

Abstract
Converting T cells into tumor cell killers by grafting them with a chimeric antigen receptor (CAR) has shown promise as a cancer immunotherapeutic. However, the inability of these cells to actively migrate and extravasate into tumor parenchyma has limited their effectiveness in vivo. Here we report the construction of a chimeric antigen receptor containing an echistatin as its targeting moiety (eCAR). As echistatin has high binding affinity to αvβ3 integrin that is highly expressed on the surface of endothelial cells of tumor neovasculature, T cells engrafted with eCAR (T-eCAR) can efficiently lyse human umbilical vein endothelial cells and tumor cells that express αvβ3 integrin when tested in vitro. Systemic administration of T-eCAR led to extensive bleeding in tumor tissues with no evidence of damage to blood vessels in normal tissues. Destruction of tumor blood vessels by T-eCAR significantly inhibited the growth of established bulky tumors. Moreover, when T-eCAR was co-delivered with nanoparticles in a strategically designed temporal order, it dramatically increased nanoparticle deposition in tumor tissues, pointing to the possibility that it may be used together with nanocarriers to increase their capability to selectively deliver antineoplastic drugs to tumor tissues.
Keywords: chimeric antigen receptor, tumor neovasculature, echistatin, T cells, αvβ3 integrin
Introduction
Antigen specific T cells recognize their targets through the binding of T cell receptors (TCRs) with antigenic peptides presented by the major histocompatibility complex (MHC) molecules. In the presence of co-stimulatory molecules, this binding leads to T cell activation and subsequent lysing of the target cells 1. As most tumor associated antigens are self-proteins to which the immune system has developed tolerance, one of the major challenges facing cancer immunotherapy is the difficulty in generating sufficient number of high affinity tumor-specific T cells 2. In recent years, genetic modification of T cells by engrafting them with a chimeric antigen receptor (CAR) has been explored as a strategy to circumvent this difficulty 3. CARs are generated by joining a single chain antibody (scFv) to an intracellular signaling domain, usually the zeta-chain of the TCR/CD3 complex. The most recent construction of CARs also contains a co-stimulatory molecule such as CD28 or 41BB that can improve effector cell survival and proliferation 4. For cancer therapy, CARs have at least 3 major advantages over a natural TCR. First, the antigen-binding affinity of a scFv is usually much stronger than the binding moiety of most TCRs. High affinity binding to target cells is desirable for efficient T cell activation. Second, due to the nature of scFv-mediated antigen binding, recognition of CAR-engrafted T cells is non-MHC restricted and independent of antigen processing. This widens their application in patients with different MHC haplotypes. Third, due to the nature of non-MHC restriction, CAR-engrafted T cells can recognize and destroy tumor cells with down regulated expression of MHC, one of the most important mechanisms by which tumor cells escape conventional cancer immunotherapy. CARs constituted with scFvs against a variety of tumor-associated antigens have been reported 5. Encouraging preclinical data have prompted a series of clinical trials using adoptive transfer of T cells engrafted with these CARs for treatment of tumors of different tissue origins including melanoma 6, lymphoma 7, 8, neuroblastoma 9 and colorectal cancer 10. Many of these trials have shown measurable responses, and in some cases, with complete remission of the established tumors.
However, despite these impressive progresses, genetic modification of T cells with CARs still faces some formidable hurdles. One concern is a lack of proliferation and persistence of these modified T cells after adoptive transfer 11, 12. Strategies to overcome this deficiency include engineering effector cells to express cytokines (such as IL-2 or IL-15) which promote cell proliferation and survival 13, 14, or engrafting CAR to virus-specific cytotoxic T lymphocytes (CTLs) that can persist due to proper co-stimulation during their generation 9, 15. Additional concerns with CAR-engrafted T cells are that they do not actively migrate to the tumor site and they also lack an active mechanism to extravasate into the tumor interstitium. One strategy that has been reported to circumvent the problem with cell migration is to engineer T cells expressing a chemokine receptor that can respond to a tumor-associated chemokine milieu 3, 16. Although this strategy improves effector cell migration to tumor sites, it does little to promote extravasation, which is needed for eventual contact of the effector cells with tumor cell targets.
Almost all CARs are constructed for their direct binding with tumor-associated antigens expressed on the surface of tumor cells. Here we report the construction of a CAR that targets tumor-associated neovasculature instead. It was constructed by linking a peptide sequence from echistatin to the zeta chain. Echistatin is a 49 amino acid disintegrin that can be found in Echis carinatus venom. It has a strong binding affinity to αvβ3 17, while the latter is highly expressed on the surface of activated endothelial cells of neovasculature 18. We chose to use the DNA sequence encoding a modified form of echistatin, in which the 28th amino acid methionine was replaced with Leu to reduce its binding affinity to α5β1 19. T cells genetically engrafted with this echistatin-containing CAR (T-eCAR) were able to efficiently lyse human umbilical vein endothelial cells and tumor cells that express αvβ3 integrin when tested in vitro. Systemic administration of T-eCAR led to extensive bleeding in tumor tissues with no evidence of damage to blood vessels in normal tissues, and significant tumor shrinkage was observed in the T-eCAR-treated group. Moreover, when T-eCAR was co-delivered with nanoparticles, it dramatically increased the deposition of the latter to tumor tissues, indicating that T-eCAR may be used together with nanocarriers to increase their capability to selectively deliver antineoplastic drugs to tumor tissues.
Materials and Methods
Cell lines
Human umbilical vein endothelial cells (HUVEC) and the murine melanoma cell line B16-F0 were obtained from ATCC (Manassas, VA). HUVEC were cultured in ATCC-formulated Dulbecco’s Modified Eagle’s Medium (DMEM; Catalog No. 30-2002) with 20% fetal bovine serum (FBS) and B16-F0 cells were grown in 10% FBS DMEM with 100 μg/ml streptomycin and 100 U/ml penicillin. B16-GFPluc cells were established in our lab by co-transfecting pIR-eGFP-luc and pCMV-piggyBac plasmids into B16-F0 followed by flow cytometry sorting and single cell cloning as previously described 20.
Retroviral vector construction and production
The construction of retroviral vectors is schematically presented in Fig. 1A. The coding sequences for Leu-28-echistatin (MECESGPCCRNCKFLKEGTICKRARGDDLDDYCNGKTCDCPRNPHKGPAT; GenBank: M27213.1) and Myc-tag (EQKLISEEDL) were synthetized by IDT (Integrated DNA Technologies, Coralville, Iowa) containing the restriction sites XhoI and NcoI. This construct was then cloned into the vector SFG-FRG5-CD28-Zeta 21, by replacing the HER2 ScFv coding sequence. A signal peptide (SP) has been added to the 5′ of the fusion gene. Additionally, a c-Myc tag (c-Myc) has been inserted between echistatin and CD28 to facilitate the detection of eCAR expression. The construct was named eCAR. For constructing SFG-GFP, the GFP gene minus the stop codon was similarly inserted into SFG-FRG5-CD28-Zeta.
Fig. 1. Construction and expression of eCAR.

A. Schematic illustration of eCAR in a retroviral vector construct. The 5′ and 3′ long terminal repeats (LTR) of the retroviral vector are labeled. The coding sequences for echistatin (Echi), CD28 (containing a transmembrane domain) and Zeta chain are labeled. A signal peptide (SP) has been added to the 5′ of the fusion gene. A c-Myc tag (c-Myc) has been inserted between echistatin and CD28 to facilitate the detection of eCAR expression. The control SFG-GFP retroviral vector was constructed in same way, with the GFP gene replacing echistatin-coding sequence. B. Transduction efficiency of eCAR. Splenocytes were transduced with eCAR or SFG-GFP retroviruses, or mock transduced. Cells were then stained with PE-conjugated anti-c-Myc antibody for two-color flow cytometry analysis to detect expression of GFP or eCAR.
To prepare viral stocks, the retroviral vector constructs were transfected into the retrovirus packaging cell line Platinum-E 22, using the FuGENE® 6 transfection reagent (Roche Applied Science Indianapolis, IN). Supernatants were harvested 48 and 72 h later and filtered through 0.45 μm filters. The purified supernatants were combined and titrated on 293 cells to determine the virus yield.
Transduction of murine splenocytes with retroviral vectors
Splenocytes were harvested from C57BL/6 mice and cultured with RPMI 1640 medium supplemented with 25 mM HEPES, 200 nM L-glutamine, 10% FBS, 1% MEM non-essential amino acids, 1 mM sodium pyruvate, 50 μM β-mercaptoethanol, 100 μg/ml streptomycin and 100 U/ml penicillin. Cells in suspension (2×106/ml) were stimulated with concanavalin A (2 μg/ml; Sigma, St. Louis, MO) and murine IL-2 (1 ng/ml; ProSpec, East Brunswick, NJ) for 24 h before they were transferred to RetroNectin (Takara Bio. Inc., Shiga, Japan) coated non–tissue culture 24-well plates for transduction with eCAR or SFG-GFP retroviruses. The transduced splenocytes were then cultured for 48 hours in fresh medium supplemented with 10 ng/ml of murine IL-2.
Flow cytometry analysis for eCAR and GFP expression
Splenocytes transduced with eCAR retrovirus were washed once with PBS containing 2% fetal bovine serum before they were incubated for 30 min at 4 °C with Mouse BD Fc Block (BD Biosciences, San Jose, CA) that contains rat anti-mouse CD16/CD32 antibody. After washing with PBS twice, cells were stained with PE-conjugated Myc-tag mouse antibody (Cell signaling, Danvers, MA) or isotype antibody for 30 min at 4°C in dark. The cells were washed twice before used for analysis. SFG-GFP transduced cell were used directly for analysis without any staining. Both cell preparations were then analyzed on BD FACSAria™ II (BD Biosciences, San Jose, California), with data analysis on >10,000 events. For determining αvβ3 integrin expression, HUVEC or B16-F0 cells were stained with 10 μg of fluorescein isothiocyanate (FITC) – conjugated Arginine-Glycine-Aspartic Acid (RGD) Peptide (AnaSpec, Fremont, CA) in 100 μl 1% FBS-PBS for 30 min at 4 °C. After washed 3 times with PBS, cells were analyzed with the same BD FACSAria™ II.
Cytotoxicity assay of retrovirus transduced splenocytes
The cytotoxicity of the retrovirus-transduced splenocytes on target cells was assayed by either visualization or by a recently reported nonradioactive quantitative measurement 20. For the visualization detection, 5×104 target cells well were initially seeded to 48-well plates. Retrovirus-transduced splenocytes (effector cells) were added 24 h later at effector to target (E:T) ratios ranging from 20:1 to 2.5:1. Cells were fixed 24 or 48 h later and stained with 1% crystal violet in 20% ethanol for visualization and imaging under a light microscope.
For quantitative measurement of cytotoxicity of the retrovirus-transduced splenocytes, 1×104 target cells were seeded on 96 well plates first. Effector cells were added 24 h later at E:T ratios ranging from 20:1 to 2.5:1. Forty-eight h later, media was removed and cells were rinse with PBS. Then, 50 μl of the Bright-Glo™ (Luciferase Assay System, Promega, Madison, WI) was added to each well. Plates were gently shaken for 2 minutes for the cells to be completely lysed. The cell lysates were then transferred into 96-well opaque plates for luminescence measurements with SpectraMax® multi-mode microplate reader (Molecular Devices, Sunnyvale, CA). Cytotoxic activity was calculated by the formula: Cell killing (%) = [1 − (reading of well with effector-cell)/(reading of well without effector cell)] × 100.
Measurement of cytokine release
Splenocytes were obtained from C57BL/6 donors. They were either untransduced (UT), or transduced with SFG-GFP, eCAR (T-eCAR) or Her2CAR (T-Her2CAR). The details of Her2CAR construction have been reported in our previous publication 20. To measure cytokine release during CAR-mediated cytolysis, HUVEC or Her2-expressing 4T1-Her2 were mixed with the corresponding T-CARs at a 1:5 ratio in 48-well plates. As all the T-CARs were prepared from splenocytes obtained from C57BL/6 mice, they presented as allogeneic effector T cells for the 4T1-Her2 target. The culture supernatants were collected after 24 h incubation. The quantity of IL-2 and IFN-γ was determined by ELISA as per the manufacturer’s instructions (R&D Systems, Minneapolis, MN).
Animal experiments
All animal experiments were carried out at the Animal Care Operations facility of the University of Houston, and approved by the university’s Institutional Animal Care and Use Committee (IACUC). For establishing tumors, 1×105 B16-F0 murine melanoma cells were implanted into the right flank of 6- to 8- week old male immunocompetent C57BL/6 mice (Taconic Farms, Hudson, NY). When tumors became palpable (around day 5), mice were intravenously injected with either eCAR or SFG-GFP retrovirus-transduced splenocytes (4×106 in 100 μl RPMI 1640) or PBS (n=10 mice per group). Tumor sizes were measured twice a week until the end of the experiment. Tumor volume was calculated by the following formula: tumor volume (mm3) = [length (mm)] × [width (mm)]2 × 0.52.
To determine the effect of retrovirus-transfected splenocytes on tumor blood vessels, mice bearing sizable B16-F0 tumors (approximately 8 mm in diameter) were intravenously injected with either eCAR or SFG-GFP retrovirus-transduced splenocytes (5×106 in 100μl RPMI 1640) or PBS (n=3 mice each group). Mice were humanely sacrificed 3 days later and their tumors excised. Tumors were fixed in 10% formalin for 24 h and then in 70% ethanol for another 24 h. This was followed by dehydration overnight in the Shandon Excelsior ES Tissue processor TM (Thermo Scientific, Waltham, MA). Successive 5 μm thick sections were cut and dehydrated in xylene and in decreasing ethanol concentrations (100% to 50%). Sections were then stained with hematoxylin and eosin for observation and micrograph under the microscope.
To investigate nanoparticle delivery following tumor blood vessel destruction, eCAR or SFG-GFP retrovirus-transduced splenocytes were intravenously injected into tumor-bearing mice as described above. Forty-eight h later, mice received intravenous injection of DSPC/CHOL/mPEG2000-DSPE liposome nanoparticles (100 μm in size) labeled with Rhodamine DHPE (FormuMax Scientific, Inc. Palo Alto, CA), at a dose of 10mg/kg diluted in 100μl PBS. Twenty-four h after liposome injection, mice were sacrificed and tumors as well as major organs including lungs, kidneys and liver were collected. The collected tumors and organs were fixed in 4% paraformaldehyde at 4°C for 24 h and then treated with 25% sucrose for another 24 h at 4°C before they were embedded in OCT. Consecutive 5 μm thick cryo-sections were prepared for observation and micrographed under the fluorescence microscope (Olympus BX51). The intensity of rhodamine image was quantitated with MicroSuite™ FIVE software. Briefly, five areas were randomly clicked in each slide to obtain the reading of intensity value. A total of three slides (one from each animal) were subjected for quantification to obtain the mean value of each treatment group.
Statistical analysis
All quantitative data are reported as mean ± SD. Statistical analysis was made for multiple comparisons using analysis of variance and Student’s t-test. P value < 0.05 was considered to be statistically significant.
Results
Construction of a CAR that can target tumor neovasculature
The detailed composition of the final construction is depicted in Fig. 1A. The DNA sequence encoding a modified form of echistatin, in which the 28th amino acid methionine was replaced with Leu to reduce its binding affinity to non-αvβ3 integrins 19, was synthesized and inserted into a retroviral vector, SFG-FRG5-CD28-Zeta 21, to replace the HER2 scFv contained within the vector. A 12 amino acid c-Myc tag was inserted between the echistatin and CD28 sequences during the vector construction to facilitate the detection of eCAR expression. The final construction is designated eCAR (for echistatin CAR) and its composition is illustrated in Fig. 1A.
To determine gene transduction efficiency of eCAR, splenocytes were transduced with either eCAR or a GFP-containing retrovirus (SFG-GFP) that was constructed by replacing the HER2 scFv in SFG-FRG5-CD28-Zeta with the GFP gene. Mock-transduced cells were included as a negative control. The cells were stained with PE-conjugated anti-c-Myc antibody before they were analyzed by two-color flow cytometry for detection of both GFP and eCAR. Almost half of the eCAR-transduced splenocytes were PE positive, while neither SFG-GFP-transduced nor control cells showed any significant PE staining (Fig. 1B). On the other hand, a high percentage of the SFG-GFP-transduced cells were GFP positive, while neither the eCAR-transduced cells nor the control cells were detectable at this wavelength. These results show that splenocytes can be efficiently engrafted with eCAR by the retrovirus construct.
T cells engrafted with eCAR can selectively kill human umbilical vein endothelial cells efficiently
We first examined the expression of αvβ3 integrin on human umbilical vein endothelial cells (HUVEC) by staining them with FITC-conjugated RGD Peptide. Flow cytometry analysis showed that αvβ3 integrin is highly expressed on the majority of HUVEC (Fig. 2A). To test the ability of T cells engrafted with eCAR (T-eCAR) to kill target cells expressing αvβ3 integrin, we mixed the effector cells with HUVEC at different ratios. Cells were stained 24 h later with crystal violet to determine cell viability. While splenocytes engrafted with SFG-GFP did not show any significant toxicity on HUVEC, T-eCAR completely lysed all the cells, even in the well with the lowest effector to target ratio (Fig. 2B). We also measured cytokine release during T-eCAR-mediated killing of HUVEC, and compared the result with that obtained from Her2-CAR-mediated killing of Her2-expressing tumor cells. The results show that both IL-2 and interferon-γ (IFN-γ) were sufficiently released at almost equivalent level during cytolysis mediated by these two CARs (Figs. 2C and 2D), indicating that they share the same killing mechanism. Although we did not specify which subset of T cells are the effectors of T-eCAR, previous studies have shown that both CD4 and CD8 T cells engrafted with antigen-specific TCRs can act as effector cells to efficiently kill tumor cells 23, 24. As such, we believe the same principle may apply here.
Fig. 2. Cytolytic effect of T-eCAR against HUVEC.

A. Flow cytometry analysis of αvβ3 expression on HUVEC. HUVECs were stained with FITC-conjugated RGD peptide before they were analyzed by flow cytometry. B. Cytolysis of HUVEC by T-eCAR. Splenocytes obtained from C57BL/6 donors were transduced with retroviruses containing either eCAR (T-eCAR) or the control SFG-GFP construct, in which the echistatin was replaced with the GFP gene. The cells were then mixed with HUVEC at the indicated ratio for 24 h. Then the T cells in suspension were removed by washing and the remaining cell monolayer was stained with crystal violet to determine cell viability. The control well represents HUVEC alone. Note that live cells are positively stained. C. Quantification of IL-2 release during T-eCAR and T-Her2CAR mediated killing of target cells. D. Quantification of IFN-γ release during T-eCAR and T-Her2CAR mediated killing of target cells.
In addition to activated endothelial cells, some tumor cells have also been reported to express elevated level of αvβ3 integrin that has been found to be associated with tumor metastasis 25, 26. Among the tumor cells that have been reported to have high level of αvβ3 integrin expression is the B16 murine melanoma cell line 27. We thus first determined the expression of αvβ3 integrin on B16-F0 cells by flow cytometry after staining them with FITC-conjugated RGD Peptide. The result showed that indeed high percentage of B16-F0 expresses αvβ3 integrin (Fig. 3A). We then analyzed the ability of T-eCAR to kill a B16 cell line that has been stably transduced with a fusion gene containing GFP and luciferase (B16-GFPluc). This cell lines would allow the killing effect of B16-GFPluc by T-eCAR to be conveniently assessed in two ways: direct visualization of GFP and a non-radioactive quantitative assay of cytolysis by measuring luciferase activity 20. Visualization by fluorescent microscopy showed that incubation of B16-GFPluc cells with T-eCAR (E:T ratio = 5) significantly reduced the number of GFP positive cells in the well while splenocytes transduced with the control SFG-GFP construct showed little or no effect on the integrity of the cell monolayer (Fig. 3B). The quantitative measurement of luciferase activity showed that T-eCAR killed over 60% of B16-GFPluc cells even at the lowest E:T ratio (2.5:1), while T cells engrafted with SFG-GFP construct only caused a moderate lysis of the B16 cells at the highest E:T ratio (10:1) (Fig. 3C). Together, these results suggest the T-eCAR has the ability to simultaneously destroy tumor neovasculature as well as tumor parenchyma if the tumor cells express elevated level of αvβ3 integrin.
Fig. 3. Cytolytic effect of T-eCAR against tumor cells expressing elevated level of αvβ3.

A. Flow cytometry analysis of αvβ3 expression on B16-F0 cells, the parental line of B16-GFPluc. B16-F0 cells were stained with FITC-conjugated RGD peptide before they were analyzed by flow cytometry. B and C. Cytolytic effect of T-eCAR against B16-GFPluc. Splenocytes transduced with retroviruses containing either eCAR or the control SFG-GFP construct were mixed B16-GFPluc at 10:1, 5:1 and 2.5:1 ratio and the mixed cells were cultured for 48 h before they were visualized or collected for measurement of luciferase activity. B shows a typical micrograph from each well where effector and target cells were mixed at 5:1 ratio. C shows the quantitative measurement of luciferase activity for determining cytolytic activity of T-eCAR. The effector and target cells were mixed at the indicated ratios and cells were collected 48 h later for determining luciferase activity. The control well contains tumor cells only. The percentage of cell killing was calculated by the formula: Cell killing (%) = [1 − (reading of well with effector-cell)/(reading of well without effector cell)] × 100. ★p<0.05 as compared with SFG-GFP transduced effector cells.
In vivo administration of T-eCAR induces extensive bleeding in tumor but not in normal tissue
To investigate the effect of T-eCAR on tumor blood vessels in vivo, we established tumor mass on the right flank of the syngeneic C57BL/6 mice through subcutaneous implantation of 2×105 freshly harvested B16 cells. Once the tumors reached approximately 8 mm in diameter, mice received intravenous injection via the tail vein of 5×106 splenocytes transduced either with eCAR or SFG-GFP. Another group of mice received PBS only as a negative control. Mice were euthanized at day 3 after adoptive cell transfer. Tumors and normal organ tissues were collected for preparation of tissue sections after paraffin embedding. There was extensive bleeding in tumors treated with T-eCAR (Fig. 4A). Tumor parenchyma was filled with red blood cells and other blood cell components. Tumors treated with SFG-GFP-transduced splenocytes did not show the same scale of bleeding. As the only difference between e-CAR and SFG-GFP constructs is that the latter has the echistatin coding sequence replaced by the GFP gene, these results thus suggest that the observed tumor blood vessel destruction after T-eCAR administration was mainly due to the incorporated echistatin sequence in eCAR enabling it to bind to neovasculature-associated αvβ3 integrin and trigger the T cell-mediated killing effect. Examination of normal organ tissues, including those from lung, liver and kidney, did not reveal any significant bleeding (Fig. 4B). This further indicates that the observed bleeding in T-eCAR-treated tumors derives from the selective destruction of tumor vessels by the introduced T cells. The failure to detect any noticeable bleeding in normal organ tissues also eases the concern that the echistatin contained within eCAR may affect the function of platelets, leading to a general bleeding.
Fig. 4. Selective destruction of tumor blood vessels after systemic administration of T-eCAR.

B16 murine melanoma was initially established at the right flank of C57BL/6 mice. When tumors reached the approximate size of 8 mm in diameter, 5×106 of T-eCAR or T cells transduced with SFG-GFP were adoptively transferred by systemic infusion through the tail vein. A third group of mice were given PBS as a control. Tumors (A) and normal organ tissues (B) were collected three days later for histological examination after H&E staining.
Adoptive transfer of T-eCAR significantly inhibits the growth of established solid tumor
To determine the consequence of tumor blood vessel destruction by T-eCAR, we conducted another in vivo experiment by initially implanting 1×105 B16 tumor cells to the right flank of C57BL/6 mice. Five days later, when the tumors became palpable, mice received an intravenous infusion of PBS, or 4×106 splenocytes transduced with either eCAR or SFG-GFP. Tumor growth was essentially unhalted in mice treated with PBS or splenocytes transduced with SGF-GFP (Fig. 5). By day 28 after the start of the treatment, the tumors in both groups reached a large size and the animals had to be euthanized due to reaching the preset endpoint. In contrast, administration of T-eCAR effectively slowed down the tumor growth. By day 42, the tumors in the T-eCAR- treated group were still relatively small and most of the animals were still alive. These results show that T-eCAR-mediated destruction of tumor blood vessels can lead to significant therapeutic benefit against established solid tumors.
Fig. 5. Therapeutic effect of T-eCAR against established solid tumor.

Syngeneic murine melanoma was established by subcutaneously implanting 1×105 B16 cells to the right flank of C57BL/6 mice. Five days later, 4×106 T-eCAR or T cells transduced with SFG-GFP were infused systemically. Mice in the third group were given PBS only. Tumors were measured weekly for determination of tumor volume. ★p<0.05, +p<0.01 as compared with SFG-GFP and PBS.
Tumor blood vessel destruction by T-eCAR can increase deposition of systemically delivered nanoparticle to tumor tissues
In recent years, nanoparticles have been extensively explored as a promising delivery vehicle for chemotherapeutics. Nanoparticles have the potential to override the poor biopharmaceutical properties of many small-molecule drugs and alter their pharmacokinetics. However, despite the tremendous promise, nanoparticle-mediated antineoplastic drug delivery has been less optimal than anticipated. The significant bleeding seen in malignant tissues (Fig. 4) after infusion of T-eCAR prompted us to explore the possibility of using this approach as a means to enhance nanoparticle delivery to tumors following their systemic administration. We initially gave T-eCAR or T cells transduced with the SFG-GFP control construct to tumor bearing mice as described in Fig. 4. This was followed by intravenous administration of rhodamine-labeled liposome nanoparticles 48 h later. Animals were euthanized one day after liposome nanoparticle administration. Tumors and major organs were collected for preparation of frozen sections that were then used for fluorescent microscopic examination. In tumor sections prepared from mice receiving SFG-GFP transduced T cells, rhodamine staining was only sparsely seen across the tissue section. This is in contrast to tumors from mice receiving T-eCAR, where there was widespread rhodamine staining across the entire tumor parenchyma (Fig. 6A). Visible blood vessels seem to have broken area (indicated by white arrows). Quantification of rhodamine in tumor tissues confirmed that there was a significant difference between SFG-GFP and T-eCAR treatment (Fig. 6B). Examination of tissue sections from normal organs reveals very little evidence of rhodamine deposition except in the lung, where blood vessels were seen to be lightly stained with rhodamine. This observation is consistent with the early report that liposome nanoparticles have the tendency to get trapped in the lung after systemic delivery 28. Despite this mechanic trap in the lung, there is little evidence for parenchyma distribution of nanoparticles in the lung. This, in combination with the failure to detect any significant bleeding in the lung (and other normal organ tissues) in Fig. 4B, supports this assumption. Together these results suggest that: 1) despite the reported presence of enhanced permeability and retention (EPR) effect in tumor tissues, it does not allow efficient deposition of nanoparticles to enter into tumor interstitium after their systemic delivery, 2) T-eCAR does not cause significant damage to blood vessels in normal organ tissues despite its potent destructive effect on tumor neovasculature, and 3) tumor blood vessel destruction mediated by T-eCAR allows systemically delivered nanoparticles to efficiently enter deep into tumor parenchyma.
Fig. 6. T-eCAR enhances tumor distribution of rhodamine-labeled nanoparticles following their systemic delivery.
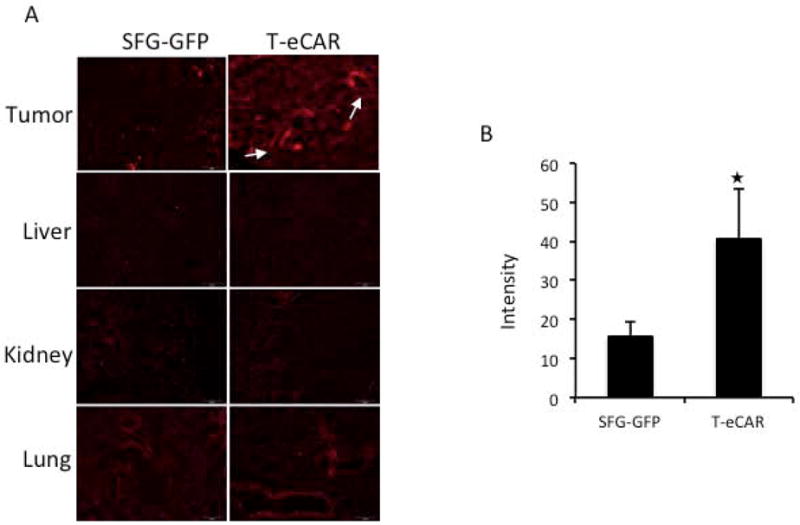
Murine melanoma was established at the right flank of C57BL/6 mice. Once tumor reached the approximate size of 8 mm in diameter, mice received intravenous infusion of 4×106 T-eCAR or SFG-GFP-transduced splenocytes. This was followed by intravenous administration of rhodamine-labeled liposome nanoparticles at a dose of 10 mg/kg 48 h later. A. Tumors and major organ tissues were collected two days after nanoparticle administration for cryo-fluorescent imaging. Visible blood vessels on T-eCAR treated tumor section are indicated by white arrows. B. The rhodamine image contained in tumor tissues was quantitated using the MicroSuite™ FIVE software. The results, given by the software as value intensity, represent the mean value of 15 randomly chosen areas across three slides, one from each tumor sample. ★p<0.01, as compared with SFG-GFP.
Discussion
Modification of T cells through engraftment of CAR has been shown to be a feasible approach for cancer immunotherapy in both preclinical studies and in clinical trials. However, one of the remaining hurdles is the lack of the ability of these cells to actively migrate and extravasate to tumor parenchyma. To overcome this obstacle, we have constructed a CAR that targets tumor neovasculature. For this purpose, we chose to link a modified form of echistatin sequence to the zeta chain. Wild type echistatin has a strong binding affinity to three members of the integrin family, αvβ3, αIIbβ3 and α5β1. Both αvβ3 and αIIbβ3 have a narrow distribution, with αvβ3 predominately expressed on the surface of activated endothelial cells and αIIbβ3 mainly associated with platelets. The other member of the three, α5β1, is more widely distributed 29, however, an early study showed that replacement of Met28 with Leu selectively decreases echistatin’s ability to bind to α5β1 19. The echistatin contained within eCAR has this replacement, and it thus has reduced ability to bind to α5β1 integrin. Nevertheless, it can still bind to αIIbβ3 in addition to αvβ3. As αIIbβ3 is highly expressed on platelets, one concern was that the abundant presence of platelets in the blood stream might prevent T-eCAR from efficient interaction with αvβ3 integrin on tumor neovasculature. However, our in vivo data showed that T-eCAR functioned effectively at destroying tumor blood vessels after their systemic delivery, indicating that the interference from platelet-associated αIIbβ3 is minimal. Part of the reason for this is probably because αIIbβ3 integrin maintains in an inactive conformation on the surface of resting platelets and has limited binding affinity to the RGD moiety within echistatin 30. Regardless, our data demonstrate that echistatin incorporated into a CAR can confer T cells with the ability to attack tumor blood vessels effectively after systemic administration to tumor-bearing hosts.
A single adoptive transfer of T-eCAR produced a substantial therapeutic effect against established melanoma. Considering that the destroyed tumor blood vessels were likely to grow back sooner or later, the observed therapeutic effect was a bit surprising. The following factors might have contributed to the apparent significant therapeutic outcome. First, eCAR construct contains the CD28 costimulatory molecule in addition to echistatin (Fig. 1A). This might have allowed the adoptively transferred T-eCAR to persist in the adoptively transferred host for a significant period of time, providing a prolonged therapeutic window. This has been demonstrated with CARs targeting other tumor molecules in clinical settings 31, 32. Second, as the B16 melanoma cells express relatively high level of αvβ3 integrin 33, the tumor cells themselves can be effectively lysed by T-eCAR (Fig. 3). As such, the adoptively transferred T-eCAR can attack both tumor blood vessels and the tumor cells. More importantly, the initial tumor blood vessel destruction might have allowed the efficient infiltration of the T-eCAR to tumor parenchyma to be in proximate contact with tumor cells. This avoids the need for active extravasation, a characteristic that T-eCAR otherwise may not possess. The combination of blood vessel destruction and the subsequent T-eCAR infiltration to directly kill tumor cells might have synergized for a better therapeutic effect than either action alone. Nevertheless, it is anticipated that the action of T-eCAR alone is unlikely to produce a long-lasting therapeutic outcome. One way to maximize the effect of T-eCAR would be to combine it with antiangiogenic agents, which can prevent new tumor blood vessel formation following T-eCAR administration. Such a combination may produce a synergistic effect, as T-eCAR mediated tumor blood vessel destruction can convert the relatively slow process of tumor angiogenesis into an acute event that would maximize the therapeutic response to antiangiogenic compounds.
The widespread blood cell leakage into tumor parenchyma after administration of T-eCAR prompted us to explore the possibility of utilizing this to enhance nanoparticle drug delivery. In recent years, nanoparticles have been extensively explored as a promising delivery vehicle for chemotherapeutics. The preferential biodistribution and retention of nanoparticles to malignant tissues relies on the poorly organized, often leaky blood vessels and lack of lymphatics within solid tumors, a feature termed the enhanced permeability and retention (EPR) effect 34. However, the ability of EPR to facilitate nanoparticle delivery to solid tumors remains controversial. This is because the same nature of leaky blood vessels and lack of lymphatic drainage in tumor tissues also contributes to another tumor-associated physical characteristic, i.e. high tumor interstitial fluid pressure (TIFP). Indeed, high TIFP is a common feature for many solid tumors 35. Normal interstitial fluid pressure is typically between −3 to 3 mmHg. But TIFP is significantly higher, ranging from 5 to 40 mmHg. In some extreme cases, it can reach 100 mmHg 36, 37. Clinically, a high TIFP is marked by reduced delivery and uptake of anticancer agents, and hence, lack of therapeutic effects 38. Consequently, it has been proposed that normalization of blood vessel or elevation of plasma colloid osmotic pressure is needed to reverse this effect for enhanced drug delivery to tumor tissues 39, 40. Most interestingly, it has been recently reported that normalization of tumor blood vessels improves the delivery of nanoparticles in a size-dependent way 41. The contradictory nature of the co-existence of EPR and high TIFP within the tumor microenvironment may partly explain the suboptimal delivery efficacy with nanoparticles despite their enormous promise. Our data show that T-eCAR-mediated blood vessel destruction allows the subsequently delivered nanoparticles to efficiently deposit in tumor tissues efficiently. This opens the possibility of combining T-eCAR with nanoparticle-mediated drug delivery of many antineoplastic small molecules.
Finally, examination on major organs after systemic delivery of rhodamine-labeled nanoparticles following T-eCAR infusion did not reveal any significant blood vessel leakage. This observation was made in the same animals that showed significant damage to tumor blood vessels from T-eCAR administration. This, in combination with the failure to detect any significant bleeding in the lung and other normal organ tissues, suggests that despite its potent effect on tumor neovasculature, T-eCAR dose not cause significant off-target effect to blood vessels in normal tissues. This indicates that, although endothelial cells of certain normal blood vessels express αvβ3 integrin, the level may be below the threshold that is readily detectable by T-eCAR.
Novelty and Impact Statement.
Current chimeric antigen receptors (CARs) exclusively target tumor cells. We report the construction of eCAR that targets tumor blood vessels instead, aiming to overcome the difficulty of CAR-modified T cells to penetrate tumor parenchyma. T cells engrafted with eCAR (T-eCAR) can efficiently lyse HUVECs and its in vivo administration results in significant tumor shrinkage. Moreover, its codelivery with nanoparticles increases the tumor deposition of the latter, indicating a combinatorial strategy for selective antineoplastic drug delivery.
Acknowledgments
We thank Dr. Nabil Ahmed for providing the SFG-FRG5-CD28-Zeta construct, Jeffrey Spencer and Jon Pierson for their careful reading of the manuscript. This work was supported by the National Cancer Institute grants R01CA106671 and R01CA132792 and also by a grant from the William and Ella Owens Medical Research Foundation (to XZ).
Footnotes
Conflict of financial interest: We wish to declare that we do not have any financial conflict of interest related to the presented studies.
References
- 1.van der Merwe PA, Davis SJ. Molecular interactions mediating T cell antigen recognition. Annu Rev Immunol. 2003;21:659–84. doi: 10.1146/annurev.immunol.21.120601.141036. [DOI] [PubMed] [Google Scholar]
- 2.Kammertoens T, Blankenstein T. Making and circumventing tolerance to cancer. Eur J Immunol. 2009;39:2345–53. doi: 10.1002/eji.200939612. [DOI] [PubMed] [Google Scholar]
- 3.Jena B, Dotti G, Cooper LJ. Redirecting T-cell specificity by introducing a tumor-specific chimeric antigen receptor. Blood. 2010;116:1035–44. doi: 10.1182/blood-2010-01-043737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, Varela-Rohena A, Haines KM, Heitjan DF, Albelda SM, Carroll RG, Riley JL, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A. 2009;106:3360–5. doi: 10.1073/pnas.0813101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davies DM, Maher J. Adoptive T-cell immunotherapy of cancer using chimeric antigen receptor-grafted T cells. Arch Immunol Ther Exp (Warsz) 2010;58:165–78. doi: 10.1007/s00005-010-0074-1. [DOI] [PubMed] [Google Scholar]
- 6.Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, Wunderlich JR, Nahvi AV, Helman LJ, Mackall CL, Kammula US, Hughes MS, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29:917–24. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, Maric I, Raffeld M, Nathan DA, Lanier BJ, Morgan RA, Rosenberg SA. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–33. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, Huls MH, Liu E, Gee AP, Mei Z, Yvon E, Weiss HL, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–70. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, Davis JL, Morgan RA, Merino MJ, Sherry RM, Hughes MS, Kammula US, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2011;19:620–6. doi: 10.1038/mt.2010.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park JR, Digiusto DL, Slovak M, Wright C, Naranjo A, Wagner J, Meechoovet HB, Bautista C, Chang WC, Ostberg JR, Jensen MC. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther. 2007;15:825–33. doi: 10.1038/sj.mt.6300104. [DOI] [PubMed] [Google Scholar]
- 12.Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, White DE, Wunderlich JR, Canevari S, Rogers-Freezer L, Chen CC, Yang JC, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12:6106–15. doi: 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsu C, Hughes MS, Zheng Z, Bray RB, Rosenberg SA, Morgan RA. Primary human T lymphocytes engineered with a codon-optimized IL-15 gene resist cytokine withdrawal-induced apoptosis and persist long-term in the absence of exogenous cytokine. J Immunol. 2005;175:7226–34. doi: 10.4049/jimmunol.175.11.7226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quintarelli C, Vera JF, Savoldo B, Giordano Attianese GM, Pule M, Foster AE, Heslop HE, Rooney CM, Brenner MK, Dotti G. Co-expression of cytokine and suicide genes to enhance the activity and safety of tumor-specific cytotoxic T lymphocytes. Blood. 2007;110:2793–802. doi: 10.1182/blood-2007-02-072843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Terakura S, Yamamoto TN, Gardner RA, Turtle CJ, Jensen MC, Riddell SR. Generation of CD19-chimeric antigen receptor modified CD8+ T cells derived from virus-specific central memory T cells. Blood. 2012;119:72–82. doi: 10.1182/blood-2011-07-366419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, Heslop HE, Brenner MK, Dotti G, Savoldo B. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. 2009;113:6392–402. doi: 10.1182/blood-2009-03-209650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar CC, Nie H, Rogers CP, Malkowski M, Maxwell E, Catino JJ, Armstrong L. Biochemical characterization of the binding of echistatin to integrin alphavbeta3 receptor. J Pharmacol Exp Ther. 1997;283:843–53. [PubMed] [Google Scholar]
- 18.Cai W, Chen X. Anti-angiogenic cancer therapy based on integrin alphavbeta3 antagonism. Anticancer Agents Med Chem. 2006;6:407–28. doi: 10.2174/187152006778226530. [DOI] [PubMed] [Google Scholar]
- 19.Wierzbicka-Patynowski I, Niewiarowski S, Marcinkiewicz C, Calvete JJ, Marcinkiewicz MM, McLane MA. Structural requirements of echistatin for the recognition of alpha(v)beta(3) and alpha(5)beta(1) integrins. J Biol Chem. 1999;274:37809–14. doi: 10.1074/jbc.274.53.37809. [DOI] [PubMed] [Google Scholar]
- 20.Fu X, Tao L, Rivera A, Williamson S, Song XT, Ahmed N, Zhang X. A simple and sensitive method for measuring tumor-specific T cell cytotoxicity. PLoS One. 2010;5:e11867. doi: 10.1371/journal.pone.0011867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahmed N, Ratnayake M, Savoldo B, Perlaky L, Dotti G, Wels WS, Bhattacharjee MB, Gilbertson RJ, Shine HD, Weiss HL, Rooney CM, Heslop HE, et al. Regression of experimental medulloblastoma following transfer of HER2-specific T cells. Cancer Res. 2007;67:5957–64. doi: 10.1158/0008-5472.CAN-06-4309. [DOI] [PubMed] [Google Scholar]
- 22.Morita S, Kojima T, Kitamura T. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 2000;7:1063–6. doi: 10.1038/sj.gt.3301206. [DOI] [PubMed] [Google Scholar]
- 23.Frankel TL, Burns WR, Peng PD, Yu Z, Chinnasamy D, Wargo JA, Zheng Z, Restifo NP, Rosenberg SA, Morgan RA. Both CD4 and CD8 T cells mediate equally effective in vivo tumor treatment when engineered with a highly avid TCR targeting tyrosinase. J Immunol. 2010;184:5988–98. doi: 10.4049/jimmunol.1000189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kerkar SP, Sanchez-Perez L, Yang S, Borman ZA, Muranski P, Ji Y, Chinnasamy D, Kaiser AD, Hinrichs CS, Klebanoff CA, Scott CD, Gattinoni L, et al. Genetic engineering of murine CD8+ and CD4+ T cells for preclinical adoptive immunotherapy studies. J Immunother. 2011;34:343–52. doi: 10.1097/CJI.0b013e3182187600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hieken TJ, Farolan M, Ronan SG, Shilkaitis A, Wild L, Das Gupta TK. Beta3 integrin expression in melanoma predicts subsequent metastasis. J Surg Res. 1996;63:169–73. doi: 10.1006/jsre.1996.0242. [DOI] [PubMed] [Google Scholar]
- 26.Duan X, Jia SF, Zhou Z, Langley RR, Bolontrade MF, Kleinerman ES. Association of alphavbeta3 integrin expression with the metastatic potential and migratory and chemotactic ability of human osteosarcoma cells. Clin Exp Metastasis. 2004;21:747–53. doi: 10.1007/s10585-005-0599-6. [DOI] [PubMed] [Google Scholar]
- 27.Gong W, Zhang GM, Liu Y, Lei Z, Li D, Yuan Y, Huang B, Feng ZH. IFN-gamma withdrawal after immunotherapy potentiates B16 melanoma invasion and metastasis by intensifying tumor integrin alphavbeta3 signaling. Int J Cancer. 2008;123:702–8. doi: 10.1002/ijc.23553. [DOI] [PubMed] [Google Scholar]
- 28.Liu Y, Mounkes LC, Liggitt HD, Brown CS, Solodin I, Heath TD, Debs RJ. Factors influencing the efficiency of cationic liposome-mediated intravenous gene delivery. Nat Biotechnol. 1997;15:167–73. doi: 10.1038/nbt0297-167. [DOI] [PubMed] [Google Scholar]
- 29.Cox D, Brennan M, Moran N. Integrins as therapeutic targets: lessons and opportunities. Nat Rev Drug Discov. 2010;9:804–20. doi: 10.1038/nrd3266. [DOI] [PubMed] [Google Scholar]
- 30.Kurtz L, Kao L, Newman D, Kurtz I, Zhu Q. Integrin alphaIIbbeta3 inside-out activation: an in situ conformational analysis reveals a new mechanism. J Biol Chem. 2012;287:23255–65. doi: 10.1074/jbc.M112.360966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kowolik CM, Topp MS, Gonzalez S, Pfeiffer T, Olivares S, Gonzalez N, Smith DD, Forman SJ, Jensen MC, Cooper LJ. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res. 2006;66:10995–1004. doi: 10.1158/0008-5472.CAN-06-0160. [DOI] [PubMed] [Google Scholar]
- 32.Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, Kamble RT, Bollard CM, Gee AP, Mei Z, Liu H, Grilley B, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121:1822–6. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smolarczyk R, Cichon T, Graja K, Hucz J, Sochanik A, Szala S. Antitumor effect of RGD-4C-GG-D(KLAKLAK)2 peptide in mouse B16(F10) melanoma model. Acta Biochim Pol. 2006;53:801–5. [PubMed] [Google Scholar]
- 34.Greish K. Enhanced permeability and retention (EPR) effect for anticancer nanomedicine drug targeting. Methods Mol Biol. 2010;624:25–37. doi: 10.1007/978-1-60761-609-2_3. [DOI] [PubMed] [Google Scholar]
- 35.Heldin CH, Rubin K, Pietras K, Ostman A. High interstitial fluid pressure - an obstacle in cancer therapy. Nat Rev Cancer. 2004;4:806–13. doi: 10.1038/nrc1456. [DOI] [PubMed] [Google Scholar]
- 36.Curti BD, Urba WJ, Alvord WG, Janik JE, Smith JW, 2nd, Madara K, Longo DL. Interstitial pressure of subcutaneous nodules in melanoma and lymphoma patients: changes during treatment. Cancer Res. 1993;53:2204–7. [PubMed] [Google Scholar]
- 37.Rofstad EK, Ruud EB, Mathiesen B, Galappathi K. Associations between radiocurability and interstitial fluid pressure in human tumor xenografts without hypoxic tissue. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16:936–45. doi: 10.1158/1078-0432.CCR-09-2718. [DOI] [PubMed] [Google Scholar]
- 38.Jain RK. Transport of molecules, particles, and cells in solid tumors. Annu Rev Biomed Eng. 1999;1:241–63. doi: 10.1146/annurev.bioeng.1.1.241. [DOI] [PubMed] [Google Scholar]
- 39.Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, Jain RK. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. 2011;91:1071–121. doi: 10.1152/physrev.00038.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hofmann M, McCormack E, Mujic M, Rossberg M, Bernd A, Bereiter-Hahn J, Gjertsen BT, Wiig H, Kippenberger S. Increased plasma colloid osmotic pressure facilitates the uptake of therapeutic macromolecules in a xenograft tumor model. Neoplasia. 2009;11:812–22. doi: 10.1593/neo.09662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chauhan VP, Stylianopoulos T, Martin JD, Popovic Z, Chen O, Kamoun WS, Bawendi MG, Fukumura D, Jain RK. Normalization of tumour blood vessels improves the delivery of nanomedicines in a size-dependent manner. Nat Nanotechnol. 2012;7:383–8. doi: 10.1038/nnano.2012.45. [DOI] [PMC free article] [PubMed] [Google Scholar]