

Abstract
The synthesis and characterization of Fe diphosphineborane complexes are described in the context of N2 functionalization chemistry. Iron aminoimides can be generated at RT under 1 atm N2 and are shown to react with E–H bonds from PhSiH3 and H2. The resulting products derive from delivery of the E fragment to Nα and the H atom to B. The flexibility and lability of the Fe–BPh interactions in these complexes engender this reactivity.
Dinitrogen functionalization reactions using synthetic Fe complexes typically employ reductants in conjunction with electrophilic reagents;1 this strategy has allowed for the reliable functionalization of Nβ in terminal Fe–N2 complexes (Scheme 1). Whereas early-metal N2 complexes have displayed rich reactivity with non-polar E–H (E = H, Si) bonds,2 such reactivity using Fe has little precedent. Addition of H2 to diiron-bridged nitrides has been demonstrated,3 and in a recent report the nitrides were derived from reductive cleavage of N2.4 The hydrogenolysis of the terminal Fe imide functional group has likewise been established,5 though these imides were not prepared from N2. As such, we sought to generate Fe aminoimides from N2 that could undergo subsequent E–H bond addition across the Fe≡NNR2 linkage as a method for Nα functionalization (Scheme 1).
Scheme 1.

In this context, we and others have studied Fe platforms that can accommodate both N2 and terminal imide ligands,1a, 1c, 5a, 6 and we have recently reported that an Fe aminoimide complex 1 (Scheme 2) can be derived from N2.1c Given previous studies that demonstrate H2 addition across the M–B bonds in related Fe and Ni complexes,7 we expected that the Fe–B bond in 1 could facilitate an E–H activation step. However, 1 does not react with either H2 (1 atm) or PhSiH3 at RT. At more elevated temperatures (see SI), 1 decomposes and no tractable products were identified in the presence of H2 or PhSiH3. We therefore explored the development of a diphosphineborane Fe system ((DPB)Fe) that might be more reactive than the triphosphineborane Fe system ((TPB)Fe) featured in 1. We now describe new (DPB)Fe≡NNR2 complexes that react with non-polar E–H bonds at RT, thereby enabling the one-pot transformation of free N2 to an Fe hydrazido(−) species—the first such complex to be derived from N2.
Scheme 2.

As an entry to useful (DPB)Fe synthons, we found that reductive metallation of isopropyl- and phenyl-substituted DPB ligands8 2a and 2b (iPrDPB = PhB(o-iPr2PC6H4)2 and PhDPB = PhB(o-Ph2PC6H4)2, respectively, Scheme 3) with FeBr2 and 1.0 equiv Na/Hg in C6H6 allows for (DPB)FeBr complexes 3a and 3b to be isolated in 84% and 64% yield, respectively. Brown 3a and 3b are pseudo-tetrahedral, S = 3/2 complexes (μeff = 3.8 and 3.6 μB in C6D6 at RT, respectively) that feature η2-BC interactions that have been previously observed in Cu and Ni complexes of this ligand class.7,9 The structures determined by XRD analysis (Figure 1 and Table 1; see SI for the XRD structure of 3b) show close Fe–B (2.3243(11) and 2.330(4) Å, respectively) and Fe–Cipso contacts (2.2605(9) and 2.193(3) Å, respectively), supporting the formulation of the η2-BC ligand as both a donor via a filled π-arene orbital and an acceptor via an empty p orbital on boron. In solution, 3a and 3b are Cs-symmetric as indicated by their 1H NMR spectra.
Scheme 3.

Figure 1.

Displacement ellipsoid (50%) representations of 3a (left) and 4 (right). PiPr2 groups are truncated and H atoms are omitted for clarity.
Table 1.
Selected bond lengths (Å)
| Fe–B | Fe–Cipso | Fe–Cortho | |
|---|---|---|---|
| 3a | 2.3242(11) | 2.2605(9) | 2.5483(11) |
| 4: FeA a | 2.3739(7) | 2.2516(6) | 2.2714(7) |
| 4: FeB a | 2.3136(7) | 2.2133(6) | 2.6642(7) |
| 6ab | 2.3768(6) | 2.1492(5) | 2.3403(6) |
| 2.4288(7) | 2.1440(6) | 2.2266(6) | |
| 7 | 2.859(5) | - | - |
Two unique Fe atoms per molecule.
Two molecules per asymmetric unit.
Further reduction of 3a with 1.0 equiv Na/Hg under 1 atm N2 results in the formation of the dinuclear, bridging N2 complex (iPrDPB)Fe(μ-1,2-N2)Fe(iPrDPB) (4). The 1H NMR spectrum of 4 in C6D6 indicates that each Fe center is equivalent and has local Cs symmetry. Solution- and solid-state IR spectra of 4 lack an N–N stretch, suggesting that the complex maintains its pseudocentrosymmetric, dinuclear structure in solution. The RT solution magnetic moment is 4.6 μB, somewhat higher than the spin-only value of 4.0 μB expected for two uncoupled S = 1 Fe centers.10 The two pseudo-tetrahedral Fe centers in 4 have different local geometries in the solid state. The geometry about one of the Fe centers (FeA, Figure 1) is distinguished by a short Fe–Cortho contact and relatively long Fe–B and Fe–Cispo distances (Table 1). The other Fe center (FeB) displays somewhat shorter Fe–B and Fe–Cipso distances and a negligible Fe–Cortho interaction. The phenyl ring bound to FeA exhibits alternating C–C bond lengths (between 1.4303(10) and 1.3711(12) Å; see SI) whereas this asymmetry is negligible for the phenyl ring bound to FeB; these metrics indicate that back-donation to the arene ring is more significant for FeA and back-donation to the B atom is more significant for FeB. Since both (DPB)Fe fragments are equivalent in solution, the Fe–BCC interaction must be highly flexible and the solid-state bond metrics reflect the large range of local geometries available to the Fe centers.
Performing an identical reduction using 1.0 equiv Na/Hg with the phenyl derivative 3b does not trigger N2 binding but generates the brown, diamagnetic complex 5 that contains an η7-BPh interaction; this coordination mode is to our knowledge unprecedented in the metalborane literature. The XRD structure of 5 shows tight Fe–η7-BPh distances (Figure 2). The bound Cipso atom is significantly pyramidalized as indicated by the sum of the two BCC and one CCC angles (342°). Further showing the significant geometrical distortion of the bound arene is the acute BCipsoCpara angle of 127.71(8)°. The η7-BPh binding mode is maintained in solution based on the significantly upfield-shifted aryl resonances in the 1H NMR spectrum (3.63 (Hortho), 3.24 (Hmeta), and 6.25 (Hpara) ppm) and the 13C NMR spectrum (106.77 (Cispo), 99.41 (Cortho), 86.36 (Cmeta), and 78.73 (Cpara) ppm).
Figure 2.

Displacement ellipsoid (50%) representations of 5 (left) and 6a (right). PR2 groups are truncated. H atoms and solvent molecules are omitted for clarity. For 6a: Only one of the two molecules per asymmetric unit is shown. Selected distances (Å) for 5: Fe–B = 2.2667(13), Fe–Cipso = 1.9669(11), Fe–Cortho = 2.090 (avg), Fe–Cmeta = 2.169 (avg), Fe–Cpara = 2.1933(11).
Each of 3a, 3b, 4, and 5 serves as a precursor to an Fe aminoimide complex derived from N2. In a nearly identical procedure for generating 1,1c diamagnetic 6a and 6b may be accessed by stirring 3a or 3b with 1.1 equiv 1,2-bis(chlorodimethylsilyl)ethane and 3.1 equiv Na/Hg in THF under 1 atm N2. Alternatively, 4 or 5 may be employed as starting materials in conjunction with 2.1 equiv Na/Hg. Both 6a and 6b are green in solution and dichroic green/brown when crystalline. The 1H NMR spectrum of 6a reveals its Cs symmetry in solution. In addition, the 1H resonances of 6a attributed to the bound aryl ring are shifted upfield (5.10 (Hortho), 6.49 (Hmeta), and 4.72 (Hpara) ppm); though this effect is not observed for 6b perhaps due to attenuated backbonding 6b due to the less electron-rich metal center.
The solid-state structures of 6a (Figure 2) and 6b (see SI) are similar. For 6a, two molecules are in the asymmetric unit. The short Fe–N distances (1.6607(5) and 1.6657(5) Å for 6a; 1.6592(7) Å for 6b) are consistent with other trigonal Fe(NR) linkages and imply an Fe≡NNR triple bond.11 The bound arenes display alternating bond lengths that vary between ca. 1.36 and 1.44 Å (see SI). Density functional theory calculations (see SI) support the formulation of 6a and 6b as typical pseudotetrahedral d6 Fe imides11 that are similar to 1 except that one phosphine donor in 1 has been replaced by the η3-BCC interaction in 6a and 6b. Although the presence of an Fe–B bond is not required for the stability of pseudotetrahedral d6 Fe imides,11–12 DFT calculations on 6a, 1,1c and related Fe imides6 show some degree of Fe–B σ bonding. Quantifying the extent of Fe–B bonding in these complexes thermodynamically is difficult because the boranes are contained within the cage structures of the ligands.
The reactions of E–H bonds with aminoimides 6a and 6b were next examined. We were satisfied to observe that, in contrast to 1, the room temperature addition of 1.1 equiv PhSiH3 to 6a readily generates a new, orange species identified as the trisilylhydrazido(−) product 7 resulting from hydrosilylation of the Fe–N bond with delivery of SiH2Ph to Nα and H to B. To our knowledge, this is the first Fe hydrazido(−) complex to be derived from N2, thereby adding to the body of previously-reported mononuclear Fe hydrazido(−) model complexes.13 Having established this elementary step, we sought to combine the formation of 6a with its subsequent hydrosilylation into a single procedure. Accordingly, 7 may be generated in one pot from 3a or 4 (eq. 1).
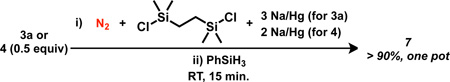 |
(1) |
For complex 7, an intense IR signal corresponding to the Si–H stretch is observed at 2090 cm−1 and a broad, intense IR stretch corresponding to the Fe–H–B functional group is observed at ca. 2000 cm−1. The solution magnetic moment (μeff = 5.0 μB, C6D6, RT) indicates an S = 2 spin state. The N–N bond is elongated from 1.326 Å (avg.) in 6a to 1.492(4) Å in 7 (Figure 3). Although both distances are consistent with N–N single bonds, the comparatively short bond in 6a is due to the sp hybridization of Nα and some degree of N–N multiple bond character. The very long N–N bond in 7 (longer than that of free N2H4) is likely due to a high degree of steric pressure exerted by its bulky Si and Fe substituents. The sum of the CBC angles is 334°, reflecting the tetrahedral geometry of the borohydride ligand.
Figure 3.

Displacement ellipsoid (50%) representation of 7 (left) and 8 (right). PR2 groups are truncated. H atoms not located in the difference map and solvent molecules are omitted for clarity.
Addition of 1 atm H2 at 50 °C to a C6H6 solution of 6b results in a pale brown solution from which colorless solids can be isolated that are identified as the S = 2 product 8 (μeff = 4.8 μB, C6D6, RT). Its IR spectrum contains a peak assigned to an N–H stretch at 3343 cm−1; the corresponding D2 addition product shows ν(N–D) at 2476 cm−1 (2441 cm−1 calc.). The broad, intense ν(Fe–H-B) stretch is observed at ca. 2100 cm−1 and is also sensitive to isotopic labeling (ν(Fe-D–B) ≈ 1550 cm−1). The connectivity of 8 was established by XRD analysis (Figure 3). In addition to the formation of new N–H and Fe–H–B bonds, the structure of 8 reflects cleavage of the N–N bond, rearrangement of the azadisilacyclopentane ring, and formal oxidation of one of the phosphines. This overall transformation is sufficiently complex that we felt it worthwhile to put forth a tentative mechanism (Scheme 4). The hydrogenation of 6b to form A is analogous to the formation of 7 from 6a. The hydrazine rearrangement step to form B has precedent for closely-related disilylhydrazines.14 Intermediate B may be sufficiently unencumbered to allow for η2-NN binding to give C which could lead to direct N–N cleavage and group transfer to a phosphine. Alternatively, the N–N bond in C may be cleaved to form an Fe(IV) imide15 D which then undergoes group transfer to give 8. Cleavage of the N–N bond of N2-derived ligands is rare for Fe1c, 4 and has been observed for bimetallic early metal complexes.2h, 16 In addition, the transformation of 6b to 8 is, to our knowledge, only the second example of N2 functionalization with H2 at a well-defined Fe complex.4
Scheme 4.

In conclusion, we have reported the generation of Fe aminoimides from N2 that undergo subsequent addition of non-polar E–H bonds. The significant flexibility of the Fe–BPh interactions facilitate both the initial formation of the Fe aminoimide as well as the E–H activation step. Whereas previous functionalization reactions of terminal Fe–N2 fragments allow for derivatization of Nβ, this report demonstrates that E–H addition to an unsaturated Fe–N bond is a viable strategy for Nα functionalization.
Supplementary Material
ACKNOWLEDGMENT
We acknowledge the NIH (GM070757) and the Beckman Institute for funding and thank Lawrence Henling and Dr. Jens Kaiser for assistance with XRD studies. We also acknowledge the Gordon and Betty Moore Foundation, the Beckman Institute, and the Sanofi-Aventis BRP at Caltech for their support of the Molecular Observatory at Caltech. SSRL is operated for the DOE and supported by its Office of Biological and Environmental Research, and by the NIH, NIGMS (including P41GM103393), and the NCRR (P41RR001209).
Footnotes
ASSOCIATED CONTENT
Supporting Information. Experimental details, spectra, DFT calculations, and XRD tables. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Sources
No competing financial interests have been declared.
REFERENCES
- 1.(a) Betley TA, Peters JC. J. Am. Chem. Soc. 2003;125:10782–10783. doi: 10.1021/ja036687f. [DOI] [PubMed] [Google Scholar]; (b) Lee Y, Mankad NP, Peters JC. Nature Chemistry. 2010;2:558–565. doi: 10.1038/nchem.660. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Moret M-E, Peters JC. J. Am. Chem. Soc. 2011;133:18118–18121. doi: 10.1021/ja208675p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Yuki M, Tanaka H, Sasaki K, Miyake Y, Yoshizawa K, Nishibayashi Y. Nature Communications. 2012;3:1254–1256. doi: 10.1038/ncomms2264. [DOI] [PubMed] [Google Scholar]
- 2.(a) Fryzuk MD, Love JB, Rettig SJ, Young VG. Science. 1997;275:1445–1447. [Google Scholar]; (b) Fryzuk MD, MacKay BA, Patrick BO. J. Am. Chem. Soc. 2003;125:3234–3235. doi: 10.1021/ja034303f. [DOI] [PubMed] [Google Scholar]; (c) Pool JA, Lobkovsky E, Chirik PJ. Nature. 2004;427:527–530. doi: 10.1038/nature02274. [DOI] [PubMed] [Google Scholar]; (d) Pool JA, Bernskoetter WH, Chirik PJ. J. Am. Chem. Soc. 2004;126:14326–14327. doi: 10.1021/ja045566s. [DOI] [PubMed] [Google Scholar]; (e) Hirotsu M, Fontaine PP, Epshteyn A, Sita LR. J. Am. Chem. Soc. 2007;129:9284–9285. doi: 10.1021/ja072248v. [DOI] [PubMed] [Google Scholar]; (f) Hirotsu M, Fontaine PP, Zavalij PY, Sita LR. J. Am. Chem. Soc. 2007;129:12690–12692. doi: 10.1021/ja0752989. [DOI] [PubMed] [Google Scholar]; (g) Pun D, Bradley CA, Lobkovsky E, Keresztes I, Chirik PJ. J. Am. Chem. Soc. 2008;130:14046–14047. doi: 10.1021/ja8048332. [DOI] [PubMed] [Google Scholar]; (h) Semproni SP, Lobkovsky E, Chirik PJ. J. Am. Chem. Soc. 2011;133:10406–10409. doi: 10.1021/ja2042595. [DOI] [PubMed] [Google Scholar]
- 3.Brown SD, Mehn MP, Peters JC. J. Am. Chem. Soc. 2005;127:13146–13147. doi: 10.1021/ja0544509. [DOI] [PubMed] [Google Scholar]
- 4.Rodriguez MM, Bill E, Brennessel WW, Holland PL. Science. 2011;334:780–783. doi: 10.1126/science.1211906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Brown SD, Peters JC. J. Am. Chem. Soc. 2004;126:4538–4539. doi: 10.1021/ja0399122. [DOI] [PubMed] [Google Scholar]; (b) Bart SC, Lobkovsky E, Bill E, Chirik PJ. J. Am. Chem. Soc. 2006;128:5302–5303. doi: 10.1021/ja057165y. [DOI] [PubMed] [Google Scholar]
- 6.Moret M-E, Peters JC. Angew. Chem. Int. Ed. 2011;50:2063–2067. doi: 10.1002/anie.201006918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Harman WH, Peters JC. J. Am. Chem. Soc. 2012;134:5080–5082. doi: 10.1021/ja211419t. [DOI] [PubMed] [Google Scholar]; (b) Fong H, Moret M-E, Lee Y, Peters JC. submitted. [Google Scholar]
- 8.(a) Bontemps S, Gornitzka H, Bouhadir G, Miqueu K, Bourissou D. Angew. Chem. Int. Ed. 2006;45:1611–1614. doi: 10.1002/anie.200503649. [DOI] [PubMed] [Google Scholar]; (b) Sircoglou M, Bontemps S, Mercy M, Saffon N, Takahashi M, Bouhadir G, Maron L, Bourissou D. Angew. Chem. Int. Ed. 2007;46:8583–8586. doi: 10.1002/anie.200703518. [DOI] [PubMed] [Google Scholar]
- 9.(a) Emslie DJH, Cowie BE, Kolpin KB. Dalton Trans. 2012;41:1101. doi: 10.1039/c1dt11271f. [DOI] [PubMed] [Google Scholar]; (b) Sircoglou M, Bontemps S, Mercy M, Miqueu K, Ladeira S, Saffon N, Maron L, Bouhadir G, Bourissou D. Inorg. Chem. 2010;49:3983–3990. doi: 10.1021/ic901896z. [DOI] [PubMed] [Google Scholar]
- 10.(a) Smith JM, Lachicotte RJ, Pittard KA, Cundari TR, Lukat-Rodgers G, Rodgers KR, Holland PL. J. Am. Chem. Soc. 2001;123:9222–9223. doi: 10.1021/ja016094+. [DOI] [PubMed] [Google Scholar]; (b) Hendrich MP, Gunderson W, Behan RK, Green MT, Mehn MP, Betley TA, Lu CC, Peters JC. Proc. Natl. Acad. Sci. U.S.A. 2006;103:17107–17112. doi: 10.1073/pnas.0604402103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Field LD, Guest RW, Turner P. Inorg. Chem. 2010;49:9086–9093. doi: 10.1021/ic101646p. [DOI] [PubMed] [Google Scholar]
- 11.Saouma CT, Peters JC. Coordination Chemistry Reviews. 2011;255:920–937. doi: 10.1016/j.ccr.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown SD, Peters JC. J. Am. Chem. Soc. 2005;127:1913–1923. doi: 10.1021/ja0453073. [DOI] [PubMed] [Google Scholar]
- 13.Selected examples: Smith JM, Lachicotte RJ, Holland PL. J. Am. Chem. Soc. 2003;125:15752–15753. doi: 10.1021/ja038152s. Crossland JL, Balesdent CG, Tyler DR. Dalton Trans. 2009:4420. doi: 10.1039/b902524c. Saouma CT, Kinney RA, Hoffman BM, Peters JC. Angew. Chem. Int. Ed. 2011;50:3446–3449. doi: 10.1002/anie.201006299.
- 14.Pitt CG, Skillern KR. Inorganic and Nuclear Chemistry Letters. 1966;2:237–241. [Google Scholar]
- 15.Thomas CM, Mankad NP, Peters JC. J. Am. Chem. Soc. 2006;128:4956–4957. doi: 10.1021/ja0604358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Selected examples: Peters JC, Cherry J-PF, Thomas JC, Baraldo L, Mindiola DJ, Davis WM, Cummins CC. J. Am. Chem. Soc. 1999;121:10053–10067. Fryzuk MD. Acc. Chem. Res. 2009;42:127–133. doi: 10.1021/ar800061g. Knobloch DJ, Lobkovsky E, Chirik PJ. Nature Chemistry. 2009;2:30–35. doi: 10.1038/nchem.477. Knobloch DJ, Lobkovsky E, Chirik PJ. J. Am. Chem. Soc. 2010;132:10553–10564. doi: 10.1021/ja104025v.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.