the culture medium to induce total cell death in the appropriate wells. Twenty-four hours after incubation at 37°C, cellular viability was photo-documented and assessed via the in vitro thiazolyl blue tetrazolium bromide (MTT) toxicology assay as previously described [21] or cells were harvested for immunoblot analysis.
Galactose Challenge
HT-22 neural cells were plated at a density of 25,000 cells/well in 6-well plates and 5,000 cells/well in 24-well plates and grown at 37°C with 5% CO2 overnight. The following day, cells were switched into 100% galactose medium (Glucose-free DMEM, 11966-025, supplemented with 25 mM galactose, 25 mM HEPES, and 0.01% BSA) or maintained in original growth media. As a positive control, a 1 M sorbitol solution was prepared in the culture medium to induce total cell death in the appropriate wells. Cells were incubated in treatment medium for 72 h. HT-22 cells from 24-well plates were photo-documented and assessed for cell viability and proliferation with an in vitro MTT toxicology assay as previously described [21], or fixed for immunostaining. HT-22 cells prepared in 6-well plates were photo-documented and then harvested for immunoblot analysis.
Immunocytochemistry
Immunocytochemistry was performed essentially as we have previously described [10]. Briefly, cells were fixed in 4% para-formaldehyde for 10 min followed by permeabilization in 0.1% Triton X-100 for 5 min. Cells were then washed 3 times with 1x PBS and blocked in 8% BSA diluted in 1x PBS for 25 min. After blocking, cells were incubated simultaneously in anti-rabbit Ki67 antibody (1:200) and anti-mouse β-tubulin (1:200) in 1% BSA at 4°C overnight. Following primary antibody incubation, cells were washed 3 times in 1x PBS and incubated simultaneously in Cy3 anti-rabbit (1:500) and Cy2 anti-mouse (1:500) secondary antibodies diluted in 1% BSA for 60 min at room temperature. Cells were then washed 5 times in 1x PBS and incubated in 1.4 μM DAPI for 10 min. After 3 additional washes in 1x PBS, coverslips were mounted in Prolong Gold and fluorescence was visualized with a Zeiss Axioplan microscope equipped with an Apotome optical sectioning slider.
Measurement of F2t-Isoprostanes
Lipid peroxidation was assessed through quantification of F2t-isoprostanes (F2t-IsoPs), prostaglandin-like molecules generated from free radical-mediated peroxidation of arachidonic acid [22]. F2t-IsoPs are measured using gas chromatography–mass spectrometry as previously described [23–25]. Briefly, cells were harvested in 6-well plates after either a 24 h glutamate exposure or a 72 h galactose exposure, and 500 μL of the lysate was mixed with methanol containing 0.05% butylated hydroxy-toluene to prevent auto-oxidation. The remaining lysate was saved for protein assay to normalize loading concentrations. F2t-IsoPs esterified to phospholipids were hydrolyzed by chemical saponification, after which total F2t-IsoPs were extracted using C-18 and silica Sep-Pak cartridges, purified by thin-layer chromatography, converted to pentaflurobenzyl ester trimethylsilyl ether derivatives, and quantified by stable isotope dilution techniques using gas chromatography/negative ion chemical ionization mass spectrometry. [2H4]-8-iso-PGF2 (m/z 573) was used as an internal standard. F2t-IsoPs are detected at m/z 569.
ATP Measurements
Measurement of ATP content was performed after 24 h galactose challenge and 24 h glutamate toxicity via bioluminescent detection of light in the presence of luciferin. Briefly, in a 6-well plate, cells were washed twice with 1x PBS and then incubated in 2 mL of 1x PBS containing 300 μL of Nucleotide Releasing Agent provided in the ViaLight HS Kit. After a 10 min incubation, 180 μl of the cell suspension along with 20 μl of ATP Monitoring Reagent were added to a 96-well, transparent bottom, white plate. Addition of this reagent leads to the formation of light from the interaction of the enzyme luciferase with ATP present in the cell and luciferin. Following a 2 min incubation, measurements can be taken using a SPECTRAfluor Plus Tecan plate reader with an integration time of 1000 ms and a gain of 150. The resulting bioluminescent measurements are related to the ATP concentration, which can be quantitatively determined via an ATP standard curve and adjusted to total protein concentrations following protein assay. Measurements were obtained in triplicate for each sample and ATP levels are expressed as the mean Relative Luminescence Units ± standard deviation.
Analysis and Statistics
Except where otherwise noted, data were summarized and are represented as mean ± SEM. The statistical significance of differences between means was assessed using one-way ANOVA at the 95% level (p<0.05), followed by either Bonferroni or Turkey multiple-comparison post-tests using GraphPad Prism software. Sample size (n) indicates separate, individual experiments.
Western blots were analyzed semi-quantitatively to determine the mean relative densities of each protein band in comparison to its control condition counterpart (NIH Image, Scion Image J). The rectangular selection tool was used to select vertical lane sections of equal area to encompass the control and experimental bands. Image J plotted each selection as a representation of the relative density of the contents of the rectangle over each lane and each peak was analyzed for area and for its percent of the total area from all peaks being compared. Relative densities were then calculated from the percent values, setting each control condition peak to a relative density of 1 [26, 27].
Results
Ischemic preconditioning of primary neuronal cultures results in decreased IDH3 When rats receive a brief (10 min) MCAO 1–7 days prior to a permanent MCAO, they are afforded neuroprotective effects as evidenced by a 50% reduction in infarct volume [10]. Proteomic and immunoblot analysis of these preconditioned samples also revealed a 30% reduction in IDH3 expression compared to controls. In this study, we utilize a previously established, powerful and reproducible in vitro primary neuronal PC culture model [10, 16, 28] in which we now confirm our in vivo results and report a substantial (34 ± 11%) reduction in IDH3 expression (Fig. 1A–B) following PC as compared to controls. In addition, to demonstrate that our PC paradigm does not result in cell death, neurons were photo-documented each day throughout the entire experiment. As shown in Figure 1C, control neurons have complex arbors, phase bright somas and little debris that would be suggestive of cell death. This morphological integrity was also confirmed 24 h after the first 5 min period of OGD (Fig. 1D), 24 h after the second period of 5 min OGD (Fig. 1E) and 24 h after the final 5 min period of OGD (Fig. 1F).
Figure 1. Preconditioning Results in a Decreased Expression of IDH3.
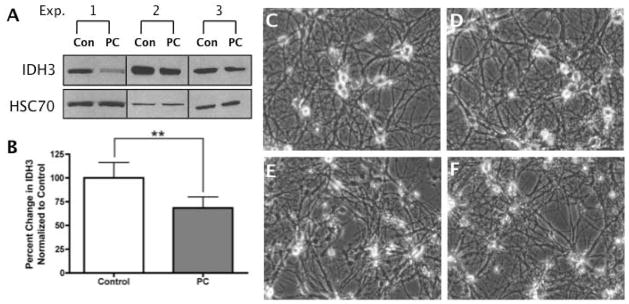
Primary neuronal cultures were exposed to 5 min of OGD on three consecutive days. Whole cell extracts were harvested 24 h following the third PC session. Fifteen μg of total protein was separated on 4–12% Bis-Tris gels, transferred onto Hybond PVDF membranes, and probed with antibodies specific for IDH3 and HSC70 (loading control). (A) A decrease in IDH3α expression was observed after 3 days of PC (n=3) and quantified (B). Neurons were photo-documented prior to PC (C), 24h after 1 day of PC (D), 24 h after 2 days of PC (E), and 24 h following day 1, 2, and 3 of PC (F) in order to demonstrate that there was no change in cell viability as a result of three consecutive days of PC. These images are in keeping with our previous use of this model [14].
These images are in keeping with our previous data in which this multi-day PC paradigm causes no appreciable changes in cellular viability compared to control cells yet provides a 45% increase in survival when PC cells are subjected to a normally lethal, 90 min OGD challenge [16]. Given that ischemia is comprised of an excitotoxic, oxidative challenge and an energetic stress, we next set out to determine which of these components is the main contributor to changes in IDH3 expression.
Oxidative stress does not affect IDH expression
OGD induces intense energetic and oxidative stress [16]. To identify the biological trigger associated with decreased IDH expression, we parsed the stress of OGD into two discreet stimuli. We first utilized the HT-22 neural cell line and glutamate toxicity. HT-22 cells undergo oxidative toxicity when exposed to glutamate due to blockage of glutamate-cystine antiporters in the plasma membrane. Without cystine as a precursor to cysteine, GSH cannot be generated. This severe GSH deficiency as well as the >400 fold increase in ROS levels in this paradigm ultimately leads to cell death [18, 29]. Therefore, HT-22 neural cells were exposed to 3 mM glutamate, a concentration known to induce substantial oxidative stress in this cell line [13].
After 24 h of glutamate exposure, immunoblot analysis revealed that there was no change in any of the IDH isoforms or in downstream signaling molecules such as α-KG or HIF1α (Fig. 2A, Fig. S1). Cell viability was assessed visually (Fig. 2C–D) and via MTT assays (Fig. 2B), and was not found to be statistically different between controls and glutamate treated cells.
Figure 2. Oxidative Stress Alone Does Not Alter IDH Expression.

HT-22 neural cells were exposed to 3 mM glutamate for 24 h. Whole cell extracts were harvested at 24 h and 10 μg of total protein was probed with antibodies specific for IDH1, IDH2, IDH3, α-KG, HIF1α, and HSC70. (A) IDH1, IDH2, IDH3, α-KG, and HIF1α showed no change in expression compared to control lysates (n=3). (B) Cell viability was assessed and quantified by MTT assay (n=3), showing this oxidative challenge was non-lethal (p>0.05). The data are expressed as a percent survival compared to sorbitol (“Total Kill”) and were post-hoc analyzed using a one-way ANOVA and Turkey’s multiple comparison post-hoc analysis. HT-22 cells were photo documented following treatment to visually assess cellular viability: both the control condition (C) and the 3mM glutamate condition (D) illustrate negligible changes in cellular viability while the sorbitol condition (E) exhibits appreciable cell death.
The purity of the oxidative stressor was assessed via ATP assays, as well as F2t-IsoP analysis, an accurate and reliable index of oxidative stress [30]. Compared to control, F2t-IsoP levels increased 4-fold (p<0.05) after 24 h glutamate exposure, confirming that glutamate exposure is indeed an intense oxidative stress (Fig. 4A) even though cell viability was unaffected. In addition, ATP analysis 24 h after glutamate exposure revealed no significant differences between control and glutamate conditions, confirming that glutamate exposure does not act as an energetic stressor (Fig. 4B). These results suggest that sub-lethal glutamate exposure in HT-22 cells results in a substantial amount of oxidative stress, while not affecting energetic homeostasis or viability, nor eliciting any changes in IDH. Therefore, we next sought to determine if an energetic challenge might be the root cause of PC-induced changes in IDH expression.
Figure 4. Galactose Challenge Induces an Energetic Stress While Glutamate Toxicity Induces an Oxidative Stress.

HT-22 cells were exposed to either 3 mM glutamate for 24 h or 72 h 25 mM galactose and were either harvested and prepared for F2t-IsoP measurements or incubated with Nucleotide Releasing Agent for ATP measurements. (A) The amount of F2t-IsoPs normalized to total protein in glutamate-treated cells differs significantly from control cells, demonstrating that glutamate toxicity induces oxidative stress (n=6, ***p<0.05). (B) The amount of ATP normalized to total protein in glutamate-exposed cells does not differ from control cells, showing no change in energetic status after glutamate toxicity (n=5, p>0.05). (C) In galactose- treated cells, the amount of F2t-IsoP did not differ significantly from control cells, demonstrating limited oxidative stress due to galactose challenge (n=4, p>0.05). (D) The amount of ATP normalized to total protein in galactose-treated cells was less than that of control cells; however, the difference was not significant (n=5, p>0.1). The data were analyzed for significance using tailed t-tests.
The energetic stress of galactose challenge increases IDH1, 2, and 3 expression and prevents neural cell proliferation
Cell lines in culture rely heavily upon the glucose supplied in their medium to generate ATP via glycolysis, a process that occurs outside of the mitochondria and independent of oxidative phosphorylation. However, when glucose is substituted with galactose (25 mM) cultured cells are forced to generate ATP via mitochondrial oxidative phosphorylation [31, 32], which is energetically unfavorable as there is no catabolic pathway to metabolize galactose. Preliminary experiments demonstrated that a 25 mM galactose challenge caused negligible cell death after 24 and 48 h in HT-22 neural cells (data not shown). Therefore, a 72 h galactose challenge was chosen to create a chronic, sub-lethal energetic stress.
Immunoblot analysis following 72 h of galactose challenge revealed a 5-fold increase in IDH1 expression and nearly a 2-fold increase in IDH3 expression, while the expression of IDH2 increased to 28 times that of the control conditions. An increase in α-KG (1.44-fold) and a subsequent decrease in HIF1α (0.59-fold) were also observed (Fig. 3A, Fig. S1). Moreover, the energetic stress of galactose challenge effectively prevented cell proliferation as demonstrated by MTT assays (Fig. 3B). These results were validated through immunocytochemistry where Ki-67, a marker of mitotic activity [33], shows significant localization to DAPI stained nuclei in control cells (Fig. 3C) but is nearly absent in the galactose treated cells (Fig. 3D).
Figure 3. Energetic Challenge in Neural Cells Increases Expression of IDH1, 2, and 3 and Inhibits Proliferation.
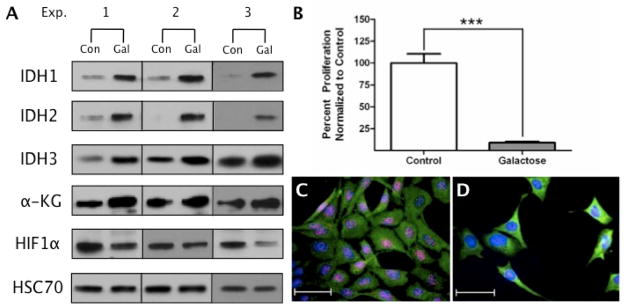
HT-22 cells were exposed to glucose-free medium substituted with 25 mM galactose for 72 h. Whole cell extracts were harvested at 72 h and 10 μg of protein was probed with antibodies specific for IDH1, IDH2, IDH3α, α-KG, HIF1α, and HSC70. (A) IDH1, IDH2, IDH3, and α-KG all demonstrated significant increases in expression compared to control lysates, while a decrease in HIF1α was observed (n=3). (B) At 72 h, the proliferative status of the 25 mM galactose condition compared to the 25 mM glucose control was assessed via MTT assays. Data are expressed as percent of control and were analyzed using a Student’s t-test (***p<0.0001). At 72 h, cells were fixed and further processed for immunocytochemistry. Ki67 (red) was used as a marker of mitotically active cells, β-tubulin (green) was used as a cytoarchitectural marker, and DAPI (blue) was utilized as a label of cell nuclei. (C) The control condition shows a predominance of Ki67 within the nuclei of the cells while (D) the 25 mM galactose condition has minimal Ki67 staining, demonstrating galactose challenge halts cell proliferation.
To confirm that galactose challenge is a solely energetic stress, F2t-IsoPs levels were measured and normalized for proliferation differences. The amount of F2t-IsoPs was found to be comparable in both control and galactose treated cells, indicating that the amount of oxidative stress induced by galactose challenge was negligible (Fig. 4C). In addition, ATP assays conducted following 24 h galactose challenge demonstrated a significant decrease in ATP levels compared to controls when normalized for proliferation differences, confirming galactose challenge as an energetic stress (Fig. 4D). These results suggest that changes in IDH expression elicited following neuronal PC are caused by the energetic component of ischemia.
Discussion
The goal of this study was to identify events that result in energetic compensation and protection in response to mild ischemic stress. Based upon our proteomics analysis, we found a significant decrease in IDH3 expression following PC in both in vivo and in vitro model systems. In addition, by experimentally isolating the two major components of ischemia, oxidative and energetic stress [34], we were able to identify a key biological trigger of neuroadaptation. Changes in IDH isoforms were found specifically in response to galactose challenge, establishing that these enzymes play a unique role in regulating neural cell responses to energetic stress. These data demonstrate that altering the available energetic environment offers a rapid and sustained means to maximize substrate utilization in neural cells. Based on prior knowledge of glioblastomas and these PC models, we hypothesize that the IDHs act as neuronal energetic sensors and appear to serve as critical gatekeeping molecules at the level of the mitochondria in order to regulate neural cell survival (Fig 5).
Figure 5. The IDHs: Energetic Gate-Keepers.

Situated within the Kreb's cycle, IDH has the ability to influence ATP and ROS production via aerobic respiration and thus is poised to serve as a critical control point during times of hypoxic stress. Utilizing the galactose challenge as an energetic stressor and glutamate toxicity as an oxidative stress, we have demonstrated that IDH expression is rapidly and consistently increased in response to energetic challenges. By moving neural cells to glycolytic means to produce ATP in stress, neurons may enhance survival of secondary stress. Moreover, limiting carbohydrates with galactose rapidly brought aerobic respiration online whereas changes in other modulators of aerobic respiration were not observed.
In response to energetic, oxidative and ionic stressors, neurons must integrate multiple signals to make decisions regarding cell fate. The phenomenon of PC is premised on the ability of acute, sublethal stresses to activate pathways and integrate signals in order to increase survival in response to a subsequent more severe stress. Calcium (Ca2+), ROS, and HIF1α have all been implicated as key players in PC and its resultant neuroprotection [35–37]. Our lab and others have demonstrated that the production of ROS is a requisite event in the cytoprotective PC program used by neurons and other tissues/cell types [36, 38–41]. ROS scavengers, such as free radical spin traps, therefore block PC in models of ischemic stroke [10, 41].
Mitochondrial signaling has gained much interest in terms of PC effects because this unique organelle lies at the apex of many of the processes and molecules that have been identified as key components of neuroprotection following PC [6, 42–44]. Although the activity of critical Krebs cycle enzymes have been analyzed [4, 45, 46], their expression levels are not often considered in the context of neuroprotection [7, 47].
While there are no known allosteric regulators of IDH1 or IDH2, there are many regulators of IDH3 [48], including Ca2+, citrate, ADP, and Mn2+ [49–51], many of which are impacted by PC [16, 41]. In non-neuronal cells, changes in the activity of IDH1 and IDH2 were suggested to support antioxidant defenses by increasing NADPH production while decreasing IDH3 activity. This was thought to provide a means in which to reduce flux through oxidative phosphorylation and influence the production of ROS [52]. Additionally, ROS inhibit the activity of IDH1 and IDH2 presumably through the oxidative modification of proline, lysine, arginine, and histidine residues into carbonyl derivatives [53]. Yang and colleagues showed that the free radical, nitric oxide, generated S-nitrosothiol adducts on Cys305 and Cys387, thereby inactivating IDH1 and IDH2 [54]. Since influx of Ca2+ and production of ROS are associated with mitochondrial stress and pathological conditions such as stroke, allosteric regulation by Ca2+ and ROS serves as an important control point for neuronal energetic status [55, 56]. The specific molecules impacted by these environments and the physiological stressors, which elicit these effects in vivo remain unknown.
We leveraged the ability that HT-22 cells have to undergo oxidative stress-induced toxicity upon exposure to glutamate, which blocks glutamate-cystine antiporters in the plasma membrane, resulting in GSH deficiency and oxidative cell death [18, 29]. Our data demonstrate that glutamate exposure in this cell line is a major oxidative stress associated with significant increases in F2t-IsoPs, the gold standard for assessing oxidative injury [57, 58]. Moreover, no changes in ATP were observed suggesting that this model induces negligible energetic stress. Following glutamate exposure, HT-22 cells demonstrate no changes in any of the IDH isoforms or downstream signaling molecules, suggesting that the changes in IDH3 seen with PC are not due to an oxidative stress response and may instead occur in response to the energetic status of a cell.
As IDH3 expression is subject to substrate limitation as well as allosteric Ca2+ control in PC and neuroprotection [55], it is likely that IDH isoforms serve as important energetic sensors for the cell. This idea is supported by our PC data in which we see a selective reduction of IDH3, an effect also observed in a PC model of rabbit ventricular myocytes by Arrell and colleagues [47]. Downregulation of IDH3 in response to this sublethal, PC stress may therefore prove beneficial to the cell through reduction of ROS due to decreased electron transport chain activity as well as through reductions in α-KG, which would promote stabilization of HIF1α. This model would therefore incorporate mechanisms employed by glioblastomas that possess IDH1 or 2 mutations.
Given that cancer cells rely almost exclusively on glycolysis for ATP production as explained by the Warburg and Crabtree effects [31], immortalized cell lines are primed to not only survive, but also to continue multiplying in hypoxic and acidic environments [59]. However, when these cells are maintained in a glucose-free medium containing galactose, the ability to undergo efficient glycolysis is lost because there is no catabolic pathway to metabolize galactose, and the cells are forced to generate ATP solely via oxidative phosphorylation within the mitochondria [32], creating a significant energetic stress. Our results indicate that galactose challenge in HT-22 neural cells is indeed a specific energetic stress as ATP levels were decreased with galactose treatment while no changes were noted in regards to F2t-IsoPs, suggesting that oxidative stress in this system was negligible. This energetic challenge resulted in increases in all three IDH isoforms, affecting downstream signaling molecules including α-KG and HIF1α and the inhibition of cellular proliferation. This suggests that neural cells increase IDH1, 2, and 3 to intensify flux through the Krebs cycle and thus maximize ATP production to promote a more favorable environment for survival following an energetic stress. Therefore, the IDH isoforms act as selective monitors of cellular energetics at the level of the mitochondria playing a key role in determining the fate of a cell and potentially representing a novel group of neurotherapeutic targets.
While all three isoforms of IDH catalyze the same fundamental reaction of converting isocitrate to α-KG, each has a unique role in maintaining redox and energetic status within cells that contributes to normal physiology and pathophysiology when aberrantly expressed. IDH3, the main isoform involved in the Krebs cycle within the mitochondrial matrix, is an NAD+-dependent heterotetramer with abundant expression in stroke-prone regions such as the cerebellum, cortex, thalamus, and hippocampus [60–62]. Within the hypothalamus, hippocampus, and medulla, homodimers of NADP+-dependent IDH1 localize to the cytosol and produce the byproduct, NADPH, a reducing equivalent that aids in the regeneration of the major cellular antioxidant, glutathione, via glutathione reductase. Thus, through the production of NADPH, IDH1 can assist the cell in lowering levels of ROS.
Yang and colleagues reported a reduction in IDH1 expression with age, further supporting an antioxidant role for IDH enzymes as antioxidants are known to decline over time [63]. Homodimers of IDH2 are also NADP+-dependent and produce NADPH, but localize to the mitochondria of neurons within the thalamus, pons, medulla, and hypothalamus [62]. Since NADPH cannot cross the mitochondrial membrane, IDH2 may have a similar antioxidant effect as IDH1 within the mitochondrial matrix [64]. In fact, studies have reported that cells deficient in IDH1 or IDH2 have an increased susceptibility to lipid peroxidation, oxidative DNA damage, and intracellular peroxide generation as well as decreased survival after oxidant exposure [53]. Given that the oxidative stress induced by glutamate exposure did not alter the expression level of any IDH isoform in HT-22 cells, IDH expression is not likely influenced solely by a selective oxidative stress. Therefore, IDH1, 2, and 3 may serve more so as critical gatekeeping molecules in response to energetic stressors.
Clinically, the significance of the IDHs in energy metabolism can be seen through the pathology associated with mutated isoforms. For instance, a homozygous mutation in the IDH3β subunit is observed in retinitis pigmentosa. This loss-of-function mutation only manifests in the retina where IDH1, IDH2, or a combination of both are unable to compensate for the loss of IDH3 within the Krebs Cycle [65]. Additionally, heterozygous mutations at Arg132 of IDH1 are found in 50–94% of grades 2 and 3 gliomas as well as secondary glioblastomas. This mutation has also been reported in a minor percentage of AML cases. Interestingly, the IDH2 analog of Arg132, Arg172, has also been reported to be mutated in grades 2 and 3 gliomas as well as AML [64]. IDH1 and IDH2 mutations lead to increased survival of glioblastoma cells in hypoxic environments. Research has shown that IDH mutants not only produce less α-KG but that they also produce the potential oncometabolite, 2-hydroxy-gluturate (2-HG) [66]. Though the mechanism of 2-HG action is unclear, some have hypothesized that 2-HG plays a role in increasing ROS damage and competitively inhibiting metabolic enzymes including N-acetyl-aspartyl-glutamate [67, 68] and prolyl hydroxylases based on their structural similarity to α-KG [69].
In future studies, we seek to determine whether decreases in IDH3 following PC and energetic challenge are due to decreases in protein expression, increases in protein degradation or as a result of altered mitochondrial biogenesis [41, 70, 71] [72]. Our preliminary data with α-KG and cytochrome oxidase in which expression levels of these enzymes are unchanged, suggest that there are no net changes in mitochondrial numbers but future analysis of the transcription factor peroxisome proliferator-activated receptor gamma coactivator 1-alpha could be used to assess mitochondrial biogenesis following PC [73, 74]. Additionally, analysis of IDH mRNA transcripts would reveal if the changes reported are simply an independent transcriptional increase within the Krebs cycle.
Ongoing experiments in our lab include further investigation into the role of the IDH isoforms in primary neuronal cultures following PC in order to identify how downstream signaling molecules such as α-KG and HIF1α respond to a combination of both energetic and oxidative stressors. We hope that these types of experiments will allow us to identify novel ways in which to regulate the IDH isoforms to promote neuronal survival.
Supplementary Material
Acknowledgments
The authors would like to thank Mrs. Jacquelynn Brown for her technical expertise, Mr. Tommy Saborido for editorial assistance, and Ms. Kylie Beck for graphical work. The authors greatly appreciate statistical analysis provided by Dr. Gregg Stanwood and graphical support provided by NICHID grant P30HD15052 from the Vanderbilt Kennedy Center for Research on Human Development. This work was supported by NINDS grant NS050396 (BAM), Vanderbilt Brain Institute Scholar Awards (AMP and BNL-M), and an American Heart Association Pre-doctoral Fellowship 12PRE11640010 (AMP). Financial support was also provided by a grant from the JB Marshall Neurovascular Therapeutics Program at Vanderbilt University (BAM) and the Vanderbilt Clinical Neuroscience Scholars Program (AMP). This content is solely the responsibility of the authors and does not necessarily represent the official views of NINDS or the NIH.
List of Abbreviations
- AML
Acute Myeloid Leukemia
- BSA
Bovine serum albumin
- GSH
Glutathione
- HIF
Hypoxia Inducible Factor
- HSC
Heat Shock Cognate
- IDH
Isocitrate Dehydrogenase
- F2t-IsoPs
F2t-Isoprostanes
- MCAO
Middle Cerebral Artery Occlusion
- α-KG
α-ketoglutarate
- MEM
Minimal essential medium
- MTT
(3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- OGD
Oxygen Glucose Deprivation
- PBS
Phosphate buffered saline
- PC
Preconditioning
- ROS
Reactive Oxygen Species
- TBS-Tween
Tris-buffered saline solution containing 0.1% Tween 20
Footnotes
CONFLICT OF INTEREST
The authors declare they have no conflict of interest
References
- 1.Krebs HA. The citric acid cycle and the szent-gyorgyi cycle in pigeon breast muscle. Biochem J. 1940;34(5):775–9. doi: 10.1042/bj0340775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garrett RH, Grisham CM. Biochemistry. 2007 [Google Scholar]
- 3.Thompson CB. Metabolic enzymes as oncogenes or tumor suppressors. N Engl J Med. 2009;360(8):813–5. doi: 10.1056/NEJMe0810213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Briere JJ, Favier J, Gimenez-Roqueplo AP, Rustin P. Tricarboxylic acid cycle dysfunction as a cause of human diseases and tumor formation. Am J Physiol Cell Physiol. 2006;291(6):C1114–20. doi: 10.1152/ajpcell.00216.2006. [DOI] [PubMed] [Google Scholar]
- 5.Malthankar-Phatak GH, Patel AB, Xia Y, Hong S, Chowdhury GM, Behar KL, Orina IA, Lai JC. Effects of continuous hypoxia on energy metabolism in cultured cerebro-cortical neurons. Brain Res. 2008;1229:147–54. doi: 10.1016/j.brainres.2008.06.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dirnagl U, Meisel A. Endogenous neuroprotection: Mitochondria as gateways to cerebral preconditioning? Neuropharmacology. 2008;55(3):334–44. doi: 10.1016/j.neuropharm.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 7.Scornavacca G, Gesuete R, Orsini F, Pastorelli R, Fanelli R, de Simoni MG, Airoldi L. Proteomic analysis of mouse brain cortex identifies metabolic down-regulation as a general feature of ischemic pre-conditioning. J Neurochem. 2012;122(6):1219–29. doi: 10.1111/j.1471-4159.2012.07874.x. [DOI] [PubMed] [Google Scholar]
- 8.Zeiger SL, Brown JE, McLaughlin BA. Proteomic identification and biochemical linkage of isocitrate dehydrogenase as a novel mediator of preconditioning. Society for Neuroscience; Chicago, IL: 2009. [Google Scholar]
- 9.Miller SLH, Brown JE, McLaughlin BA. New York Academy of Sciences: Mitochondria and Oxidative Stress in Neurodegenerative Disorders. New York, NY: 2007. Protective pathways activated by mitochondrial dysfunction in preconditioned neurons. [Google Scholar]
- 10.McLaughlin B, Hartnett KA, Erhardt JA, Legos JJ, White RF, Barone FC, Aizenman E. Caspase 3 activation is essential for neuroprotection in preconditioning. Proc Natl Acad Sci U S A. 2003;100(2):715–20. doi: 10.1073/pnas.0232966100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stetler RA, Gao Y, Zhang L, Weng Z, Zhang F, Hu X, Wang S, Vosler P, Cao G, Sun D, Graham SH, Chen J. Phosphorylation of hsp27 by protein kinase d is essential for mediating neuroprotection against ischemic neuronal injury. J Neurosci. 2012;32(8):2667–82. doi: 10.1523/JNEUROSCI.5169-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McLaughlin BA, Nelson D, Silver IA, Erecinska M, Chesselet MF. Methylmalonate toxicity in primary neuronal cultures. Neuroscience. 1998;86(1):279–90. doi: 10.1016/s0306-4522(97)00594-0. [DOI] [PubMed] [Google Scholar]
- 13.Stankowski JN, Zeiger SL, Cohen EL, DeFranco DB, Cai J, McLaughlin B. C-terminus of heat shock cognate 70 interacting protein increases following stroke and impairs survival against acute oxidative stress. Antioxid Redox Signal. 2010;14(10):1787–801. doi: 10.1089/ars.2010.3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Y, Stevens B, Chang J, Milbrandt J, Barres BA, Hell JW. Ns21: Re-defined and modified supplement b27 for neuronal cultures. J Neurosci Methods. 2008;171(2):239–47. doi: 10.1016/j.jneumeth.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McLaughlin BA, Nelson D, Erecinska M, Chesselet MF. Toxicity of dopamine to striatal neurons in vitro and potentiation of cell death by a mitochondrial inhibitor. J Neurochem. 1998;70(6):2406–15. doi: 10.1046/j.1471-4159.1998.70062406.x. [DOI] [PubMed] [Google Scholar]
- 16.Zeiger SL, McKenzie JR, Stankowski JN, Martin JA, Cliffel DE, McLaughlin B. Neuron specific metabolic adaptations following multi-day exposures to oxygen glucose deprivation. Biochim Biophys Acta. 2010;1802(11):1095–104. doi: 10.1016/j.bbadis.2010.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maher P. The effects of stress and aging on glutathione metabolism. Ageing Res Rev. 2005;4(2):288–314. doi: 10.1016/j.arr.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 18.van Leyen K, Siddiq A, Ratan RR, Lo EH. Proteasome inhibition protects ht22 neuronal cells from oxidative glutamate toxicity. J Neurochem. 2005;92(4):824–30. doi: 10.1111/j.1471-4159.2004.02915.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ratan RR, Murphy TH, Baraban JM. Oxidative stress induces apoptosis in embryonic cortical neurons. J Neurochem. 1994;62(1):376–9. doi: 10.1046/j.1471-4159.1994.62010376.x. [DOI] [PubMed] [Google Scholar]
- 20.Davis JB, Maher P. Protein kinase c activation inhibits glutamate-induced cytotoxicity in a neuronal cell line. Brain Res. 1994;652(1):169–73. doi: 10.1016/0006-8993(94)90334-4. [DOI] [PubMed] [Google Scholar]
- 21.Musiek ES, Breeding RS, Milne GL, Zanoni G, Morrow JD, McLaughlin B. Cyclopentenone isoprostanes are novel bioactive products of lipid oxidation which enhance neurodegeneration. J Neurochem. 2006;97(5):1301–13. doi: 10.1111/j.1471-4159.2006.03797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Musiek ES, McLaughlin B, Morrow JD. Electrophilic cyclopentenone isoprostanes in neurodegeneration. J Mol Neurosci. 2007;33(1):80–6. doi: 10.1007/s12031-007-0042-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Milne GL, Sanchez SC, Musiek ES, Morrow JD. Quantification of f2-isoprostanes as a biomarker of oxidative stress. Nat Protoc. 2007;2(1):221–6. doi: 10.1038/nprot.2006.375. [DOI] [PubMed] [Google Scholar]
- 24.Milne GL, Yin H, Brooks JD, Sanchez S, Jackson Roberts L, 2nd, Morrow JD. Quantification of f2-isoprostanes in biological fluids and tissues as a measure of oxidant stress. Methods Enzymol. 2007;433:113–26. doi: 10.1016/S0076-6879(07)33006-1. [DOI] [PubMed] [Google Scholar]
- 25.Fessel JP, Porter NA, Moore KP, Sheller JR, Roberts LJ., 2nd Discovery of lipid peroxidation products formed in vivo with a substituted tetrahydrofuran ring (isofurans) that are favored by increased oxygen tension. Proc Natl Acad Sci U S A. 2002;99(26):16713–8. doi: 10.1073/pnas.252649099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gassmann M, Grenacher B, Rohde B, Vogel J. Quantifying western blots: Pitfalls of densitometry. Electrophoresis. 2009;30(11):1845–55. doi: 10.1002/elps.200800720. [DOI] [PubMed] [Google Scholar]
- 27.Raband WS. Imagej. National Institutes of Health; Bethesda, Maryland, USA: 1997–2012. http://imagej.nih.gov/ij/( [Google Scholar]
- 28.McLaughlin B. The kinder side of killer proteases: Caspase activation contributes to neuroprotection and cns remodeling. Apoptosis. 2004;9(2):111–21. doi: 10.1023/B:APPT.0000018793.10779.dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ratan RR, Murphy TH, Baraban JM. Macromolecular synthesis inhibitors prevent oxidative stress-induced apoptosis in embryonic cortical neurons by shunting cysteine from protein synthesis to glutathione. J Neurosci. 1994;14(7):4385–92. doi: 10.1523/JNEUROSCI.14-07-04385.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kadiiska MB, Gladen BC, Baird DD, Graham LB, Parker CE, Ames BN, Basu S, Fitzgerald GA, Lawson JA, Marnett LJ, Morrow JD, Murray DM, Plastaras J, Roberts LJ, 2nd, Rokach J, Shigenaga MK, Sun J, Walter PB, Tomer KB, Barrett JC, Mason RP. Biomarkers of oxidative stress study iii. Effects of the nonsteroidal anti-inflammatory agents indomethacin and meclofenamic acid on measurements of oxidative products of lipids in ccl4 poisoning. Free Radic Biol Med. 2005;38(6):711–8. doi: 10.1016/j.freeradbiomed.2004.10.024. [DOI] [PubMed] [Google Scholar]
- 31.Marroquin LD, Hynes J, Dykens JA, Jamieson JD, Will Y. Circumventing the crabtree effect: Replacing media glucose with galactose increases susceptibility of hepg2 cells to mitochondrial toxicants. Toxicol Sci. 2007;97(2):539–47. doi: 10.1093/toxsci/kfm052. [DOI] [PubMed] [Google Scholar]
- 32.Swiss R, Will Y. Assessment of mitochondrial toxicity in hepg2 cells cultured in high-glucose- or galactose-containing media. Curr Protoc Toxicol. 2011;Chapter 2(Unit 2):20. doi: 10.1002/0471140856.tx0220s49. [DOI] [PubMed] [Google Scholar]
- 33.Majdzadeh N, Wang L, Morrison BE, Bassel-Duby R, Olson EN, D'Mello SR. Hdac4 inhibits cell-cycle progression and protects neurons from cell death. Dev Neurobiol. 2008;68(8):1076–92. doi: 10.1002/dneu.20637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taoufik E, Probert L. Ischemic neuronal damage. Curr Pharm Des. 2008;14(33):3565–73. doi: 10.2174/138161208786848748. [DOI] [PubMed] [Google Scholar]
- 35.Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL, MacDonald JF, Wemmie JA, Price MP, Welsh MJ, Simon RP. Neuroprotection in ischemia: Blocking calcium-permeable acid-sensing ion channels. Cell. 2004;118(6):687–98. doi: 10.1016/j.cell.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 36.Yang Q, Dong H, Deng J, Wang Q, Ye R, Li X, Hu S, Dong H, Xiong L. Sevoflurane preconditioning induces neuroprotection through reactive oxygen species-mediated up-regulation of antioxidant enzymes in rats. Anesth Analg. 2011;112(4):931–7. doi: 10.1213/ANE.0b013e31820bcfa4. [DOI] [PubMed] [Google Scholar]
- 37.Liu J, Narasimhan P, Yu F, Chan PH. Neuroprotection by hypoxic preconditioning involves oxidative stress-mediated expression of hypoxia-inducible factor and erythropoietin. Stroke. 2005;36(6):1264–9. doi: 10.1161/01.STR.0000166180.91042.02. [DOI] [PubMed] [Google Scholar]
- 38.Cohen MV, Yang XM, Downey JM. Acidosis, oxygen, and interference with mitochondrial permeability transition pore formation in the early minutes of reperfusion are critical to postconditioning's success. Basic Res Cardiol. 2008;103(5):464–71. doi: 10.1007/s00395-008-0737-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wiegand F, Liao W, Busch C, Castell S, Knapp F, Lindauer U, Megow D, Meisel A, Redetzky A, Ruscher K, Trendelenburg G, Victorov I, Riepe M, Diener HC, Dirnagl U. Respiratory chain inhibition induces tolerance to focal cerebral ischemia. J Cereb Blood Flow Metab. 1999;19(11):1229–37. doi: 10.1097/00004647-199911000-00007. [DOI] [PubMed] [Google Scholar]
- 40.Wada K, Miyazawa T, Nomura N, Tsuzuki N, Nawashiro H, Shima K. Preferential conditions for and possible mechanisms of induction of ischemic tolerance by repeated hyperbaric oxygenation in gerbil hippocampus. Neurosurgery. 2001;49(1):160–6. doi: 10.1097/00006123-200107000-00025. discussion 166–7. [DOI] [PubMed] [Google Scholar]
- 41.Brown JE, Zeiger SL, Hettinger JC, Brooks JD, Holt B, Morrow JD, Musiek ES, Milne G, McLaughlin B. Essential role of the redox-sensitive kinase p66shc in determining energetic and oxidative status and cell fate in neuronal preconditioning. J Neurosci. 2010;30(15):5242–52. doi: 10.1523/JNEUROSCI.6366-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Penna C, Perrelli MG, Pagliaro P. Mitochondrial pathways, permeability transition pore, and redox signaling in cardioprotection: Therapeutic implications. Antioxid Redox Signal. 2012 doi: 10.1089/ars.2011.4459. [DOI] [PubMed] [Google Scholar]
- 43.Perez-Pinzon MA, Stetler RA, Fiskum G. Novel mitochondrial targets for neuroprotection. J Cereb Blood Flow Metab. 2012;32(7):1362–76. doi: 10.1038/jcbfm.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Quarrie R, Lee DS, Steinbaugh G, Cramer B, Erdahl W, Pfeiffer DR, Zweier JL, Crestanello JA. Ischemic preconditioning preserves mitochondrial membrane potential and limits reactive oxygen species production. J Surg Res. 2012 doi: 10.1016/j.jss.2012.05.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gibson GE, Kingsbury AE, Xu H, Lindsay JG, Daniel S, Foster OJ, Lees AJ, Blass JP. Deficits in a tricarboxylic acid cycle enzyme in brains from patients with parkinson's disease. Neurochem Int. 2003;43(2):129–35. doi: 10.1016/s0197-0186(02)00225-5. [DOI] [PubMed] [Google Scholar]
- 46.Parihar MS, Brewer GJ. Mitoenergetic failure in alzheimer disease. Am J Physiol Cell Physiol. 2007;292(1):C8–23. doi: 10.1152/ajpcell.00232.2006. [DOI] [PubMed] [Google Scholar]
- 47.Arrell DK, Elliott ST, Kane LA, Guo Y, Ko YH, Pedersen PL, Robinson J, Murata M, Murphy AM, Marban E, Van Eyk JE. Proteomic analysis of pharmacological preconditioning: Novel protein targets converge to mitochondrial metabolism pathways. Circ Res. 2006;99(7):706–14. doi: 10.1161/01.RES.0000243995.74395.f8. [DOI] [PubMed] [Google Scholar]
- 48.Sazanov LA, Jackson JB. Proton-translocating transhydrogenase and nad- and nadp-linked isocitrate dehydrogenases operate in a substrate cycle which contributes to fine regulation of the tricarboxylic acid cycle activity in mitochondria. FEBS Lett. 1994;344(2–3):109–16. doi: 10.1016/0014-5793(94)00370-x. [DOI] [PubMed] [Google Scholar]
- 49.Soundar S, O'Hagan M, Fomulu KS, Colman RF. Identification of mn2+-binding aspartates from alpha, beta, and gamma subunits of human nad-dependent isocitrate dehydrogenase. J Biol Chem. 2006;281(30):21073–81. doi: 10.1074/jbc.M602956200. [DOI] [PubMed] [Google Scholar]
- 50.McCormack JG, Denton RM. The role of ca2+ ions in the regulation of intramitochondrial metabolism and energy production in rat heart. Mol Cell Biochem. 1989;89(2):121–5. [PubMed] [Google Scholar]
- 51.Reitman ZJ, Yan H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: Alterations at a crossroads of cellular metabolism. J Natl Cancer Inst. 2010;102(13):932–41. doi: 10.1093/jnci/djq187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mailloux RJ, Beriault R, Lemire J, Singh R, Chenier DR, Hamel RD, Appanna VD. The tricarboxylic acid cycle, an ancient metabolic network with a novel twist. PLoS One. 2007;2(8):e690. doi: 10.1371/journal.pone.0000690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee SM, Koh HJ, Park DC, Song BJ, Huh TL, Park JW. Cytosolic nadp(+)-dependent isocitrate dehydrogenase status modulates oxidative damage to cells. Free Radic Biol Med. 2002;32(11):1185–96. doi: 10.1016/s0891-5849(02)00815-8. [DOI] [PubMed] [Google Scholar]
- 54.Yang ES, Richter C, Chun JS, Huh TL, Kang SS, Park JW. Inactivation of nadp(+)-dependent isocitrate dehydrogenase by nitric oxide. Free Radic Biol Med. 2002;33(7):927–37. doi: 10.1016/s0891-5849(02)00981-4. [DOI] [PubMed] [Google Scholar]
- 55.Kristian T, Gido G, Kuroda S, Schutz A, Siesjo BK. Calcium metabolism of focal and penumbral tissues in rats subjected to transient middle cerebral artery occlusion. Exp Brain Res. 1998;120(4):503–9. doi: 10.1007/s002210050424. [DOI] [PubMed] [Google Scholar]
- 56.Kristian T, Siesjo BK. Calcium in ischemic cell death. Stroke. 1998;29(3):705–18. doi: 10.1161/01.str.29.3.705. [DOI] [PubMed] [Google Scholar]
- 57.Roberts LJ, 2nd, Milne GL. Isoprostanes. J Lipid Res. 2009;50(Suppl):S219–23. doi: 10.1194/jlr.R800037-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Il'yasova D, Morrow JD, Ivanova A, Wagenknecht LE. Epidemiological marker for oxidant status: Comparison of the elisa and the gas chromatography/mass spectrometry assay for urine 2,3-dinor-5,6-dihydro-15-f2t-isoprostane. Ann Epidemiol. 2004;14(10):793–7. doi: 10.1016/j.annepidem.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 59.Diaz-Ruiz R, Rigoulet M, Devin A. The warburg and crabtree effects: On the origin of cancer cell energy metabolism and of yeast glucose repression. Biochim Biophys Acta. 1807(6):568–76. doi: 10.1016/j.bbabio.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 60.Nekrutenko A, Hillis DM, Patton JC, Bradley RD, Baker RJ. Cytosolic isocitrate dehydrogenase in humans, mice, and voles and phylogenetic analysis of the enzyme family. Mol Biol Evol. 1998;15(12):1674–84. doi: 10.1093/oxfordjournals.molbev.a025894. [DOI] [PubMed] [Google Scholar]
- 61.Cankaya M, Hernandez AM, Ciftci M, Beydemir S, Ozdemir H, Budak H, Gulcin I, Comakli V, Emircupani T, Ekinci D, Kuzu M, Jiang Q, Eichele G, Kufrevioglu OI. An analysis of expression patterns of genes encoding proteins with catalytic activities. BMC Genomics. 2007;8:232. doi: 10.1186/1471-2164-8-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Science, A.I.f.B. Allen brain atlas resources. 2009 http://www.brain-map.org.
- 63.Yang S, Liu T, Li S, Zhang X, Ding Q, Que H, Yan X, Wei K, Liu S. Comparative proteomic analysis of brains of naturally aging mice. Neuroscience. 2008;154(3):1107–20. doi: 10.1016/j.neuroscience.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 64.Qi F, Chen X, Beard DA. Detailed kinetics and regulation of mammalian nad-linked isocitrate dehydrogenase. Biochim Biophys Acta. 2008;1784(11):1641–51. doi: 10.1016/j.bbapap.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hartong DT, Dange M, McGee TL, Berson EL, Dryja TP, Colman RF. Insights from retinitis pigmentosa into the roles of isocitrate dehydrogenases in the krebs cycle. Nat Genet. 2008;40(10):1230–4. doi: 10.1038/ng.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Prensner JR, Chinnaiyan AM. Metabolism unhinged: Idh mutations in cancer. Nat Med. 2011;17(3):291–3. doi: 10.1038/nm0311-291. [DOI] [PubMed] [Google Scholar]
- 67.Reitman ZJ, Jin G, Karoly ED, Spasojevic I, Yang J, Kinzler KW, He Y, Bigner DD, Vogelstein B, Yan H. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc Natl Acad Sci U S A. 2011;108(8):3270–5. doi: 10.1073/pnas.1019393108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ferroli P, Acerbi F, Finocchiaro G. From standard treatment to personalized medicine: Role of idh1 mutations in low-grade glioma evolution and treatment. World Neurosurg. 2010;73(4):234–6. doi: 10.1016/j.wneu.2010.02.050. [DOI] [PubMed] [Google Scholar]
- 69.Frezza C, Tennant DA, Gottlieb E. Idh1 mutations in gliomas: When an enzyme loses its grip. Cancer Cell. 2010;17(1):7–9. doi: 10.1016/j.ccr.2009.12.031. [DOI] [PubMed] [Google Scholar]
- 70.Yin W, Signore AP, Iwai M, Cao G, Gao Y, Chen J. Rapidly increased neuronal mitochondrial biogenesis after hypoxic-ischemic brain injury. Stroke. 2008;39(11):3057–63. doi: 10.1161/STROKEAHA.108.520114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huang C, Andres AM, Ratliff EP, Hernandez G, Lee P, Gottlieb RA. Preconditioning involves selective mitophagy mediated by parkin and p62/sqstm1. PLoS One. 2011;6(6):e20975. doi: 10.1371/journal.pone.0020975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kleman AM, Brown JE, Zeiger SL, Hettinger JC, Brooks JD, Holt B, Morrow JD, Musiek ES, Milne GL, McLaughlin B. p66shc's role as an essential mitophagic molecule in controlling neuronal redox and energetic tone. Autophagy. 2010;6(7):948–9. doi: 10.4161/auto.6.7.13007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator pgc-1. Cell. 1999;98(1):115–24. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 74.Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12(8):2245–56. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.