

Abstract
Severe burn induces rapid skeletal muscle proteolysis after the injury that persists for up to one year and results in skeletal muscle atrophy despite dietary and rehabilitative interventions. The purpose of this research was to determine acute changes in gene expression of skeletal muscle mass regulators post-burn injury. Biopsies were obtained from the vastus lateralis of a non-burned leg of eight burned subjects (6M, 2F: 34.8 ± 2.7 years: 29.9 ± 3.1% total body surface area burn) at 5.1 ± 1.1 days post-burn injury and from matched controls. mRNA expression of cytokines and receptors in the tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) families, and the ubiquitin proteasome E3 ligases, atrogin-1 and MuRF1, was determined. TNF receptor 1A was over 3.5 fold higher in burn. Expression of TNF-like weak inducer of apoptosis and its receptor were over 1.6 and 6.0-fold higher in burn. IL-6, IL-6 receptor, and glycoprotein 130, were elevated in burned subjects with IL-6 receptor over 13-fold higher. Suppressor of cytokine signaling-3 was also elevated in burn nearly 6-fold. Atrogin-1 and MuRF1, were more than 4- and 3-fold higher in burn. These results demonstrate for the first time that severe burn in humans has a remarkable impact on gene expression in skeletal muscle of a non-burned limb of genes that promote inflammation and proteolysis. Because these changes likely contribute to the acute skeletal muscle atrophy in areas not directly affected by the burn, in the future it will be important to determine the responsible systemic cues.
Keywords: skeletal muscle atrophy, tumor necrosis factor alpha like weak inducer of apoptosis, atrogenes, inflammation
Introduction
Following severe burn injury, hypermetabolism contributes to the accelerated breakdown of skeletal muscle resulting in considerable muscle atrophy. The breakdown of skeletal muscle throughout the body, including muscle not directly burned, results in mass and functional deficits that persist for months or even years post-injury [1]. Loss of lean mass can also contribute to clinical complications already of paramount concern to burn patients, including increasing infectious complications and impairing wound healing [2–4]. Despite advancements in surgical, dietary, and rehabilitative interventions, the optimal treatment to prevent post-burn skeletal muscle atrophy and its associated complications has not been determined, in part because of a lack of understanding of the molecular etiology of burn-induced muscle atrophy. Prior studies in humans have documented skeletal muscle atrophy and the metabolic response post-burn injury [1, 5–8], and important animal research has also been performed to better understand some of the molecular and cellular responses to burn injury in skeletal muscle [9, 10]. However, no studies exist on human skeletal muscle molecular responses to burn injury.
Skeletal muscle atrophy is regulated at the molecular level by a number of different mediators involved in both protein synthesis and degradation [11]. We have previously shown that there is no significant drop in several important signaling molecules that regulate muscle protein synthesis in human skeletal muscle acutely after burn [12], implicating that increased proteolysis is driving muscle atrophy in the acute phase post-burn injury. In fact, we have shown a significant increase in muscle degradation signaling through calpain activity and ubiquitination which is consistent with animal studies. However, skeletal muscle specific atrophy genes, atrogin-1 and MuRF-1, which are upregulated in multiple atrophy conditions [13–17] and in a burn animal model [10], have not been studied in human muscle post-burn.
Heightened inflammation signaling in skeletal muscle also contributes to muscle atrophy [18–20]. Multiple inflammatory cytokines are dramatically increased systemically in burn-injured patients [21, 22], including interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α), both of which are capable of inducing skeletal muscle proteolysis leading to atrophy [23, 24]. Limited data have been presented to verify the role of inflammation in human muscle atrophy post-burn injury, although evidence of post-burn inflammation contributing to atrophy does exist in animal models. Of particular interest in inflammation signaling and skeletal muscle is signaling via TNF-like weak inducer of apoptosis (TWEAK) and its receptor (Fn14), which was recently discovered as a potent inducer of skeletal muscle atrophy [25]. The TWEAK axis has not yet been studied in animal models or in humans in the context of burn-induced muscle proteolysis.
The incomplete understanding of the signaling mechanisms underlying human burn-induced skeletal muscle atrophy precludes sufficient treatments to prevent it. For example, Tuvdendorj et al. recently reported that protein breakdown remained elevated 6 months post-burn in children, and that the skeletal muscles remained unresponsive (net protein balance remained negative) to high dose amino acids via infusion, which has proven to be acutely anabolic in most all other conditions. Significant advancements are needed before this poorly understood proteolytic state, which is complicated by anabolic resistance, can be treated effectively [26]. The purpose of this study was thus to take an important first step toward advancing the field by determining, for the first time, a targeted transcript expression profile of putative and relatively novel proteolytic and pro-inflammatory genes in skeletal muscle in the initial days post-burn injury.
Methods
Subjects
Eight patients from the local Level 1 regional burn center were enrolled in the study. Adults with a burn injured area between 20% and 60% total body surface area (TBSA) with no other trauma related injuries, were eligible to enroll. Prior to enrollment, all subjects, or their legally authorized representatives, gave written informed consent. Utilizing a tissue bank of muscle samples from de-identified, healthy volunteers, control subjects were chosen to match burned subjects for age, sex, and body mass index. All procedures were approved by the local Institutional Review Board.
Sample Collection
Skeletal muscle biopsies were obtained during burn-related surgical procedures between 3 and 10 days after the injury event (during acute hypermetabolic phase). Percutaneous needle muscle biopsies were taken from the vastus lateralis of a non-burned thigh as previously described [27]. Muscle samples (30 mg portions) were snap frozen in liquid nitrogen and stored at −80°C until further analysis. Banked vastus lateralis muscle samples had been obtained similarly and stored identically prior to being matched to burned subjects.
RNA isolation
RNA was isolated and purified from the frozen muscle samples using Tri-Reagent (Molecular Research Center, Cincinnati, OH) and RNeasy Mini Kits (QIAGEN Inc., Valencia, CA), following the manufacturer’s instructions. RNA quantity and quality were determined using a spectrophotometer to measure absorbance at 260 nm and the 260:280 ratio, respectively (NanoDrop ND-1000. ThermoScientific. Rockford, IL).
RT-qPCR
mRNA expression of twelve targets known to be involved in muscle protein breakdown and/or inflammation were measured from burn and control muscle biopsies using RT-qPCR (StepOne System, Applied Biosystems, Foster City, CA).
cDNA was synthesized using the SuperScript VILO cDNA Synthesis kit (Invitrogen , Carlsbad, CA). The following TaqMan® Gene Expression Assays (Applied Biosystems) were used for analysis of mRNA expression; atrogin (Hs01041408_m1), MuRF1 (Hs00822397_m1), IL6 (Hs00985639_m1), IL6 receptor (Hs00794121_m1), IL6ST/gp130 (Hs00174360_m1), SOCS3 (Hs00269575_s1), TNF (Hs00174128_m1), TNF Receptor 1A (Hs00533560_m1), TWEAK (Hs00356411_m1), TWEAK Receptor (Hs0017993_m1), IGF1 (Hs01547656_m1), and IGF1 receptor (Hs00609566_m1). GAPDH (Hs02758991_g1) was used as an endogenous control. All amplifications were run in 10 µl of total volume using TaqMan® Fast Advanced Master Mix (Applied Biosystems) following the manufacturer’s instructions using the StepOne™ Real-Time PCR System (Applied Biosystems). All samples were run in triplicate. Relative amounts of target mRNA was determined using the comparative threshold cycle method (ΔΔCT). Expression of target genes was normalized to the corresponding expression level of GAPDH, which did not differ between groups. The data were analyzed using StepOne software version 2.2.2 (Applied Biosystems), and all results are expressed as the relative fold change compared to the young group at baseline.
Statistics
All values are reported as means ± SEM. Burn subjects and control groups were compared using the Wilcoxon ranked sum test. Significance was defined as p < 0.05.
Results
Burned subjects ranged in age from 24 to 47 years with an average of 34.8 ± 2.7 years and had an average BMI of 26.0 ± 1.8. The size of burns ranged from 20% – 47% TBSA, with an average burn size of 29.9 ± 3.1%. Muscle samples were collected between three and ten days post-injury (5.1 ± 1.1 days) during the hypermetabolic flow phase. Control subjects ranged in age from 25 to 47 years with an average of 33.4 ± 2.6 years and had an average BMI of 26.1 ± 0.9. No significant differences existed between burned and control subjects for age or BMI. All burned subjects survived the burn injury. Three burned patients were treated with oxandrolone and insulin and one other was treated with only insulin. All had been receiving general burn standard of care in the trauma/burn intensive care unit or trauma/burn nursing unit including nutritional support. Fluid resuscitation was based on the Parkland formula. The initial resuscitation was started at 4cc/kg/% total body surface area burn of lactated ringer’s solution and then the fluids were titrated based on urine output. The goal was an hourly urine output of 30–50 milliliters. Enteral access was obtained on admission if the patients were intubated, and then a standard high protein high calorie commercially available enteral formulation was started generally by post burn day 2. The initial goal was to deliver approximately 25 calories per kilogram of normal body weight. By the second week, indirect calorimetry was performed and the results were used to determine tube feeds. If the patient was not intubated, the patient was allowed a regular diet and calorie counts were performed to ensure adequate intake. The patients were taken to the operating room by post burn day #5 to start the excision and grafting procedures.
Muscle specific E3 ubiquitin ligases, atrogin-1 and MuRF-1 are known to induce muscle proteolysis and atrophy. In this study, atrogin-1 and MuRF-1 mRNAs were significantly higher in the skeletal muscles of burn victims in the days after injury (Figure 1). Atrogin-1 mRNA was over 4-fold higher and MuRF-1 mRNA over 3-fold higher in burned subjects than in matched controls (p < 0.05).
Figure 1.

Relative skeletal muscle mRNA expression of muscle specific E3 ligases involved in muscle atrophy. Data expressed as mean +/− SEM. * indicates statistical significance (p < 0.05).
The expression level of the inflammation-related TWEAK-Fn14 signaling axis in burn was of particular interest since it has recently been shown to be a powerful effecter of skeletal muscle atrophy [25]. TWEAK mRNA expression was 1.75-fold higher (p < 0.05) in burn and the TWEAK receptor, Fn14, was 6-fold higher (p < 0.05) in burn (Figure 2). Further, TNFα Receptor 1A was over 3.6 fold higher in burn than control (p < 0.05), while the expression of TNF-α itself did not differ between groups (Figure 3).
Figure 2.
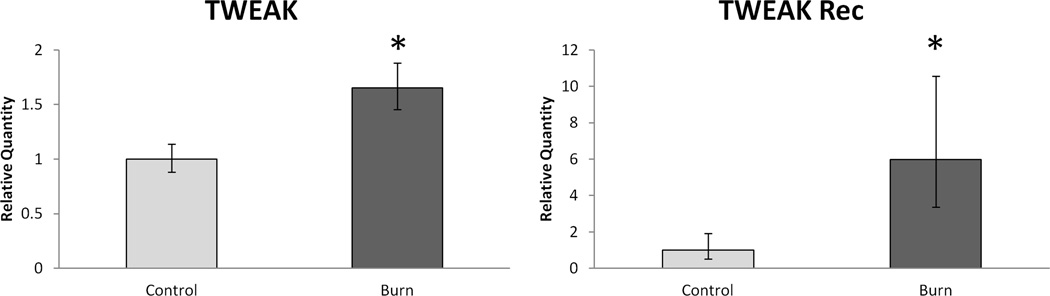
Relative skeletal muscle mRNA expression of the TWEAK/Fn 14 system. Data expressed as mean +/− SEM. * indicates statistical significance (p < 0.05).
Figure 3.
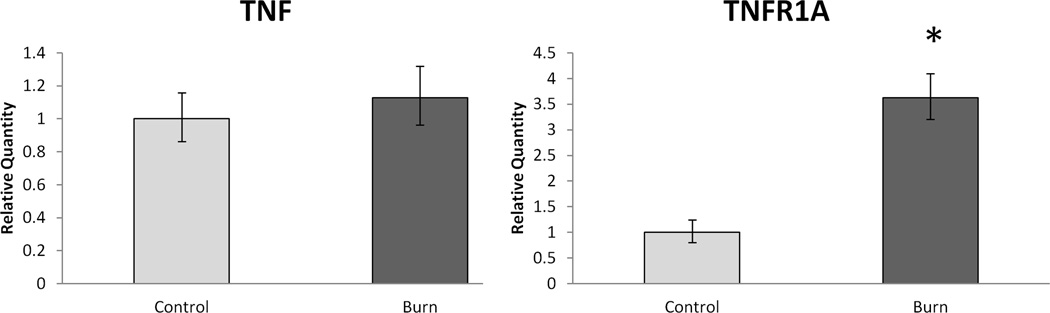
Relative skeletal muscle mRNA expression of TNF-α and TNF-α 1A Receptor. Data expressed as mean +/− SEM. * indicates statistical significance (p < 0.05).
We also determined mRNA levels of several members of the IL-6 signaling pathway (Figure 4). Muscle levels of IL-6 were nearly 3-fold higher in burn (p < 0.05) while the levels of IL-6 receptor were over 13-fold higher (p < 0.05). The common cytokine receptor signal transducer, GP130, was also higher (over 1.7-fold) in burn. In addition to the increased IL-6 signaling mRNA expression, SOCS3 was nearly 6-fold higher in burn muscle than control (p < 0.05).
Figure 4.

Relative skeletal muscle mRNA expression of members of the IL-6 signaling pathway. Data expressed as mean +/− SEM. * indicates statistical significance (p < 0.05).
Additionally, IGF1 and IGF1 receptor mRNA levels were determined. No significant difference was found for IGF1 levels, but IGF1 receptor expression was almost 2-fold higher.
Discussion
As demonstrated in the current study, severe burn injury has potent and systemic effects that markedly affect the expression of several genes in non-burned skeletal muscle that regulate protein metabolism and muscle mass including the skeletal muscle atrophy genes, atrogin-1 and MuRF-1, genes of the TWEAK/Fn14 pathway, and a number of other important inflammatory signaling processes within the muscle, all of which have been shown to affect muscle mass.
While this is the first time increases in atrogin-1 and MuRF-1 have been observed in human skeletal muscle in response to burn injury, it was not unexpected, as both of these are known to negatively influence skeletal muscle mass [28, 29], and, using a rat model, Lang et al. have previously shown the up-regulation of these genes in the days following burn injury [10]. Increased expression of atrogin-1 and/or MuRF-1 has also been shown in humans in other conditions leading to atrophy such as unloading [14, 16], aging [17], fasting [15],and in disease states such as COPD [13]. Since both are E3 ligases, atrogin-1 and MuRF-,1 ultimately exert their influence on protein breakdown through the ubiquitin-proteasome system by tagging proteins for degradation [30]. Previous work in our laboratory determined that more ubiquitinated proteins existed in the skeletal muscle of burned patients relative to matched controls [22]. The data presented in this study correspond with our previous observations and suggest that the increased atrogin-1 and MuRF-1 mRNAs are likely translated to protein and mediate proteasome activation and subsequent protein degradation. Unfortunately suitable human antibodies for atrogin-1 and MuRF-1 protein could not be found to confirm this despite a concentrated effort. However, it is likely that atrogin-1 and MuRF-1 were ultimately responsible for the increase in the number of ubiquitinated proteins. This increase could have powerful effects on skeletal muscle mass after burn injury because of their actions on proteins within the muscle. MuRF-1 is involved in the degradation of myosin heavy chain, thick, filaments [31], and atrogin-1 degrades proteins involved in protein synthesis and stem cell activation pathways, theoretically interfering with protein synthesis and hypertrophy/regeneration mechanisms [32, 33]. Nevertheless, it is worth noting that atrogin-1 and MuRF-1 are not necessarily all that is required for atrophy as muscle protein breakdown can be prevented despite high levels of MuRF-1 and atrogin-1 expression. Septic rats typically experience skeletal muscle atrophy similar to that of burned rats, but calpain inhibitors can prevent muscle atrophy in septic rats even in the presence of elevated MuRF-1 and atrogin-1 mRNA levels [34]. This is further support that the proteasome alone is unable to degrade intact myofibrils and calpain activity is necessary to initiate the process of myofibril protein breakdown before MuRF-1 can further break them down. This might also suggest calpain signaling may be involved in mediating the translation of atrogin-1 and MuRF-1 mRNAs to protein. Interestingly, we have previously shown high levels of calpain2 in skeletal muscle from burn patients [22].
The skeletal muscles of burned subjects had high mRNA levels of TWEAK and, perhaps more importantly, its receptor, Fn14. We are the first to show this in humans suffering from a known atrophy-inducing condition, but a series of elegant experiments from the laboratory of Kumar have elucidated its role in muscle atrophy using in vitro experiments and animal models [25, 35–40]. TWEAK exerts its negative effects on skeletal muscle in several ways including inhibiting the PI3K/Akt protein synthesis pathway, activating the pro-inflammatory transcription factor NFκB, activating matrix metalloproteinases, and increasing expression of MuRF-1 [41]. Based on these recent discoveries, it is likely that the high TWEAK expression in burned subject’s muscles contributes to acute post-burn muscle atrophy. As we have previously shown, serum TNF-α is increased post-burn, and in this study we observed that the mRNA expression the TNF-α receptor 1A was over 3.6 fold higher in burn than control, although muscle-specific expression of TNF-α was no different than matched controls [22]. In addition to activating NFκB, TNF-α can mediate muscle atrophy signaling by stimulating the expression of atrogin-1 through p38 MAPK [42]. In our prior work we did not find evidence of elevated NFκB signaling; however, our analysis was limited to the p50 subunit and it has been observed that differences exist in the levels of individual NFκB family members during atrophy [43]. Still, it is attractive to speculate that TNF-α or TNF-like signaling was at least partially responsible for up-regulating atrogin-1 expression and that this may have been driven by p38 MAPK. This is an important future direction, as p38 MAPK signaling can be inhibited pharmacologically..
An additional factor potentially influencing skeletal muscle loss post-burn injury is the activity of IL-6 within the skeletal muscle. We previously reported serum IL-6 levels 70-fold higher in burn patients vs. control. Here we found markedly up-regulated muscle expression of several IL-6 related transcripts including IL-6 itself, the IL-6 receptor, gp130, and SOCS3. Elevated IL-6 causes skeletal muscle atrophy in animals [8, 23], and only a modest elevation is needed to induce myofibrillar protein loss [23]. This may result at least in part from IL-6 mediated activation of atrogin-1 [44]. Strong associations exist between high IL-6 levels and muscle dysfunction in humans [45, 46], and high SOCS3 plays a role in age-related muscle atrophy [47] perhaps by its effects on growth hormone receptor signaling [48]. Despite being unable to directly correlate these expression changes with myofibrillar protein loss, the fact that we found up-regulation of all four IL-6 related genes, and that IL-6 has been shown to induce muscle atrophy [23], strongly implicates a central role of muscle IL-6 in post-burn skeletal muscle atrophy.
The gene expression findings presented here clearly show that serious burn injury markedly impacts skeletal muscle in non-burned regions. The average TBSA burn was 30%, substantially lower than the 40+% TBSA that is associated with the most dramatic and prolonged skeletal muscle atrophy [7], and 4 of the 8 burn victims were receiving either insulin, oxandrolone, or both, which are known to partially attenuate skeletal muscle catabolism post-burn[12, 49–51]. That the molecular responses were robust in spite of these factors further underscores the potent and systemic impact of burn injury on skeletal muscle.
While the importance of these findings should not be discounted, we should point out several study limitations which should be considered when interpreting the results. As with many studies of trauma victims, we were limited to a small set of subjects who fit our enrollment criteria and were able to consent. As such, the subjects were not the homogenous group that is expected of animal studies, and the group was not sufficiently large enough to divide into subsets as is seen in larger clinical trials. Measurement of muscle atrophy was not performed, so we cannot definitively prove that atrophy occurred in these particular subjects, although we have no reason to believe that our severely burned subjects would respond differently than countless other severely burned patients who have had documented skeletal muscle atrophy following similar burn injuries. The biopsy was performed on the vastus lateralis of a non-burned limb in order to prevent the confounding influence of a direct injury to the muscle and to allow for comparison of our results to other studies of human muscle atrophy, a vast majority of which also use a biopsy the vastus lateralis. The results therefore do not represent what would happen in other muscles with different physiologic requirements.
Since it is clear from our gene expression results in the current study that the process of muscle atrophy is initiated very soon after burn injury, future research should definitively determine whether these early gene expression changes initiate changes at the protein signaling level and drive burn-induced skeletal muscle atrophy. This information will lead to potential therapeutic approaches to lessen their negative effects. Victims of severe burn injury face a long and difficult recovery and rehabilitative period made even more challenging by the functional limitations subsequent to extensive skeletal muscle loss. Knowledge of the mechanisms underlying burn-induced muscle loss is critical for the development of treatments to attenuate it and allow for a quicker, more complete functional recovery.
Acknowledgments
We are indebted to the patients for their willingness to participate. The project described was supported by F32AR060670 (EKM) and a VA Merit Review Award (MMB). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Arthritis and Musculoskeletal and Skin Diseases or the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hart DW, et al. Persistence of muscle catabolism after severe burn. Surgery. 2000;128(2):312–319. doi: 10.1067/msy.2000.108059. [DOI] [PubMed] [Google Scholar]
- 2.Chandra RK. Nutrition and immunology: from the clinic to cellular biology and back again. Proc Nutr Soc. 1999;58(3):681–683. doi: 10.1017/s0029665199000890. [DOI] [PubMed] [Google Scholar]
- 3.Wilmore DW. Catabolic illness. Strategies for enhancing recovery. N Engl J Med. 1991;325(10):695–702. doi: 10.1056/NEJM199109053251005. [DOI] [PubMed] [Google Scholar]
- 4.Zaloga GP RP. Early enteral feeding improves outcome. In: JL V, editor. Yearbook of Intensive Care and Emergency Medicine. Berlin: Springer-Verlag; 1997. pp. 701–714. [Google Scholar]
- 5.Pereira C, et al. Post burn muscle wasting and the effects of treatments. Int J Biochem Cell Biol. 2005;37(10):1948–1961. doi: 10.1016/j.biocel.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 6.Sayeed MM. Signaling mechanisms of altered cellular responses in trauma, burn, and sepsis: role of Ca2+ Arch Surg. 2000;135(12):1432–1442. doi: 10.1001/archsurg.135.12.1432. [DOI] [PubMed] [Google Scholar]
- 7.Hart DW, et al. Determinants of skeletal muscle catabolism after severe burn. Ann Surg. 2000;232(4):455–465. doi: 10.1097/00000658-200010000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rutan RL, Herndon DN. Growth delay in postburn pediatric patients. Arch Surg. 1990;125(3):392–395. doi: 10.1001/archsurg.1990.01410150114021. [DOI] [PubMed] [Google Scholar]
- 9.Fang CH, et al. Protein breakdown in muscle from burned rats is blocked by insulin-like growth factor i and glycogen synthase kinase-3beta inhibitors. Endocrinology. 2005;146(7):3141–3149. doi: 10.1210/en.2004-0869. [DOI] [PubMed] [Google Scholar]
- 10.Lang CH, Huber D, Frost RA. Burn-induced increase in atrogin-1 and MuRF-1 in skeletal muscle is glucocorticoid independent but downregulated by IGF-I. Am J Physiol Regul Integr Comp Physiol. 2007;292(1):R328–R336. doi: 10.1152/ajpregu.00561.2006. [DOI] [PubMed] [Google Scholar]
- 11.Kandarian SC, Jackman RW. Intracellular signaling during skeletal muscle atrophy. Muscle Nerve. 2006;33(2):155–165. doi: 10.1002/mus.20442. [DOI] [PubMed] [Google Scholar]
- 12.Merritt EK, Cross JM, Bamman MM. Inflammatory and protein metabolism signaling responses in human skeletal muscle after burn injury. J Burn Care Res. 2012;33(2):291–297. doi: 10.1097/BCR.0b013e3182331e4b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doucet M, et al. Muscle atrophy and hypertrophy signaling in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;176(3):261–269. doi: 10.1164/rccm.200605-704OC. [DOI] [PubMed] [Google Scholar]
- 14.Gustafsson T, et al. Effects of 3 days unloading on molecular regulators of muscle size in humans. J Appl Physiol. 2010;109(3):721–727. doi: 10.1152/japplphysiol.00110.2009. [DOI] [PubMed] [Google Scholar]
- 15.Larsen AE, et al. Actions of short-term fasting on human skeletal muscle myogenic and atrogenic gene expression. Ann Nutr Metab. 2006;50(5):476–481. doi: 10.1159/000095354. [DOI] [PubMed] [Google Scholar]
- 16.Levine S, et al. Increased proteolysis, myosin depletion, and atrophic AKT-FOXO signaling in human diaphragm disuse. Am J Respir Crit Care Med. 2011;183(4):483–490. doi: 10.1164/rccm.200910-1487OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Raue U, et al. Proteolytic gene expression differs at rest and after resistance exercise between young and old women. J Gerontol A Biol Sci Med Sci. 2007;62(12):1407–1412. doi: 10.1093/gerona/62.12.1407. [DOI] [PubMed] [Google Scholar]
- 18.Degens H, Alway SE. Control of muscle size during disuse, disease, and aging. Int J Sports Med. 2006;27(2):94–99. doi: 10.1055/s-2005-837571. [DOI] [PubMed] [Google Scholar]
- 19.Spate U, Schulze PC. Proinflammatory cytokines and skeletal muscle. Curr Opin Clin Nutr Metab Care. 2004;7(3):265–269. doi: 10.1097/00075197-200405000-00005. [DOI] [PubMed] [Google Scholar]
- 20.Li H, Malhotra S, Kumar A. Nuclear factor-kappa B signaling in skeletal muscle atrophy. J Mol Med. 2008;86(10):1113–1126. doi: 10.1007/s00109-008-0373-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Finnerty CC, et al. Cytokine expression profile over time in severely burned pediatric patients. Shock. 2006;26(1):13–19. doi: 10.1097/01.shk.0000223120.26394.7d. [DOI] [PubMed] [Google Scholar]
- 22.Merritt EK, Cross JM, Bamman MM. Inflammatory and Protein Metabolism Signaling Responses in Human Skeletal Muscle After Burn Injury. J Burn Care Res. 2012;33(2):291–297. doi: 10.1097/BCR.0b013e3182331e4b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haddad F, et al. IL-6-induced skeletal muscle atrophy. J Appl Physiol. 2005;98(3):911–917. doi: 10.1152/japplphysiol.01026.2004. [DOI] [PubMed] [Google Scholar]
- 24.Warren RS, et al. The acute metabolic effects of tumor necrosis factor administration in humans. Arch Surg. 1987;122(12):1396–1400. doi: 10.1001/archsurg.1987.01400240042007. [DOI] [PubMed] [Google Scholar]
- 25.Dogra C, et al. TNF-related weak inducer of apoptosis (TWEAK) is a potent skeletal muscle-wasting cytokine. Faseb J. 2007;21(8):1857–1869. doi: 10.1096/fj.06-7537com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tuvdendorj D, et al. Skeletal muscle is anabolically unresponsive to an amino acid infusion in pediatric burn patients 6 months postinjury. Ann Surg. 2011;253(3):592–597. doi: 10.1097/SLA.0b013e31820d9a63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Evans WJ, Phinney SD, Young VR. Suction applied to a muscle biopsy maximizes sample size. Med Sci Sports Exerc. 1982;14(1):101–102. [PubMed] [Google Scholar]
- 28.Bodine SC, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294(5547):1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 29.Gomes MD, et al. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci U S A. 2001;98(25):14440–14445. doi: 10.1073/pnas.251541198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lecker SH. Ubiquitin-protein ligases in muscle wasting: multiple parallel pathways? Curr Opin Clin Nutr Metab Care. 2003;6(3):271–275. doi: 10.1097/01.mco.0000068963.34812.e5. [DOI] [PubMed] [Google Scholar]
- 31.Cohen S, et al. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J Cell Biol. 2009;185(6):1083–1095. doi: 10.1083/jcb.200901052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lagirand-Cantaloube J, et al. Inhibition of atrogin-1/MAFbx mediated MyoD proteolysis prevents skeletal muscle atrophy in vivo. PLoS One. 2009;4(3):e4973. doi: 10.1371/journal.pone.0004973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lagirand-Cantaloube J, et al. The initiation factor eIF3-f is a major target for atrogin1/MAFbx function in skeletal muscle atrophy. EMBO J. 2008;27(8):1266–1276. doi: 10.1038/emboj.2008.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fareed MU, et al. Treatment of rats with calpain inhibitors prevents sepsis-induced muscle proteolysis independent of atrogin-1/MAFbx and MuRF1 expression. Am J Physiol Regul Integr Comp Physiol. 2006;290(6):R1589–R1597. doi: 10.1152/ajpregu.00668.2005. [DOI] [PubMed] [Google Scholar]
- 35.Bhatnagar S, et al. TWEAK causes myotube atrophy through coordinated activation of ubiquitin-proteasome system, autophagy, and caspases. J Cell Physiol. 2011 doi: 10.1002/jcp.22821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dogra C, et al. Fibroblast growth factor inducible 14 (Fn14) is required for the expression of myogenic regulatory factors and differentiation of myoblasts into myotubes. Evidence for TWEAK-independent functions of Fn14 during myogenesis. J Biol Chem. 2007;282(20):15000–15010. doi: 10.1074/jbc.M608668200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumar M, et al. TNF-like weak inducer of apoptosis (TWEAK) activates proinflammatory signaling pathways and gene expression through the activation of TGF-beta-activated kinase 1. J Immunol. 2009;182(4):2439–2448. doi: 10.4049/jimmunol.0803357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mittal A, et al. The TWEAK-Fn14 system is a critical regulator of denervation-induced skeletal muscle atrophy in mice. J Cell Biol. 2010;188(6):833–849. doi: 10.1083/jcb.200909117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mittal A, et al. Genetic ablation of TWEAK augments regeneration and post-injury growth of skeletal muscle in mice. Am J Pathol. 2010;177(4):1732–1742. doi: 10.2353/ajpath.2010.100335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Panguluri SK, et al. Genomic profiling of messenger RNAs and microRNAs reveals potential mechanisms of TWEAK-induced skeletal muscle wasting in mice. PLoS One. 2010;5(1):e8760. doi: 10.1371/journal.pone.0008760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bhatnagar S, Kumar A. The TWEAK-Fn14 System: Breaking the Silence of Cytokine-Induced Skeletal Muscle Wasting. Curr Mol Med. 2012;12(1):3–13. doi: 10.2174/156652412798376107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li YP, et al. TNF-alpha acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. Faseb J. 2005;19(3):362–370. doi: 10.1096/fj.04-2364com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hunter RB, et al. Activation of an alternative NF-kappaB pathway in skeletal muscle during disuse atrophy. FASEB J. 2002;16(6):529–538. doi: 10.1096/fj.01-0866com. [DOI] [PubMed] [Google Scholar]
- 44.Baltgalvis KA, et al. Muscle wasting and interleukin-6-induced atrogin-I expression in the cachectic Apc (Min/+) mouse. Pflugers Arch. 2009;457(5):989–1001. doi: 10.1007/s00424-008-0574-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schaap LA, et al. Inflammatory markers and loss of muscle mass (sarcopenia) and strength. Am J Med. 2006;119(6):526. doi: 10.1016/j.amjmed.2005.10.049. e9-17. [DOI] [PubMed] [Google Scholar]
- 46.Schaap LA, et al. Higher inflammatory marker levels in older persons: associations with 5-year change in muscle mass and muscle strength. J Gerontol A Biol Sci Med Sci. 2009;64(11):1183–1189. doi: 10.1093/gerona/glp097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leger B, et al. Human sarcopenia reveals an increase in SOCS-3 and myostatin and a reduced efficiency of Akt phosphorylation. Rejuvenation Res. 2008;11(1):163–175B. doi: 10.1089/rej.2007.0588. [DOI] [PubMed] [Google Scholar]
- 48.Hansen JA, et al. Mechanism of inhibition of growth hormone receptor signaling by suppressor of cytokine signaling proteins. Mol Endocrinol. 1999;13(11):1832–1843. doi: 10.1210/mend.13.11.0368. [DOI] [PubMed] [Google Scholar]
- 49.Hart DW, et al. Anabolic effects of oxandrolone after severe burn. Ann Surg. 2001;233(4):556–564. doi: 10.1097/00000658-200104000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miller JT, Btaiche IF. Oxandrolone treatment in adults with severe thermal injury. Pharmacotherapy. 2009;29(2):213–226. doi: 10.1592/phco.29.2.213. [DOI] [PubMed] [Google Scholar]
- 51.Przkora R, Herndon DN, Suman OE. The effects of oxandrolone and exercise on muscle mass and function in children with severe burns. Pediatrics. 2007;119(1):e109–e116. doi: 10.1542/peds.2006-1548. [DOI] [PMC free article] [PubMed] [Google Scholar]