

Abstract
Individuals with ulcerative colitis face an increased risk of developing colorectal cancer and would benefit from early chemopreventive intervention. Results from preclinical studies in the mouse model of dextran sulfate sodium-induced colitis demonstrate that flat and polypoid colitis-associated dysplasias arise via distinct genetic pathways, impacted by the allelic status of p53. Furthermore, flat and polypoid dysplasias vary in their response to induction by the heterocyclic amine 2-amino-3-methylimidazo[4,5-f]quinoline (IQ) and inhibition by 5-aminosalicylic acid, a common therapy for the maintenance of colitis patients. These data suggest that use of combination therapy is essential for the optimal inhibition of colitis-associated colorectal cancer.
Keywords: Colorectal cancer, ulcerative colitis, chemoprevention, heterocyclic amines, 5-aminosalicylic acid, mouse
1. Introduction
Ulcerative colitis is a chronic form of inflammatory bowel disease of unknown etiology that affects approximately 700,000 individuals per year in the US [1]. Hallmarks of the disease include repeated flares of inflammation and inflammatory cell infiltration in the colorectum that severely disrupt the architecture of the colonic mucosa and promote cellular transformation [2]. Patients with ulcerative colitis face a risk of developing colorectal cancer that is significantly higher than that of the general population and increases with the extent of colonic involvement, disease severity, duration of colitis and a family history of colorectal cancer [3]. A meta-analysis of colorectal cancer risk in ulcerative colitis patients suggests a cumulative incidence of malignancy of 2% by 10 years, 8% by 20 years and 18% by 30 years of disease [4]. However, risk estimates from individual studies are as high as 27.5% after 45 years of disease [5] and 33% after 20 years for inflammatory bowel disease patients with primary sclerosing cholangitis [6]. While significant advancements have been made in managing the symptoms experienced by colitis patients, much less attention has been given to ameliorating the significant cancer risk associated with this disease.
Development of colitis-associated colorectal cancer in humans is known to progress in a stepwise manner, with the inflamed mucosa giving rise to flat and polypoid dysplasias and ultimately invasive cancers [7; 8]. While dysplasia, defined as neoplastic epithelium confined to the basement membrane, remains the most reliable marker of risk for colonic malignancy in colitis patients [9–11], current surveillance protocols fail to reliably identify the early stages of colitis-associated neoplasia [12]. In particular, detection of flat lesions that lack an elevated growth component is extremely challenging in patients with pancolitis, where as many as 50% can be missed during surveillance colonoscopy [13]. Furthermore, high-grade dysplasia or cancer was found in 27% of patients who had a colectomy within 6 months of a diagnosis of flat low-grade dysplasia [8]. Because the dysplasia-to-carcinoma sequence is not absolute [14], much controversy surrounds the management of flat low-grade dysplasias once they are identified [10]. These data, when combined with the current recommendation not to initiate endoscopic screening until at least seven years following diagnosis, speak clearly to the critical need to detect colitis-associated neoplasms more reliably and earlier when chemopreventive intervention may be efficacious.
Although significant overlap exists in the specific genetic aberrations that contribute to sporadic and colitis-associated colorectal cancer, the timing of oncogenic events in the two diseases differs dramatically. While mutation of APC represents the putative gate-keeping event in sporadic colorectal carcinogenesis, it occurs later and much less frequently in patients with ulcerative colitis (6% for colitis-associated neoplasms and 74% for sporadic tumors) [15]. In contrast to sporadic disease, alterations in p53 are not only found in the earliest colitis-associated dysplasias but may occur well in advance of the formation of dysplasia and confer cancer susceptibility [16; 17]. Disruption of the MAPK pathway through mutation of either RAS or BRAF is found in approximately 30% of all colitis-associated cancers [18].
The dextran sulfate sodium (DSS) mouse model of induced colitis represents a clinically relevant system in which to study the molecular basis of colitis-associated lesions. Similar to humans, colorectal lesions develop as both flat (nonpolypoid) and polypoid neoplasias (Fig. 1). Several observations in mice with DSS-induced colitis provide compelling evidence that these morphological subtypes of lesions arise via distinct genetic mechanisms (Table 1). First, flat dysplasias/cancers exhibit inflammation scores (a product of the intensity and extent of inflammatory infiltration) that are approximately 10-fold higher than those of polypoid lesions. Second, the majority of cancers arise from flat colonic dysplasias. Based on observations in the azoxymethane (AOM) mouse model of colon carcinogenesis, flat dysplasias are expected to progress to invasive cancers without transitioning through a polypoid intermediate [19]. Large differences in the incidence of flat colorectal cancers among various mouse strains exposed to AOM provide evidence that the development of specific morphological subtypes of lesions is genetically predetermined. Third, β-catenin is localized to the nucleus of polypoid dysplasias induced by DSS, while flat lesions exhibit membranous staining. Of note, this differential is lost when DSS is administered in combination with AOM due to the ability of AOM to induce β-catenin mutations.
Fig. 1.

Histopathology of representative colorectal lesions in Swiss Webster mice with AOM/DSS-induced colitis. A) Chronic colitis characterized by thickening of the colonic mucosa, crypt distortion, and chronic inflammation (10× magnification). B) Ulcer in a background of chronic colitis (10× magnification). C) Polypoid colitis-associated dysplasia (4× magnification). Note: The lesion projects above the mucosa (compare to panel D). D) Flat (nonpolypoid) dysplasia. Note: The flat dysplasia (solid arrows) has a contour and thickness similar to that of the adjacent nonneoplastic mucosa (open arrow) (compare to panel C).
Table 1.
Comparison of the characteristics of flat and polypoid colitis-associated colorectal lesions in the DSS model*
| Strain | Treatment | Flat | Polypoid | |
|---|---|---|---|---|
| Inflammation | SW | DSS | 10× | × |
| Nuclear β-cat | SW/C57BL/6J | DSS | − | + |
| Cancers | SW | DSS | +++ | + |
| p53 mutation | C57BL/6J (p53−/−) | DSS | + | − |
| (p53+/+ or p53+/−) | DSS | − | + | |
| β-catenin mutation | C57BL/6J (p53−/−) | DSS | + | − |
| (P53+/+, p53+/−) | DSS | − | + | |
| IQ | Apc+/Min-FCCC | DSS | Increase | No change |
| 5-ASA | SW | AOM/DSS | Decrease | Trend |
DSS – dextran sulfate sodium was added to the drinking water and administered in cycles.
Results from previous studies by this group demonstrate that loss of p53 is a critical event in the commitment of colitis-associated neoplasias to develop as flat or polypoid morphological subtypes [20]. The role of p53 in colitis-associated tumorigenesis was examined by administering DSS to C57BL/6J mice that were either wild-type for p53 or p53 deficient (heterozygous or homozygous null) for 3–4 cycles followed by 120 days of untreated water. Complete loss of p53 led to an increase in the overall incidence (2.8-fold) and multiplicity (2.7 to 4.8-fold) of colitis-associated dysplasias and cancers as compared to animals wild-type or heterozygous for p53. Interestingly, a direct correlation was observed between loss of wild-type p53 and increasing multiplicity of flat dysplasias and cancers (Fig. 2). In mice bearing two copies of wild-type p53, 100% of the dysplasias were polypoid and no cancers were observed. The profile changed in mice heterozygous for p53, where the majority of the colitis-associated cancers were flat and the dysplasias polypoid. In p53 null mice, flat lesions were predominant, with only 15% of the lesions being polypoid. Furthermore, β-catenin mutations were not found in colitis-associated colorectal lesions from p53 null mice, while β-catenin mutations were detected in 75% of the lesions from p53 wild-type mice. These data provide strong evidence that flat and polypoid colitis-associated lesions arise via independent genetic pathways.
Fig. 2.

Comparison of the morphological subtypes of colorectal dysplasias and cancers in C57B1/6J mice with DSS-induced colitis that are either wild-type (p53+/+) or deficient (p53+/− or p53−/−). A direct correlation exists between the number of alleles of wild-type p53 present and the percentage of polypoid colorectal lesions, while an inverse association is observed with flat lesions.
Consistent with differences in the genetic make-up of flat and polypoid lesions, data from this group indicate that morphological subtypes of colitis-associated lesions vary in their response to both carcinogens and chemopreventive agents. The following section describes the ability of the heterocyclic amine IQ to preferentially induce flat dysplasias while 5-aminosalicylic acid (5-ASA), a common maintenance therapy for patients with ulcerative colitis, is most effective in inhibiting flat lesions [21].
2. Contribution of meat and heterocyclic amines to colitis-associated neoplasia
Data from recent epidemiological studies [22–25] continue to confirm earlier reports of a strong association between the consumption of red meat and increased risk of colorectal cancer. This putative association is of great concern in ulcerative colitis patients who consume significantly more animal protein, dietary fat, cholesterol, cereals and simple carbohydrates/sugars in an attempt to alleviate the symptoms of their disease when in clinical remission [26]. Adoption of these dietary habits leads to a greater degree of colonic aneuploidy, an independent risk factor for the development of cancer among colitis patients [27]. Jowett and colleagues [28] reported that colitis patients who ate red and processed meats were six times more likely to relapse as compared to those who consumed much less meat. Consistent with this clinical observation, circulating levels of gamma-glutamyltransferase, a marker of oxidative stress, were found to be elevated in subjects who consumed meat; an association that remained significant after adjusting for body mass index and other life style factors [29; 30]. Similar studies examining the effect of red meat consumption on plasma levels of C reactive protein in humans have yielded both positive and negative results [29; 31; 32]. It should be noted that there are numerous substances in meat that could potentially exacerbate colorectal inflammation and oxidative stress, including saturated fats, nitrates, nitrites, polycyclic aromatic hydrocarbons and heterocyclic amines.
Heterocyclic amines (HCAs) are potent carcinogens and mutagens that are generated from the reaction of creatine, creatinine, amino acids and sugar when protein-rich foods, including meats, are cooked at a high temperature [33; 34]. The most common HCAs include 2-amino-3-methylimidazo[4,5-f]quinoline (IQ) and 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP), the latter of which induces gastrointestinal cancers when administered to animals [35–37]. Results from the Tennessee Colorectal Polyp Study, a large colonoscopy-based case-control study, indicate an association between exposure to HCAs (PhIP, MelQx and DiMelQx) in cooked meats and increased risk of colorectal adenomas (OR – 1.3–1.4; P ⩽ 0.034) [24]. The association of HCAs with colon cancer risk was stronger for patients with multiple adenomas, as compared to single adenomas, and for serrated vs. nonserrated polyps. Similar associations have been reported between PhIP and rectal adenoma (OR = 1.75, P = 0.02) [38] and MeIQx and DiMeIQx and colon cancer (HR = 1.19 and 1.17, respectively (Ptrend < 0.001)) [23].
HCAs are procarcinogens that require metabolic activation to become mutagenic and carcinogenic [39]. Cytochrome P450 enzymes, primarily CYP1A2, metabolize HCAs to N-hydroxylamines that are further converted to esters by acetyltransferase and sulfotransferase. The resulting N-acetoxy metabolites of HCAs are spontaneously converted to arylnitrenium ions (R-NH+) and react with DNA to form adducts at the 8-position carbon of guanine (N-(deoxyguanosin-8-yl)-IQ). IQ and MeIQx can also form adducts by binding to the N2 position of guanine (i.e., 5-(deoxyguanosin-N2-yl)-IQ) [40–44]. Genetic alterations linked to HCA exposure include chromosomal aberrations, sister chromatid exchange [45] and microsatellite instability [46; 47]. Mutations in cancer-related genes, including H-ras, p53, Apc and β-catenin, have also been identified in tumors induced by HCAs [46–50].
3. Ability of the heterocyclic amine IQ to induce flat (but not polypoid) colitis-associated neoplasms
Previous studies from this group demonstrated that induction of colitis (by DSS) in mice bearing an Apc mutation (Apc+/Min-FCCC) drives the progression of colorectal dysplasias to cancers, allowing an analysis of both the early and late phases of carcinogenesis [51]. The effect of IQ exposure on inflammation- associated tumorigenesis was examined in female Apc+/Min-FCCC mice (9 wks of age, N=131) that were obtained from an established colony at Fox Chase Cancer Center (FCCC) and maintained on Global 2018 diet (Harlan Teklad, Madison, WI) for the duration of the study. Mice were randomized to one of four groups (untreated (N=27); IQ alone (N=38); DSS alone (N=36); and DSS and IQ (N=30)) and treated as outlined in Fig. 3. Ulcerative colitis was induced by administering two cycles of DSS (MW 30,000–40,000) (MP Biomedicals, Solon, OH). Cycle one consisted of 3 days of 4% DSS in the drinking water followed by 18 days of untreated water. Cycle two consisted of 2 days of 4% DSS in the drinking water plus 19 days of untreated water. Animals were gavaged with either IQ (40 mg/kg body weight) (Toronto Research Chemical, Inc., Ontario, Canada) or vehicle (DMSO/saline, volume ratio 1:1) on 2, 4 and 6 days post DSS treatment during each cycle of DSS. Body weights were recorded at the beginning and end of each cycle of DSS, and prior to gavage. At the time of sacrifice (16 weeks of age), the entire colon and rectum were examined grossly, fixed in 10% formalin and processed in their entirety for histopathologic evaluation.
Fig. 3.
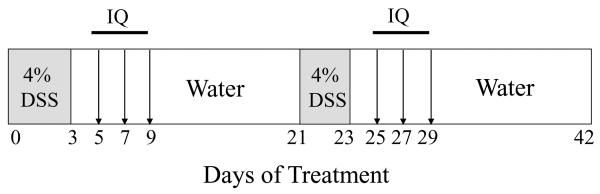
Regimen for the administration of IQ to animals with DSS-induced colitis. All animals were fed a Teklad Global 2018 diet for 5 weeks prior to the initiation of 2 cycles of DSS and for the duration of the study. DSS was added to the drinking water, and IQ (40 mg/kg body weight) was administered by gavage.
IQ was well tolerated when administered in the absence of DSS; with animals gaining weight at a rate comparable to that of untreated control mice. Body weights dropped immediately following DSS exposure but recovered over time; consistent with previous observations [21]. The rate of survival among Apc+/Min-FCCC mice treated with IQ, DSS, or DSS and IQ did not differ significantly (60.5%, 61.1% and 70%, respectively (P = 0.08)).
3.1. Incidence, multiplicity and types of dysplasia
The incidence of colorectal dysplasias in Apc+/Min-FCCC mice treated with IQ alone (65%) was comparable to that of untreated control mice (61%). Consistent with previous results [51], the incidence of dysplasias in mice administered DSS alone or in combination with IQ was 100%. No cancers were observed in either untreated mice or mice treated with IQ alone. However, exposure to IQ and DSS in combination resulted in a trend towards accelerated disease progression, increasing the incidence of cancers in DSS-treated mice from 22.7% to 37% (P = 0.3219).
The multiplicity of colonic dysplasias in Apc+/Min-FCCC mice administered IQ alone was comparable to that of untreated controls (Mean ± SEM - 0.96 ± 0.24 and 1.13 ± 0.25, respectively) (P > 0.05), with number of lesions per female as expected for this strain (Fig. 4A). Treatment with DSS led to a 30-fold increase in the number of dysplastic lesions per mouse (Mean SEM - 29.64 ± 3.00), as compared to controls (untreated or IQ alone) (P < 0.0001). Administration of IQ to Apc+/Min-FCCC mice with DSS-induced colitis further increased tumor multiplicity to 36.63 ± 2.98.
Fig. 4.

Effect of IQ on colonic tumorigenesis in APC+/Min-FCCC mice with DSS-induced colitis. A) Total tumor multiplicity is based on histopathological reviews. Asterisk: Significantly different from untreated control mice and mice treated with IQ alone (P < 0.05) as determined by the Wilcoxon 2-sample test. B) Linear regression analyses revealed trends of decreased polypoid lesions (P = 0.03) and increased flat lesions (P = 0.002) across the treatment groups.
Lesions were classified as flat or polypoid based on standardized morphology and nomenclature for the human pathology of colitis-associated colorectal neoplasia [52], as described previously [53]. Any dysplasia or cancer exhibiting an elevated growth pattern, either grossly or microscopically, was considered polypoid. Flat (nonpolypoid) lesions did not have an elevated growth component, with a height less than 2-fold that of the adjacent nonneoplastic mucosa. While polypoid lesions were the predominant subtype in both untreated (82%) and IQ-treated (no DSS) (65%) animals, the number of flat colonic lesions was 2-fold higher in mice treated with IQ alone as compared to untreated Apc+/Min-FCCC mice. In contrast, flat lesions were the major morphological subtype present in DSS-treated animals (Fig. 4B). Exposure to IQ led to a 1.3-fold increase in the percentage of flat colorectal lesions in mice with DSS-induced colitis (61%) as compared to animals administered only DSS (48%). Linear regression analyses yielded significant trends of decreased polypoid lesions (P = 0.03) and increased flat lesions (P = 0.002) across the treatment groups (untreated, IQ alone, DSS alone and DSS/IQ) as illustrated in Fig. 4B). Interestingly, all flat lesions in untreated mice and mice treated with IQ alone were microadenomas (1–2 glands). Based on the established prevalence of wild-type p53 in polypoid vs. flat lesions and the known ability of IQ to induce p53 mutations [50; 54], one can speculate that IQ alters the status of p53, thus promoting the growth of flat lesions preferentially.
3.2 Effect of IQ on the proliferation and apoptosis of colitis-associated lesions
In order to investigate the mechanism by which IQ promotes flat lesions on a background of colitis, the effect of IQ on cell proliferation (Ki-67) and apoptosis (active caspase-3) was investigated. Immunohistochemical staining was performed on specimens of the distal colon (nonneoplastic mucosa, polypoid lesions and flat lesions) from mice treated with DSS or DSS and IQ. Tissue sections were subjected to antigen retrieval (Reagent cc-1 for 30 minutes) and incubated with either primary antibody against caspase 3 (active) (AF835, R & D Systems, Minneapolis, MN 1:800 dilution) or Ki-67 (VP-K451, Vector Laboratories, Inc. Burlington, CA. 1:1500) for 1 hr at 37°C. Non-immune rabbit Ig at the same concentration as the primary antibody served as a negative control. As anticipated, the proliferative index of flat and polypoid lesions was higher than that of nonneoplastic colonic mucosa (Fig. 5). IQ increased the proliferative index of both the nonneoplastic colonic mucosa and polypoid lesions significantly (P = 0.05 and 0.017, respectively) but had no effect on cell proliferation in flat lesions (P = 0.60).
Fig. 5.

Impact of IQ exposure on cell proliferation (immunostaining for Ki-67). Values represent the number of Ki-67 positive nuclei per 300 tumor cells for polypoid or flat lesions and 6–7 normal colonic crypts for nonneoplastic mucosa (mean ± SEM. Asterisk: Significantly different from the DSS control group (P < 0.05) as determined by the Wilcoxon 2-sample test.
The number of caspase-3 (active) positive cells in the nonneoplastic inflamed colonic mucosa of animals administered DSS was very low; less than one per field (60×) (Fig. 6). In mice exposed to DSS alone or in combination with IQ, the number of apoptotic cells was at least 15-fold higher in both polypoid and flat lesions than in the nonneoplastic colonic mucosa. Treatment with IQ had no effect on the level of apoptosis in the nonneoplastic colonic mucosa. IQ significantly increased the number of caspase-3 positive cells in polypoid lesions (P = 0.002) but not in flat lesions (P = 0.13). In the case of polypoid lesions, the effect of IQ on cell proliferation is counterbalanced by an effect of this heterocyclic amine on apoptosis, resulting in a comparable multiplicity of polypoid dysplasias in animals receiving DSS alone or in combination with IQ. In contrast, flat lesions from mice treated with DSS/IQ had less apoptotic activity but a similar rate of cell proliferation as compared to mice treated with only DSS. Inhibition of apoptosis by IQ resulted in a significant increase (56%) in the number of flat lesions in mice treated with DSS and IQ (22.3%) as compared to those treated with DSS alone (14.3%; P = 0.004). These data when combined with the inability of standard white light endoscopy to reliably detect flat dysplasias and cancers speak clearly to the need to both improve current methods of clinical surveillance and establish dietary recommendations for ulcerative colitis patients who are at increased risk for colorectal cancer.
Fig. 6.

Effect of IQ on apoptosis as determined by immunohistochemistry using antibodies against caspase 3 (active form). Values represent the number of cells positive for caspase 3 per field (600×). Asterisk: Significantly different from the DSS control group (P = 0.0017) as determined by the Wilcoxon 2-sample test.
3.3 Impact of inhibition of inducible nitric oxide synthase on the formation of colitis-associated tumors
Inducible nitric oxide synthase (iNOS) is overexpressed within the colon of ulcerative colitis patients and may contribute to the development of colitis-associated neoplasia [55; 56]. Chronic activation of iNOS leads to the sustained production of nitric oxide at cytotoxic levels. The potential ability of IQ to be converted to a more potent carcinogen in the presence of free radical oxygen was investigated by administering S,S'-1,4-phenylene-bis(1,2-ethanediyl)bis-isothiourea (PBIT), an inhibitor of inducible nitric oxide synthase (iNOS), to IQ-treated mice with DSS-induced colitis. Administration of PBIT failed to decrease either the multiplicity of total lesions (DSS/IQ – 36.6 ± 3.0; DSS/IQ/PBIT – 38.4 ± 3.5) or the percentage of flat lesions (DSS/IQ – 61%); DSS/IQ/PBIT – 58%). These data suggest that IQ specifically induces flat lesions in a manner that is independent of iNOS activity.
4. 5-aminosalicylic acid is most effective in inhibiting flat colitis-associated neoplasias
5-aminosalicylic acid (5-ASA), a common therapy for the treatment of flare-ups and maintenance of disease remission in patients with ulcerative colitis, is structurally similar to aspirin. Results from several clinical studies suggest that long-term treatment with 5-ASA decreases the risk of developing colorectal cancer in patients with ulcerative colitis [57–59]. However, results from a recent comprehensive population-based analysis of long-term 5-ASA use (⩾ 5 years) by patients with inflammatory bowel disease revealed no chemoprophylactic activity against colorectal cancer [60].
5-ASA and other nonsteroidal anti-inflammatory drugs (NSAIDs) have common molecular targets including inflammation, proliferation, angiogenesis, and/or apoptosis. Specific pathways targeted by 5-ASA include TNF-α, TGF-β, NF-κB and Wnt/β-catenin signaling as well as cell cycle control, scavenging of reactive oxygen and nitrogen species and antimicrobiol activity [61]. While the growth inhibitory effects of 5-ASA are associated with downregulation of COX-2 mRNA and protein, the ability of 5-ASA to inhibit the growth of colorectal carcinoma cells that do not express COX-2 has been demonstrated [62]. Additional potential mechanisms of action of 5-ASA include enhanced apoptosis via inhibition of NF-kappaB and p38/MAP kinases, decreased Wnt/beta-catenin activity and elevation of SH-PTP-2, a phosphatase that targets and inactivates the EGF receptor [63]. Results from other studies attribute the antineoplastic activity of 5-ASA to its ability to improve the fidelity of DNA replication by reducing frameshift mutations at microsatellites [64] and inhibit the formation of reactive oxygen species by polymorphonuclear leukocytes [65; 66].
The efficacy of 5-ASA in inhibiting colitis-associated neoplasia has been evaluated in the mouse model of AOM/DSS-induced colitis [21]. Swiss Webster mice were administered 5-ASA in the drinking water at various doses starting one week prior to three cycles of DSS treatment. A 55% reduction in the size of polypoid colonic dysplasias was observed in mice receiving chronic 5-ASA at a dose (75 mg) comparable to that used for the treatment of patients with colitis. Interestingly, 5-ASA had no effect on the multiplicity of polypoid lesions. In contrast, the multiplicity of flat lesions was significantly decreased (44%) in mice administered 5-ASA at the same dose, while no reduction in lesion size was observed. Comprehensive analyses of the genome-wide RNA and microRNA expression profiles of flat and polypoid lesions are anticipated to enhance our understanding of the molecular mechanisms that underlie their differential response to 5-ASA exposure. These data strongly support the need to rationally design combination therapies for the prevention of colitis-associated cancer.
In summary, emerging data from in-depth molecular and pathological analyses continue to suggest that flat and polypoid colitis-associated colorectal neoplasms arise via distinct genetic mechanisms, with the mutational status of p53 being a major determinant of their ultimate morphology. Both the nuclear localization of β-catenin in DSS-induced polypoid lesions and the enhanced prevalence of infiltrating inflammatory cells in flat lesions provide further evidence of the inherent heterogeneity of colitis-associated neoplasms. The functional significance of the biological differences noted between flat and polypoid lesions is confirmed by their differential response to induction by the carcinogen IQ and chemoprevention by 5-ASA. These data provide unique insight into the essential need for combination therapy to target both flat and polypoid neoplasms and effectively inhibit the risk of colorectal cancer among patients with ulcerative colitis.
Acknowledgements
We wish to thank Marie Estes and Maureen Climaldi for their excellent administrative support in preparing this manuscript for publication and the Laboratory Animal Facility at Fox Chase Cancer Center for maintaining the animals required for these studies.
Role of the funding source This work was supported by grants CA072613, CA124693, and CA006927 from the National Cancer Institute and by an appropriation from the Commonwealth of Pennsylvania. The contents of this manuscript are solely the responsibility of the authors and do not necessarily represent the official views of the National Cancer Institute, or any other sponsoring organization.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement The authors have no conflicts of interest to disclose.
References
- [1].What are Crohn's & Colitis? Crohn's & Colitis Foundation of America; http://www.ccfa.org/what-are-crohns-and-colitis. Retrieved 11/27/2012. [Google Scholar]
- [2].Loftus EV., Jr. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology. 2004;126:1504–1517. doi: 10.1053/j.gastro.2004.01.063. [DOI] [PubMed] [Google Scholar]
- [3].Itzkowitz SH, Harpaz N. Diagnosis and management of dysplasia in patients with inflammatory bowel diseases. Gastroenterology. 2004;126:1634–1648. doi: 10.1053/j.gastro.2004.03.025. [DOI] [PubMed] [Google Scholar]
- [4].Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut. 2001;48:526–535. doi: 10.1136/gut.48.4.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Rutter MD, Saunders BP, Wilkinson KH, Rumbles S, Schofield G, Kamm MA, Williams CB, Price AB, Talbot IC, Forbes A. Thirty-year analysis of a colonoscopic surveillance program for neoplasia in ulcerative colitis. Gastroenterology. 2006;130:1030–1038. doi: 10.1053/j.gastro.2005.12.035. [DOI] [PubMed] [Google Scholar]
- [6].Jayaram H, Satsangi J, Chapman RW. Increased colorectal neoplasia in chronic ulcerative colitis complicated by primary sclerosing cholangitis: fact or fiction? Gut. 2001;48:430–434. doi: 10.1136/gut.48.3.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sharan R, Schoen RE. Cancer in inflammatory bowel disease. An evidence-based analysis and guide for physicians and patients. Gastroenterol. Clin. North Am. 2002;31:237–254. doi: 10.1016/s0889-8553(01)00014-0. [DOI] [PubMed] [Google Scholar]
- [8].Ullman T, Croog V, Harpaz N, Sachar D, Itzkowitz S. Progression of flat low-grade dysplasia to advanced neoplasia in patients with ulcerative colitis. Gastroenterology. 2003;125:1311–1319. doi: 10.1016/j.gastro.2003.08.023. [DOI] [PubMed] [Google Scholar]
- [9].Bernstein CN, Shanahan F, Weinstein WM. Are we telling patients the truth about surveillance colonoscopy in ulcerative colitis? Lancet. 1994;343:71–74. doi: 10.1016/s0140-6736(94)90813-3. [DOI] [PubMed] [Google Scholar]
- [10].Farraye FA, Odze RD, Eaden J, Itzkowitz SH. AGA technical review on the diagnosis and management of colorectal neoplasia in inflammatory bowel disease. Gastroenterology. 2010;138:746–774. 774, e1–4. doi: 10.1053/j.gastro.2009.12.035. [DOI] [PubMed] [Google Scholar]
- [11].Goldman H. Significance and detection of dysplasia in chronic colitis. Cancer. 1996;78:2261–2263. doi: 10.1002/(sici)1097-0142(19961201)78:11<2261::aid-cncr1>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- [12].Rutter M, Saunders B, Wilkinson K, Rumbles S, Schofield G, Kamm M, Williams C, Price A, Talbot I, Forbes A. Severity of inflammation is a risk factor for colorectal neoplasia in ulcerative colitis. Gastroenterology. 2004;126:451–459. doi: 10.1053/j.gastro.2003.11.010. [DOI] [PubMed] [Google Scholar]
- [13].Bruewer M, Krieglstein CF, Utech M, Bode M, Rijcken E, Anthoni C, Laukoetter MG, Schuermann G, Senninger N. Is colonoscopy alone sufficient to screen for ulcerative colitis-associated colorectal carcinoma? World J. Surg. 2003;27:611–615. doi: 10.1007/s00268-003-6639-y. [DOI] [PubMed] [Google Scholar]
- [14].Lewis JD. The many faces of low-grade dysplasia. Gastroenterology. 2003;125:1531–1533. doi: 10.1016/j.gastro.2003.09.006. [DOI] [PubMed] [Google Scholar]
- [15].Tarmin L, Yin J, Harpaz N, Kozam M, Noordzij J, Antonio LB, Jiang HY, Chan O, Cymes K, Meltzer SJ. Adenomatous polyposis coli gene mutations in ulcerative colitis-associated dysplasias and cancers versus sporadic colon neoplasms. Cancer Res. 1995;55:2035–2038. [PubMed] [Google Scholar]
- [16].Lashner BA, Shapiro BD, Husain A, Goldblum JR. Evaluation of the usefulness of testing for p53 mutations in colorectal cancer surveillance for ulcerative colitis. Am. J. Gastroenterol. 1999;94:456–462. doi: 10.1111/j.1572-0241.1999.877_f.x. [DOI] [PubMed] [Google Scholar]
- [17].Hussain SP, Amstad P, Raja K, Ambs S, Nagashima M, Bennett WP, Shields PG, Ham AJ, Swenberg JA, Marrogi AJ, Harris CC. Increased p53 mutation load in noncancerous colon tissue from ulcerative colitis: a cancer-prone chronic inflammatory disease. Cancer Res. 2000;60:3333–3337. [PubMed] [Google Scholar]
- [18].Aust DE, Haase M, Dobryden L, Markwarth A, Lohrs U, Wittekind C, Baretton GB, Tannapfel A. Mutations of the BRAF gene in ulcerative colitis-related colorectal carcinoma. Int. J. Cancer. 2005;115:673–677. doi: 10.1002/ijc.20925. [DOI] [PubMed] [Google Scholar]
- [19].Uronis JM, Herfarth HH, Rubinas TC, Bissahoyo AC, Hanlon K, Threadgill DW. Flat colorectal cancers are genetically determined and progress to invasion without going through a polypoid stage. Cancer Res. 2007;67:11594–11600. doi: 10.1158/0008-5472.CAN-07-3242. [DOI] [PubMed] [Google Scholar]
- [20].Chang WC, Coudry RA, Clapper ML, Zhang X, Williams KL, Spittle CS, Li T, Cooper HS. Loss of p53 enhances the induction of colitis-associated neoplasia by dextran sulfate sodium. Carcinogenesis. 2007;28:2375–2381. doi: 10.1093/carcin/bgm134. [DOI] [PubMed] [Google Scholar]
- [21].Clapper ML, Gary MA, Coudry RA, Litwin S, Chang WC, Devarajan K, Lubet RA, Cooper HS. 5-aminosalicylic acid inhibits colitis-associated colorectal dysplasias in the mouse model of azoxymethane/dextran sulfate sodium-induced colitis. Inflamm. Bowel Dis. 2008;14:1341–1347. doi: 10.1002/ibd.20489. [DOI] [PubMed] [Google Scholar]
- [22].Chan DS, Lau R, Aune D, Vieira R, Greenwood DC, Kampman E, Norat T. Red and processed meat and colorectal cancer incidence: meta-analysis of prospective studies. PLoS One. 2011;6:e20456. doi: 10.1371/journal.pone.0020456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Cross AJ, Ferrucci LM, Risch A, Graubard BI, Ward MH, Park Y, Hollenbeck AR, Schatzkin A, Sinha R. A large prospective study of meat consumption and colorectal cancer risk: an investigation of potential mechanisms underlying this association. Cancer Res. 2010;70:2406–2414. doi: 10.1158/0008-5472.CAN-09-3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Fu Z, Shrubsole MJ, Smalley WE, Wu H, Chen Z, Shyr Y, Ness RM, Zheng W. Association of meat intake and meat-derived mutagen exposure with the risk of colorectal polyps by histologic type. Cancer Prev. Res. 2011;4:1686–1697. doi: 10.1158/1940-6207.CAPR-11-0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Xu X, Yu E, Gao X, Song N, Liu L, Wei X, Zhang W, Fu C. Red and processed meat intake and risk of colorectal adenomas: A meta-analysis of observational studies. Int. J. Cancer. 2013;132:437–448. doi: 10.1002/ijc.27625. [DOI] [PubMed] [Google Scholar]
- [26].Rosman-Urbach M, Niv Y, Birk Y, Morgenstern S, Schwartz B. Relationship between nutritional habits adopted by ulcerative colitis relevant to cancer development patients at clinical remission stages and molecular-genetic parameters. Br. J. Nutr. 2006;95:188–195. doi: 10.1079/bjn20051624. [DOI] [PubMed] [Google Scholar]
- [27].Gerling M, Meyer KF, Fuchs K, Igl BW, Fritzsche B, Ziegler A, Bader F, Kujath P, Schimmelpenning H, Bruch HP, Roblick UJ, Habermann JK. High Frequency of Aneuploidy Defines Ulcerative Colitis-Associated Carcinomas: A Prognostic Comparison to Sporadic Colorectal Carcinomas. Ann. Surg. 2010;252:74–83. doi: 10.1097/SLA.0b013e3181deb664. [DOI] [PubMed] [Google Scholar]
- [28].Jowett SL, Seal CJ, Pearce MS, Phillips E, Gregory W, Barton JR, Welfare MR. Influence of dietary factors on the clinical course of ulcerative colitis: a prospective cohort study. Gut. 2004;53:1479–1484. doi: 10.1136/gut.2003.024828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Montonen J, Boeing H, Fritsche A, Schleicher E, Joost HG, Schulze MB, Steffen A, Pischon T. Consumption of red meat and whole-grain bread in relation to biomarkers of obesity, inflammation, glucose metabolism and oxidative stress. Eur. J. Nutr. 2012 doi: 10.1007/s00394-012-0340-6. e-pub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lee DH, Steffen LM, Jacobs DR., Jr. Association between serum gamma-glutamyltransferase and dietary factors: the Coronary Artery Risk Development in Young Adults (CARDIA) Study. Am. J. Clin. Nutr. 2004;79:60060–60065. doi: 10.1093/ajcn/79.4.600. [DOI] [PubMed] [Google Scholar]
- [31].Azadbakht L, Esmaillzadeh A. Red meat intake is associated with metabolic syndrome and the plasma C-reactive protein concentration in women. J. Nutr. 2009;139:335–339. doi: 10.3945/jn.108.096297. [DOI] [PubMed] [Google Scholar]
- [32].Hodgson JM, Ward NC, Burke V, Beilin LJ, Puddey IB. Increased lean red meat intake does not elevate markers of oxidative stress and inflammation in humans. J. Nutr. 2007;137:363–367. doi: 10.1093/jn/137.2.363. [DOI] [PubMed] [Google Scholar]
- [33].Felton JS, Knize MG. Heterocyclic-amine mutagens/carcinogens in foods. In: Cooper CS, Grover PL, editors. Handbook of Experimental Pharmacology. Springer-Verlag; Berlin: 1990. pp. 471–502. 1990. [Google Scholar]
- [34].Pais P, Salmon CP, Knize MG, Felton JS. Formation of mutagenic/carcinogenic heterocyclic amines in dry-heated model systems, meats, and meat drippings. J. Agric. Food Chem. 1999;47:1098–1108. doi: 10.1021/jf980644e. [DOI] [PubMed] [Google Scholar]
- [35].Ito N, Hasegawa R, Sano M, Tamano S, Esumi H, Takayama S, Sugimura T. A new colon and mammary carcinogen in cooked food, 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) Carcinogenesis. 1991;12:1503–1506. doi: 10.1093/carcin/12.8.1503. [DOI] [PubMed] [Google Scholar]
- [36].Ochiai M, Imai H, Sugimura T, Nagao M, Nakagama H. Induction of intestinal tumors and lymphomas in C57BL/6N mice by a food-borne carcinogen, 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine. Jpn J. Cancer Res. 2002;93:478–483. doi: 10.1111/j.1349-7006.2002.tb01281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ohgaki H, Takayama S, Sugimura T. Carcinogenicities of heterocyclic amines in cooked food. Mutat. Res. 1991;259:399–410. doi: 10.1016/0165-1218(91)90130-e. [DOI] [PubMed] [Google Scholar]
- [38].Ferrucci LM, Sinha R, Huang WY, Berndt SI, Katki HA, Schoen RE, Hayes RB, Cross AJ. Meat consumption and the risk of incident distal colon and rectal adenoma. Br. J. Cancer. 2012;106:608–616. doi: 10.1038/bjc.2011.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sugimura T, Wakabayashi K, Nakagama H, Nagao M. Heterocyclic amines: Mutagens/carcinogens produced during cooking of meat and fish. Cancer Sci. 2004;95:290–299. doi: 10.1111/j.1349-7006.2004.tb03205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hein DW, Rustan TD, Ferguson RJ, Doll MA, Gray K. Metabolic activation of aromatic and heterocyclic N-hydroxyarylamines by wild-type and mutant recombinant human NAT1 and NAT2 acetyltransferases. Arch. Toxicol. 1994;68:129–133. doi: 10.1007/s002040050045. [DOI] [PubMed] [Google Scholar]
- [41].Minchin RF, Reeves PT, Teitel CH, McManus ME, Mojarrabi B, Ilett KF, Kadlubar FF. N-and O-acetylation of aromatic and heterocyclic amine carcinogens by human monomorphic and polymorphic acetyltransferases expressed in COS-1 cells. Biochem. Biophys. Res. Commun. 1992;185:839–844. doi: 10.1016/0006-291x(92)91703-s. [DOI] [PubMed] [Google Scholar]
- [42].Snyderwine EG, Yamashita K, Adamson RH, Sato S, Nagao M, Sugimura T, Thorgeirsson SS. Use of the 32P-postlabeling method to detect DNA adducts of 2-amino-3-methylimidazolo[4,5-f]quinoline (IQ) in monkeys fed IQ: identification of the N-(deoxyguanosin-8-yl)-IQ adduct. Carcinogenesis. 1988;9:1739–1743. doi: 10.1093/carcin/9.10.1739. [DOI] [PubMed] [Google Scholar]
- [43].Turesky RJ, Rossi SC, Welti DH, Lay JO, Jr., Kadlubar FF. Characterization of DNA adducts formed in vitro by reaction of N-hydroxy-2-amino-3-methylimidazo[4,5-f]quinoline and N-hydroxy-2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline at the C-8 and N2 atoms of guanine. Chem. Res. Toxicol. 1992;5:479–490. doi: 10.1021/tx00028a005. [DOI] [PubMed] [Google Scholar]
- [44].Illig R, Largo RH, Weber M, Augsburger T, Lipp A, Wissler D, Perrenoud AE, Torresani T. Sixty children with congenital hypothyroidism detected by neonatal thyroid: mental development at 1, 4, and 7 years: a longitudinal study. Acta Endocrinol. Suppl. 1986;279:346–353. doi: 10.1530/acta.0.112s346. [DOI] [PubMed] [Google Scholar]
- [45].Sugimura J, Tamura G, Suzuki Y, Fujioka T. Allelic loss on chromosomes 3p, 5q and 17p in renal cell carcinomas. Pathol. Int. 1997;47:79–83. doi: 10.1111/j.1440-1827.1997.tb03724.x. [DOI] [PubMed] [Google Scholar]
- [46].Nagao M, Ushijima T, Toyota M, Inoue R, Sugimura T. Genetic changes induced by heterocyclic amines. Mutat. Res. 1997;376:161–167. doi: 10.1016/s0027-5107(97)00039-0. [DOI] [PubMed] [Google Scholar]
- [47].Toyota M, Ushijima T, Kakiuchi H, Canzian F, Watanabe M, Imai K, Sugimura T, Nagao M. Genetic alterations in rat colon tumors induced by heterocyclic amines. Cancer. 1996;77:1593–1597. doi: 10.1002/(SICI)1097-0142(19960415)77:8<1593::AID-CNCR26>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- [48].Dashwood RH, Suzui M, Nakagama H, Sugimura T, Nagao M. High frequency of beta-catenin (ctnnb1) mutations in the colon tumors induced by two heterocyclic amines in the F344 rat. Cancer Res. 1998;58:1127–1129. [PubMed] [Google Scholar]
- [49].Yu M, Snyderwine EG. H-ras oncogene mutations during development of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP)-induced rat mammary gland cancer. Carcinogenesis. 2002;23:2123–2128. doi: 10.1093/carcin/23.12.2123. [DOI] [PubMed] [Google Scholar]
- [50].Makino H, Ishizaka Y, Tsujimoto A, Nakamura T, Onda M, Sugimura T, Nagao M. Rat p53 gene mutations in primary Zymbal gland tumors induced by 2-amino-3-methylimidazo[4,5-f]quinoline, a food mutagen. Proc. Natl. Acad. Sci. USA. 1992;89:4850–4854. doi: 10.1073/pnas.89.11.4850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Cooper HS, Everley L, Chang WC, Pfeiffer G, Lee B, Murthy S, Clapper ML. The role of mutant Apc in the development of dysplasia and cancer in the mouse model of dextran sulfate sodium-induced colitis. Gastroenterology. 2001;121:1407–1416. doi: 10.1053/gast.2001.29609. [DOI] [PubMed] [Google Scholar]
- [52].Riddell RH, Goldman H, Ransohoff DF, Appelman HD, Fenoglio CM, Haggitt RC, Ahren C, Correa P, Hamilton SR, Morson BC, et al. Dysplasia in inflammatory bowel disease: standardized classification with provisional clinical applications. Hum. Pathol. 1983;14:931–968. doi: 10.1016/s0046-8177(83)80175-0. [DOI] [PubMed] [Google Scholar]
- [53].Cooper HS, Murthy S, Kido K, Yoshitake H, Flanigan A. Dysplasia and cancer in the dextran sulfate sodium mouse colitis model. Relevance to colitis-associated neoplasia in the human: a study of histopathology, B-catenin and p53 expression and the role of inflammation. Carcinogenesis. 2000;21:757–768. doi: 10.1093/carcin/21.4.757. [DOI] [PubMed] [Google Scholar]
- [54].Fujimoto Y, Hampton LL, Snyderwine EG, Nagao M, Sugimura T, Adamson RH, Thorgeirsson SS. p53 gene mutation in hepatocellular carcinoma induced by 2-amino-3-methylimidazo[4,5-f]quinoline in nonhuman primates. Jpn. J. Cancer Res. 1994;85:506–509. doi: 10.1111/j.1349-7006.1994.tb02387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Jaiswal M, LaRusso NF, Gores GJ. Nitric oxide in gastrointestinal epithelial cell carcinogenesis: linking inflammation to oncogenesis. Am. J. Physiol. Gastrointest. Liver Physiol. 2001;281:G626–34. doi: 10.1152/ajpgi.2001.281.3.G626. [DOI] [PubMed] [Google Scholar]
- [56].Wink DA, Vodovotz Y, Laval J, Laval F, Dewhirst MW, Mitchell JB. The multifaceted roles of nitric oxide in cancer. Carcinogenesis. 1998;19:711–721. doi: 10.1093/carcin/19.5.711. [DOI] [PubMed] [Google Scholar]
- [57].Eaden J, Abrams K, Ekbom A, Jackson E, Mayberry J. Colorectal cancer prevention in ulcerative colitis: a case-control study. Aliment Pharmacol Ther. 2000;14:145–53. doi: 10.1046/j.1365-2036.2000.00698.x. [DOI] [PubMed] [Google Scholar]
- [58].van Staa TP, Card T, Logan RF, Leufkens HG. 5-Aminosalicylate use and colorectal cancer risk in inflammatory bowel disease: a large epidemiological study. Gut. 2005;54:1573–1578. doi: 10.1136/gut.2005.070896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Velayos FS, Terdiman JP, Walsh JM. Effect of 5-aminosalicylate use on colorectal cancer and dysplasia risk: a systematic review and metaanalysis of observational studies. Am. J. Gastroenterol. 2005;100:1345–1353. doi: 10.1111/j.1572-0241.2005.41442.x. [DOI] [PubMed] [Google Scholar]
- [60].Bernstein CN, Nugent Z, Blanchard JF. 5-aminosalicylate is not chemoprophylactic for colorectal cancer in IBD: a population based study. Am. J. Gastroenterol. 2011;106:731–736. doi: 10.1038/ajg.2011.50. [DOI] [PubMed] [Google Scholar]
- [61].Lyakhovich A, Gasche C. Systematic review: molecular chemoprevention of colorectal malignancy by mesalazine. Aliment. Pharmacol. Ther. 2010;31:202–209. doi: 10.1111/j.1365-2036.2009.04195.x. [DOI] [PubMed] [Google Scholar]
- [62].Stolfi C, Fina D, Caruso R, Caprioli F, Sarra M, Fantini MC, Rizzo A, Pallone F, Monteleone G. Cyclooxygenase-2-dependent and -independent inhibition of proliferation of colon cancer cells by 5-aminosalicylic acid. Biochem. Pharmacol. 2008;75:668–676. doi: 10.1016/j.bcp.2007.09.020. [DOI] [PubMed] [Google Scholar]
- [63].Monteleone G, Franchi L, Fina D, Caruso R, Vavassori P, Monteleone I, Calabrese E, Naccari GC, Bellinvia S, Testi R, Pallone F. Silencing of SH-PTP2 defines a crucial role in the inactivation of epidermal growth factor receptor by 5-aminosalicylic acid in colon cancer cells. Cell Death Differ. 2006;13:202–211. doi: 10.1038/sj.cdd.4401733. [DOI] [PubMed] [Google Scholar]
- [64].Gasche C, Goel A, Natarajan L, Boland CR. Mesalazine improves replication fidelity in cultured colorectal cells. Cancer Res. 2005;65:3993–3997. doi: 10.1158/0008-5472.CAN-04-3824. [DOI] [PubMed] [Google Scholar]
- [65].McKenzie SM, Doe WF, Buffinton GD. 5-aminosalicylic acid prevents oxidant mediated damage of glyceraldehyde-3-phosphate dehydrogenase in colon epithelial cells. Gut. 1999;44:180–185. doi: 10.1136/gut.44.2.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Miyachi Y, Yoshioka A, Imamura S, Niwa Y. Effect of sulphasalazine and its metabolites on the generation of reactive oxygen species. Gut. 1987;28:190–195. doi: 10.1136/gut.28.2.190. [DOI] [PMC free article] [PubMed] [Google Scholar]