

Abstract
Disruption of copper homeostasis has been implicated in Alzheimer’s disease (AD) during the last two decades; however, whether copper is a friend or a foe is controversial. Within a genetically tractable Drosophila AD model, we manipulated the expression of human high affinity copper importer orthologous in Drosophila to explore the in vivo roles of copper ions in the development of AD. We found that inhibition of Ctr1C expression by RNAi in Aβ-expressing flies significantly reduced copper accumulation in the brains of the flies as well as ameliorating neurodegeneration, enhancing climbing ability and prolonging lifespan. Interestingly, Ctr1C inhibition led to a significant increase in higher molecular weight Aβ42 forms in brain lysates, while it was accompanied by a trend of decreased expression of amyloid-β degradation proteases (including NEP1-3 and IDE) with age and reduced Cu-Aβ interaction-induced oxidative stress in Ctr1C RNAi flies. Similar results were obtained from inhibiting another copper importer Ctr1B and overexpressing a copper exporter DmATP7 in the nervous system of AD flies. These results imply that copper may play a causative role in developing AD, as either Aβ oligomers or aggregates were less toxic in a reduced copper environment or one with less copper binding. Early manipulation of brain copper uptake can have a great effect on Aβ pathology.
Keywords: Alzheimer's disease, Drosophila, Copper, Amyloid-β, Neurodegeneration, High affinity copper importer, Ctr1, DmATP7
1. Introduction
Copper serves as a catalytic center in a broad range of proteins and enzymes, e.g. cytochrome c oxidase, superoxide dismutase, laccase, and ceruloplasmin that are essential for the survival of all living organisms. However, copper is highly toxic when free Cu2+ ions are present, due to its capacity to mediate the formation of reactive oxygen species via Fenton reactions (Halliwell and Gutteridge, 1990). Therefore, copper homeostasis is strictly controlled within an organism’s cells. Copper dys-homeostasis has been found to induce many human disorders, such as Menkes and Wilson’s diseases (Lutsenko and Petris, 2003), Prion disease (Jones et al., 2004), Parkinson’s disease (Bharathi et al., 2007) and Alzheimer’s disease (Adlard and Bush, 2006; Strozyk et al., 2009).
Alzheimer’s disease (AD) is characterized pathologically by cerebral deposition of extracellular amyloid-β (Aβ) plaques. Considerable evidence indicates that the formation of senile plaques is mediated by endogenous biometals, especially Cu, Zn and Fe (Bush et al., 1994; Cherny et al., 1999; Dong et al., 2003). In healthy brains, most Aβ peptides are present as soluble forms; however, Aβ plaques occur, particularly in AD patients. Aβ peptides are produced from proteolytic cleavage of the amyloid precursor protein (APP), which usually constitutes two forms, Aβ40 and Aβ42 (White et al., 1999). Aβ42 forms protofibrils and fibrils much more readily than Aβ40 and is the predominant form of the peptide found in plaques in AD brains. It is known that copper plays critical roles in Aβ plaque formation (Barnham and Bush, 2008; Lovell et al., 1998; Miller et al., 2006); however, the precise function of copper in Aβ amyloid genesis is controversial.
Overexpression of the human Aβ42 peptide in Drosophila nervous system tissues results in structural as well as behavioral phenotypes resembling several AD-like symptoms, in a doseand age-dependent manner (Finelli et al., 2004; Iijima et al., 2004). Using Aβ42-expressing flies (Iijima et al., 2008), we can screen modifiers of Aβ42-induced phenotypes, which may lead to the dissection of mechanisms of Aβ toxicity and potentially novel AD therapies.
Given the importance of copper and its close relationship with AD pathology, we explored the effect of genetically controlling the level of a copper importer on AD-like pathology within Aβ42-expressing flies. The Ctr1 family proteins are high affinity copper importers, and are conserved from yeast to humans. In the Drosophila genome, there are three Ctr1 family copper transporters, Ctr1A, Ctr1B and Ctr1C. Ctr1A is constitutively and ubiquitously expressed, and loss of Ctr1A will result in developmental arrest at an early larval stage and a general failure of copper-dependent processes (Turski and Thiele, 2007). Ctr1B is expressed exclusively during the late embryonic and larval stages of development and is responsible for copper uptake from the intestine, but is only essential for development and viability under conditions of either extremely limiting or abnormally abundant copper (Zhou et al., 2003). Zhou et al. made an initial characterization of Ctr1C in which it was shown able to complement copper import-deficient yeast and to be expressed in Drosophila third instar larvae and adult males (Zhou et al., 2003). After that no further studies were made until recently, when Steiger et al. characterized Ctr1C as a copper importer in male fertility (Steiger et al., 2010). Here, we show that RNA interference (RNAi)-induced silencing of Ctr1C in the nervous system of AD flies ameliorates the Aβ42-induced AD-like symptoms, Similar effects were also produced by silencing another copper importer Ctr1B through RNAi or over-expressing a copper exporter DmATP7 in the nervous system of AD flies. Given the simpler and time-saving of genetic studies in Drosophila than in mice, should facilitate our understanding of the role of copper and its homeostasis controlling genes in the development of human AD.
2. Materials and methods
2.1. DNA constructs and transgenic flies
Ctr1C were amplified from a Drosophila cDNA pool and cloned into the GAL4-responsive pUAST expression vector. To make the Ctr1C RNAi construct, a cDNA fragment corresponding to base pairs 1–700 of Ctr1C was PCR-amplified (forward primer, 5’-GGGTCTAGAATGGACCACCATGG-3’; reverse primer, 5’-GGGTCTAGAGCATTAGC AAAAAGGA-3’). Two copies of the PCR fragments were cloned in opposite orientations into the Xba I restriction site of the pWIZ expression vector. Transgenic strains were created by embryo injection through P-element-mediated germline transformation. Approximately 3 µg of pUAST-Ctr1C or pWIZ-Ctr1C-RNAi transgenic construct was mixed with 1 µg of helper plasmid in 10 µl of injection buffer. All transgenic flies were generated in the w1118 background following standard protocols. All general fly stocks and Gal4 lines were obtained from the Bloomington Drosophila Stock Center, which includes Actin-Gal4, elav-Gal4, da-GAL4, GMR-GAL4, A9-Gal4, and Timan-Gal4. The UAS-Aβ42 transgenic Drosophila strain was reported previously (Iijima et al, 2008). DmATP7 overexpression (Bloomington #16866) transgenic strains were acquired from the Bloomington Drosophila Stock Center. Ctr1B RNAi was obtained from the National Institute of Genetics (NIG#7459R-2)
2.2. Complementation assay
Using a pan-neuronal elav-Gal4 driver to specifically over-express Ctr1C (located on chromosome 1) in the fly nervous system, the flies will be developmentally arrested and die at the second or third instar larval stage when the larvae are fed a normal diet (NF) at 25°C. For the complementation assay, the food was supplemented with a gradient concentration of either bathocuproine disulfonate (BCS; Sigma-Aldrich number 14,662-5) or ethylenedinitrilotetraacetic acid (EDTA). BCS is a specific copper chelator used to deplete copper in the food. The number of rescued flies was scored. Survival rates were calculated relative to fully viable male elav-Gal4 flies.
2.3. Quantification of neurodegeneration
Adult fly heads were fixed in Carnoy solution (ethanol:chloroform:acetic acid, 6:3:1) overnight at 4 C°, dehydrated in serial ethanol solutions, and then embedded in paraffin and sectioned at 6 µm thickness. H&E staining was performed following standard protocols. Neurodegeneration was assessed by quantification of vacuoles with diameter greater than 3 µm in the fly brains. At least five fly brains were analyzed for each genotype.
2.4. Western blot analysis
Protein samples including SDS-soluble or SDS-insoluble but formic acid-soluble Aβ42 were prepared as previously reported (Iijima et al., 2004). Lysates from an equal number of fly heads were diluted in SDS sample buffer, separated on 10–20% Tris-Tricine gels (Invitrogen), and transferred to nitrocellulose membranes (Invitrogen). Membranes were boiled in PBS for 3 min. Membranes were blocked with 3% BSA and blotted with primary antibody. Primary antibodies used in this study were mouse anti-Aβ42 (6E10, Covance Research Products) and rabbit anti-Actin (Sigma). After washing with TBST 3 times for 5 minutes each, membranes were incubated with secondary antibodies for 1 hr at RT. Then after same 3 washes in TBST, the membranes were incubated with ECL working solution (GE healthcare) and developed on film (Kodak). Data were analyzed with ImageJ software (NIH).
2.5. Pavlovian olfactory associative memory recording
The training and testing procedures were as previously described (Tully and Quinn, 1985; Tully et al., 1994; Yin et al., 1994). During one training session, a group of 100 flies was sequentially exposed for 60 s to two odors, octanol (OCT) or methylcyclohexanol (MCH), with 45 s of fresh air in between. Flies were subjected to foot-shock (1.5 s pulses with 3.5 s intervals, 60 V) during exposure to the first odor (CS+) but not to the second (CS−). To measure “immediate memory (also referred to as “learning”)”, flies were transferred immediately after training to the choice point of a T-maze and forced to choose between the two odors for 2 min, at which time they were trapped in their respective T-maze arms, anesthetized, and counted. A performance index (PI) was calculated from the distribution of flies in the T-maze. A reciprocal group of flies was trained and tested using OCT as the CS+ and MCH as the CS+, respectively. PIs from these two groups were finally averaged for an n = 1 and multiplied by 100. A PI of 0 represented a 50:50 distribution, whereas a PI of 100 represented 100% avoidance of the shock-paired odor.
2.6. Quantitative analysis of gene expression
Total RNA was extracted from the brains of 20 adults for each sample using TRIzol® Reagent (Invitrogen) according to the manufacturers’ instructions and subjected to DNA digestion using DNAse I (Ambion) immediately. The concentration and quality of the DNAse-treated total RNA were then tested, and 1 µg total RNA from each sample was used to synthesize cDNA using the Superscript™ II Reverse Transcriptase Kit (Invitrogen) with oligo(dT) primers. For quantitative RT-PCR (qRT-PCR), real-time PCR reactions were monitored on an iCycler (Bio-Rad) by means of SYBR Green (Bio-Rad) dye. Messenger RNA expression levels were determined relative to rp49 expression by relative quantification. Primers for amplifying Ctr1C, NEP1, NEP2, NEP3 and Aβ42 are listed in Table S1. Statistical analysis was performed using the Student’s t-test.
2.7. Metal content and oxidative stress assay
For the metal content analysis, flies were reared on normal food and fly heads were collected and weighed on day 30 after eclosion. Fly heads were dissolved in 1 ml 65% HNO3, boiled in a 100 °C water bath for 10 min and diluted to 10 ml for quantitative elemental analysis with inductively coupled plasma–mass spectrometry (ICP-MS) XII (Thermo Electron Corp., Waltham, MA, USA) at the Analysis Center of Tsinghua University.
For the oxidative stress assay, flies were collected 2 days after eclosion. After being reared on normal food for 10 days, flies were transferred to filter paper supplemented with 5% sucrose and 20 mM paraquat. Mortality was recorded about every 8 hours. Each vial contained 20–25 flies, and the experiments were repeated at least three times.
2.8. Climbing assay
The climbing assay was referenced to Iijima et al. (2004). Briefly, twenty flies were placed in a plastic vial and gently tapped to the bottom. The number of flies at the top of the vial was then counted after 18 s of climbing under red light (Kodak, GBX-2, Safelight Filter). The data shown represent results from a cohort of flies with four repeats tested serially for 5–50 days. The experiment was repeated more than three times.
2.9. Longevity assay
Twenty to 23 1–2 day newly eclosed flies were placed in a food vial. Each vial was kept at 25 or 29°C, 70% humidity, under a 12-h light–dark cycle. Food vials were changed every 2–3 days, and dead flies were counted at that time. At least 150 flies were prepared from each genotype, and the experiments were carried out more than three times. Percent increases in life span were based on comparing the median survivals. Prism (GraphPad) was used for statistical analysis of lifespan data. Mantel-Cox log-rank statistical analysis was used for testing the statistical significance of the differences between the survivorship curves.
2.10. Statistical analysis
All data were analyzed by Student’s t-test or two-way ANOVA (otherwise indicated). Statistical results were presented as means ± SEM. Asterisks indicate critical levels of significance (*p<0.05, **p<0.01 and ***p<0.001).
3. Results
3.1. Ctr1C can function as a copper importer in the Drosophila brain and nervous system
To determine whether Ctr1C levels could influence Drosophila brain copper homeostasis, we first specifically over-expressed Ctr1C in the fly nervous system using a pan-neuronal elav-Gal4 driver. We found that the elav-Gal4>UAS-Ctr1C progeny were developmentally arrested and died at the second or third instar larval stage when they were fed on normal food at 25°C. To determine whether the lethality was due to copper toxicity in the flies’ nervous systems, we raised fly larvae on normal food supplemented with different concentrations of the copper chelator BCS. Fig. 1A shows that the lethality of Ctr1C over-expression in flies was rescued by supplementing with BCS, and such rescue was dose dependent and reached nearly 100% with 500 µM BCS. However, we failed to rescue the lethality by adding EDTA, which is not a copper-specific chelator. When raised at a lower temperature of 18°C, the elav-Gal4>UAS-Ctr1C flies could survive to adulthood, but had an abnormal wing phenotype (Figs. 1A) and rough eyes (Fig. 1B). Fig. S1B shows sqRT-PCR results confirming Ctr1C was overexpressed in the brains of the transgenic flies. Flies developed normally when Ctr1C was over-expressed with wing (A9-Gal4, Figs. 1A), eye (GMR-Gal4), or heart (Timan-Gal4) specific drivers. These data provide evidence that Ctr1C can function as a copper importer in fly brains, that the lethality of pan-neuronal over-expression of Ctr1C is due to copper-overloading-induced toxicity to the nervous system, and copper overloading is more toxic to the nervous system than to other tested tissues.
Fig. 1.

Ctr1C can function as a copper importer in fly brain nervous system. Specifically, over-expression of Ctr1C in the fly nervous system using a pan-neuronal elav-Gal4 driver caused copper-dependent lethality when fly larvae were raised on normal food at 25°C, but could be rescued by supplying a concentration gradient of BCS (A). Such flies could survive to adulthood when they were raised at a lower temperature (18 °C), but showed rough eyes (B). The “relative to male” is the calculation of the percentage of the number of eclosed female flies to male flies in each group. Gray column (control) represents progeny of ♂w1118×♀ elav-Gal4 crosses. Blue column represents progeny of ♂UAS-Ctr1C×♀ elav-Gal4 crosses.
3.2. Knocking down Ctr1C increases the climbing ability and lifespan of Aβ42 flies
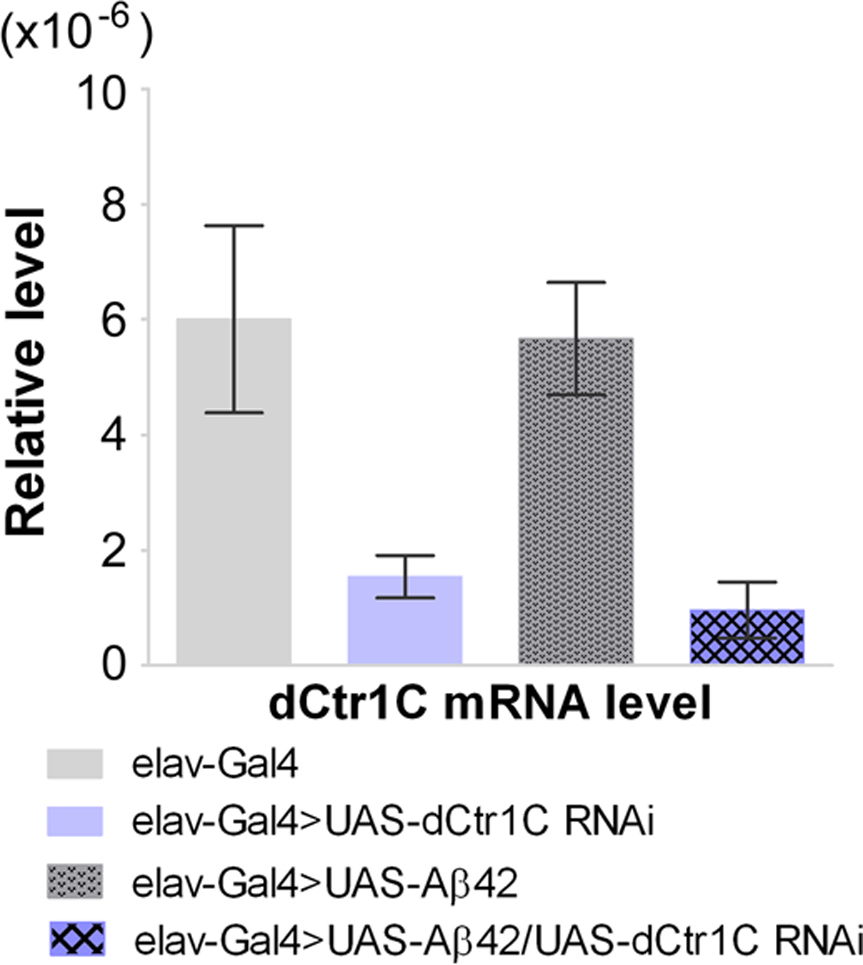
As copper has been implicated as a critical factor in the amyloid pathology of AD, we next wished to determine how the inhibition of a copper importer Ctr1C would influence Aβ42-induced pathology in a Drosophila model. Aβ42 expressing flies have been documented to display locomotor dysfunction after three weeks of age and their lifespan is significantly reduced (Finelli et al., 2004; Iijima et al., 2004; Lang et al., 2012). Fig. S2 shows that in adult fly brains either with or without Aβ42 expression, Ctr1C RNAi could lead to about a threefold reduction in Ctr1C mRNA levels compared with controls. Assays of climbing ability demonstrated that Aβ42 flies started to have a locomotor defect at 15 days of age compared to elav-Gal4 flies without expressed Aβ42 (Fig. 2A), while Aβ42 flies with Ctr1C RNAi started to have locomotor defects at 30 days of age compared to elav-Gal4 flies (Fig. 2A). In all the tested time courses, Aβ42 flies with decreased Ctr1C expression through RNAi had a delayed climbing deficit compared with age-matched Aβ42 flies (Fig. 2A). As a further control, we tested the climbing ability of transgenic flies without Aβ42 expression (Fig. 2B). No significant differences were found between age-matched elav-Gal4 and Ctr1C -RNAi flies.
Fig. 2.

Reduced Ctr1C expression can ameliorate the climbing and longevity defects of Aβ42 flies. (A) Aβ42 expression in fly brains (elav-Gal4>UAS-Aβ42) induced a climbing deficit as compared with control elav-Gal flies. Knocking down Ctr1C by RNAi (elav-Gal4>UAS-Aβ42/UAS-Ctr1C-RNAi) could significantly increase the climbing ability of Aβ42 flies. Two-way ANOVA, **p<0.01, ***p<0.001 (in comparison with elav-Gal4>UAS-Aβ42 flies). n=4 independent experiments. (B) In the absence of Aβ42 expression, no significant locomotor deficits were found among elav-Gal4 and elav-Gal4>UAS-Ctr1C-RNAi flies. n=4 independent experiments. (C–D) Ctr1C knockdown significantly lengthened the lifespan of Aβ42 flies. The percentage survivorship was plotted against age (C). Ctr1C RNAi significantly prolonged the life span of Aβ42 flies, with a 32.4% extension in median lifespan over that of Aβ42 flies at 25 °C (p<0.0001, C and D). Reported P values are from Mantel-Cox log-rank statistical analysis.
Consistent with the results obtained in the climbing assay, the lifespan of Aβ42 flies was prolonged by RNAi-based knockdown of Ctr1C expression (Fig. 2C and D). The Ctr1C RNAi transgenic line exhibited an increase of 32.4% in the median lifespan of Aβ42 flies reared at 25 °C. The result indicated that a reduction of Ctr1C expression in Aβ42 flies leads to an improved locomotor ability and a longer lifespan. The Aβ42 gene was directly under the control of elav-Gal4, and indeed we did not see changes in Aβ42 mRNA expression when the control and experimental flies had different number of UAS transgenes (Lang et al., 2012). Therefore, the results implied that the locomotion and life span defects resulting from Aβ42 toxicity were closely related to Cu status, and could be modulated through changes in Ctr1C expression level.
3.3. Knocking down Ctr1C ameliorates Aβ42-induced neurodegeneration accompanied by a reduction in copper levels
Vacuolization of brain tissues is a major hallmark of neurodegeneration in Aβ42 flies (Muqit and Feany, 2002). Next we wanted to know whether the improved locomotor ability and longer lifespan of Aβ42 flies produced by Ctr1C RNAi was associated with ameliorated brain neurodegeneration. Fig. 3A shows that in 30 day old Aβ42-expressing fly brains, there were more and larger bubbles in both the cortex and neuropil regions compared to the brains of Aβ42 flies with Ctr1C RNAi. Counting the numbers of vacuoles in the cortex and neuropil revealed that Ctr1C knockdown dramatically decreased brain vacuolization in Aβ42 flies (Fig. 3B). These results give evidence that inhibiting Ctr1C expression can ameliorate Aβ42-induced neurodegeneration in fly brains.
Fig. 3.

Specifically knocking down fly brain Ctr1C can ameliorate Aβ42-induced neurodegeneraion with a decrease in copper levels. Paraffin sections of 30-day-old fly brains were stained with H&E (A). Pan-neuronal expression of Aβ42 in fly brains induced neurodegeneration (arrowheads indicate vacuoles) in both the cortex and neuropil regions, while Ctr1C RNAi ameliorated neurodegeneration. Scale bar, 50µm. (B) is a statistical analysis of Aβ42-induced neurodegeneration bubbles. Brain sections across the mushroom body somatic region were chosen for comparison. The number of vacuoles (diameter > 3 mm) in each section was counted and summarized. Significant differences were observed between Ctr1C RNAi (elav-Gal4>UAS-Aβ42/UAS-Ctr1C RNAi) flies and control (elav-Gal4>UAS-Aβ42) flies (p<0.05). (C–D) shows metal content in brains of 30-day old flies as measured by ICP-MS. Ctr1C RNAi (elav-Gal4>UAS-Aβ42/UAS-Ctr1C RNAi) significantly slowed down brain Cu accumulation (C, p<0.05), while not significantly disturbing the Zn, Fe and Mn content of Aβ42 fly brains (D). Data are expressed as means ± SEM and analyzed by Student’s t-test. n=3 for each genotype.
Using ICP-MS, we determined the copper levels in 30-day-old fly brains when Ctr1C was knocked down by RNAi. The results showed that knocking down Ctr1C significantly reduced brain copper compared with controls (Fig. 3C, p<0.05); however, other metals, like Zn, Fe and Mn were not significantly disturbed by Ctr1C RNAi (Fig. 3D). These results indicate that Ctr1C RNAi could specifically reduce copper levels in fly brains without changing the status of other metal ions such as Zn, Fe and Mn, which have also been implicated in the pathogenesis of AD.
3.4. Knocking down Ctr1C does not rescue Aβ42-induced early memory loss
Progressive memory loss is another hallmark of Alzheimer’s disease. In Aβ42-expressing adult flies, an obvious memory defect was found at around 10 days old (Chiang et al., 2010; Iijima et al., 2004). To investigate the effect of Ctr1C knockdown on Aβ42-induced early memory loss, we used the same extensively characterized Pavlovian olfactory aversive conditioning. Ctr1C knockdown did not rescue memory loss at this stage (Fig. 4). As a control, we examined how reduction in Ctr1C expression alone might impact memory scores in the absence of Aβ42. Knocking down Ctr1C did not significantly influence the memory of normal 10-day-old male flies (Fig. 4). Therefore, reducing Cu levels through knocking down Ctr1C did not ameliorate Aβ42 toxicity on fly memory at early stages.
Fig. 4.

Inhibiting Ctr1C does not affect Aβ42-induced early memory loss. Performance indices represent the memory ability of different genotypes in 10-day-old flies. Pan-neuronal knock down of Ctr1C did not influence memory performance as well as affecting the memory defect caused by excessive Aβ42. All behavioral data are normalized to control flies. Data are expressed as means ± SEM. n=8 PIs for each genotype. One-way ANOVA, ***p<0.001. n.s., not significant.
3.5. Knocking down Ctr1C changes fly brain Aβ composition
Because Aβ42 can oligomerize and aggregate into different states (Bitan et al., 2003; Sgourakis et al., 2007), we then asked how Ctr1C RNAi affects various forms of Aβ42. Fly brain lysates were used for Western blotting analysis. The result showed that the SDS-soluble Aβ42 (low level aggregate forms) in male brains was at similar levels between Aβ42 flies with and without Ctr1C RNAi. However, dramatic increases in SDS-insoluble but formic acid-soluble Aβ42 were detected in male Aβ42 flies with Ctr1C knock down (Fig. 5A and B).
Fig. 5.

Knocking down Ctr1C promotes aggregation of higher molecular weight Aβ42 forms with a trend for decreased NEP and IDE expression levels in brains of Aβ42 flies. Representative western blotting data (A) and statistical results (B). The amount of SDS-soluble Aβ42 was equivalent in Aβ42-expressing flies with Ctr1C RNAi and those without Ctr1C RNAi. However, SDS-insoluble but formic acid (FA, 70%) soluble Aβ42 forms were dramatically increased after knocking down Ctr1C. The amount of Aβ42 and actin proteins in each well were quantified by using ImageJ software, and the data showed the relative percentage of amount of Aβ42 to actin protein. Student’s t-test, **p<0.01 (in comparison with elav-Gal4>UAS-Aβ42 flies). n=3 independent experiments. The impact of Ctr1C RNAi on NEP1(C), NEP2 (D), NEP3 (E) and IDE (F) mRNA expression levels in brains of 5 and 25-day-old flies were determined by qRT-PCR. Statistical analysis showed no significant differences for the four tested enzymes between elav-Gal4>UAS-Aβ42 and elav-Gal4>UAS-Aβ42/UAS-Ctr1C RNAi flies at day 5 after eclosion. However, at day 25 after eclosion, NEP1(C), NEP3 (E) and IDE (F) mRNA expression levels were significantly decreased in brains of Ctr1C RNAi flies (elav-Gal4>UAS-Aβ42/UAS-Ctr1C RNAi). Student’s t-test, *p<0.05, **p<0.01 (in comparison with elav-Gal4>UAS-Aβ42 flies). The relative NEP1-3 and IDE expression levels against rp49 were from three independent biological replicates and plotted with SEM (error bars).
Because of the significant increase in formic acid-soluble Aβ42 fractions brought about by Ctr1C RNAi, we then asked whether this was due to an inhibition of Aβ42 clearance. Several proteases (NEP1-3, IDE) have been proposed to act in Aβ degradation (Farris et al., 2003; Iwata et al., 2000); we thus explored whether they were affected by Ctr1C RNAi in brains of Aβ flies. We did not find any significant changes in IDE and NEP1-3 mRNA levels in young (5 days old) Aβ42 fly brains induced by Ctr1C RNAi (Fig. 5C–F). However, with ageing, IDE, NEP1 and NEP3 mRNA levels were dramatically reduced in 25-day-old Aβ42 fly brains by Ctr1C RNAi (Fig. 5C, E and F). Therefore, the increase in SDS-insoluble but formic acidsoluble Aβ42 fractions by Ctr1C RNAi possibly resulted from these reductions in degrading proteases.
3.6. Knocking down Ctr1C greatly enhances flies’ tolerance to oxidative stress
Reactive oxidative species are associated with many pathological processes, including that in Alzheimer’s disease. As copper is a redox-active agent producing ROS in vivo, to determine whether the reduction in Ctr1C expression levels in brain could alter the flies’ susceptibility to oxidative injury, we treated Ctr1C RNAi and control Aβ42 flies with paraquat, a widely used free radical generator that can increase ROS levels in vivo (Arking et al., 1991; Dias-Santagata et al., 2007). Targeted knock down of Ctr1C in the CNS of Aβ42 flies rendered them more resistant to paraquat treatment than control Aβ42 flies (Fig. 6), with a 45.5% increase in mean life span from 16.5 h to 24 h. The two survival curves were significantly different (p<0.0001).
Fig. 6.

Ctr1C RNAi significantly increased Aβ42 flies’ tolerance to oxidative stress. About 10-day old normal growing flies were treated in vials with filter paper supplemented with 5% sucrose and 20 mM paraquat. Mortality was recorded about every 8 hours and life span was measured over 45 h. Ctr1C RNAi flies (elav-Gal4>UAS-Aβ42/UAS-Ctr1C RNAi ) survived much better than control Aβ42 flies (elav-Gal4>UAS-Aβ42), with a 45.5% increase in mean life span. The two survival curves are significantly different (p<0.0001). Reported P values are from Mantel-Cox log-rank statistical analysis. The experiments were repeated at least three times.
3.7. Knocking down Ctr1B and over-expressing DmATP7 increase the climbing ability and lifespan of Aβ42 flies
To further confirm the ameliorating effects through lowering brain Cu accumulation by Ct1C RNAi in AD flies, we tested the longevity and climbing ability of AD flies through knocking down another copper importer Ctr1B or over-expressing a copper exporter DmATP7. Over-expressing Ctr1B has been reported to aggravate rough eye phenotype due to its resulted copper overloading (Saini et al., 2010), and over-expressing DmATP7 could lower copper accumulation in fly brains (Southon et al., 2010). Fig. 7A showed that the longevity of AD flies was significantly increased when Ctr1B was knocked down (p<0.0001), or DmATP7 was over-expressed (p<0.0001), with a 17.9% and a 33.3% increase in median life-span compared with control Aβ42 flies, respectively. In consistent with the longevity test, the climbing ability of AD flies was also improved significantly through knocking down Ctr1B or over-expressing DmATP7 in the nervous system (Fig. 7B). Taken together, these results corroborated that specifically lowering copper accumulation in brains of AD flies could ameliorate the AD-like symptoms in Drosophila.
Fig. 7.

Kocking down Ctr1B and over-expressing DmATP7 can ameliorate the climbing and longevity defects of Aβ42 flies. (A) Ctr1B knockdown and DmATP7 over-expression significantly lengthened the lifespan of Aβ42 flies, with a 17.9% and a 33.3% extension in median lifespan over that of Aβ42 flies at 25 °C (p<0.0001), respectively. Reported P values are from Mantel-Cox log-rank statistical analysis. The percentage survivorship was plotted against age. n=4 independent experiments. (B) The climbing deficit of Aβ42 expression flies was rescued through knocking down Ctr1B by RNAi or over-expressing DmATP7. Two-way ANOVA, ***p<0.001 (in comparison with elav-Gal4>UAS-Aβ42 flies). n=4 independent experiments.
4. Discussion
As we know, in healthy and/or young tissue, efficient homeostatic mechanisms are sufficient to maintain normal compartmentalization and release of metals. This compensatory capability of the brain is gradually lost with age, which would lead to an imbalance in brain metal levels and distribution (Bleackley and MacGillivray, 2011). In the last two decades, increasing evidences have demonstrated that disturbances in metal (especially Zn, Cu and Fe) homeostasis play critical roles in the development of AD (Adlard and Bush, 2006; Duce et al., 2010; James et al., 2012; Maynard et al., 2002; Strozyk et al., 2009). However, the exact role of metal ions in AD is still in debate; more research is urgently needed to understand how this disturbance is initiated and how it can be addressed to ameliorate the disease. Copper is an essential but very active redox agent in living cells. It has been reported that increased brain copper levels are associated with normal ageing and AD (Maynard et al, 2002). In this study, we showed that genetic inhibition of human high affinity copper importer Ctr1 orthologous, Ctr1C, in whole life-course of AD model flies ameliorated brain neurodegeneration and prolonged the flies’ life-span with enhanced climbing ability. Our results favors the foe role for increased copper accumulation in promoting brain neurodegeneration, which is also supported by data delivered from a recent review (Brewer, 2012).
Besides the oxidative toxicity of Cu itself in AD process, the level of Cu in tissues also influences the Aβ toxicity. In vivo studies have shown that the soluble oligomeric species are more neurotoxic than the insoluble amyloid fibrils, and are likely responsible for neurodegeneration/memory loss (Golde et al., 2006; Haass et al., 2007). Also some reports indicate that a significant increase in SDS insoluble but formic acid-soluble Aβ42 usually signals worsening neurodegeneration (Bieler and Soto, 2004; Horikoshi et al., 2004; Xing and Higuchi, 2002). However, the locomotor ability and longevity of Ctr1C RNAi Aβ42 flies were much improved even when the SDS insoluble but formic acid-soluble Aβ42 fractions are markedly increased in our results. These paradoxical results suggest that the Aβ toxicity may not be determined solely by the aggregation form. One possibility is that the toxicity of either Aβ42 oligomers or aggregates is mostly induced by bound Cu ions through the production of ROS radical species by redox reactions. To test this hypothesis in vivo, we treated adult flies with an in vivo ROS generator, paraquat. Surprisingly, the flies’ tolerance to paraquat challenge was much enhanced through pan-neuronal knock down of Ctr1C. These results suggested that with less copper or in a copper reduced environment, the Aβ42 soluble fractions or the increased Aβ42 aggregates (here referring only to the SDS insoluble but formic acid-soluble Aβ42 fractions) may have less toxicity. Some recent experiments using synthesized bifunctional compounds (BFCs) to treat copper-mediated Aβ aggregation or preformed Aβ fibrils also supported our observations (Geng et al., 2012; Sharma et al., 2012). BFCs could inhibit the copper mediated Aβ42 aggregation and promote disaggregation of amyloid fibrils accompanying with a dramatic reduction of H2O2 production. However, the formation of soluble Aβ42 oligomers in presence of copper ions and the compounds still lead to an increased cellular toxicity (Sharma et al., 2012), which also emphasized the toxicity of soluble Aβ42 oligomers when they are present with copper ions.
As neprilysin (NEP) and the insulin-degrading enzyme (IDE) are well known proteases involved in amyloid degradation (Sudoh et al., 2002; Carty et al., 2013; Yamamoto et al., 2013), we thus measured their mRNA level in brains of young (5-day old) and older (25-day old) flies to explore why Ctr1C RNAi led to a significant increase in SDS insoluble but 70% FA soluble Aβ42 fractions. It’s interesting that we found the expression levels of potential amyloid-β degradation proteases (including NEP1-3 and IDE) exhibited a trend of decreasing with age when Ctr1C was knocked down. We hypothesized that the decreased expression of NEP or IDE may partially explain the increased level of SDS-insoluble but FA soluble fraction of Aβ42 because less monomers are degradated. However, one previous published paper (Sudoh et al., 2002) have clear in vivo evidences that the detergent-insoluble fraction extracted with 70% formic acid decreased markedly in IDE overexpression cells, and inhibit NEP expression, the cell will accumulate less insoluble Aβ. Their results showed that both IDE and NEP could degrade SDS soluble and insoluble fractions. One would generally expect that the levels of these proteases should be increased in order to achieve greater clearance of Aβ aggregates and in turn ameliorate AD defects, but we found decreased levels of these proteases accompanied with improved longevity and ameliorated neurodegeneration. Therefore, copper-Aβ interaction-initiated oxidative stress may play a causative role in AD development.
A considerable number of reports indicate that oxidative stress arises early, before the appearance of AD symptoms, and is also a feature of mild cognitive impairment (Butterfield et al., 1999; Keller, et al., 2005; Nunomura, et al., 2001; Reddy, 2006; Yatin, et al., 1999). It has been proven that binding of Aβ42 with Cu2+ not only induces the precipitation of Aβ42 (Atwood, et al., 1998; Bush et al., 1994; Lovell et al., 1998) but also induces oxidative stress through the generation of H2O2 (Huang et al., 1999). Increased copper binding to Aβ is evident in AD (Bush et al., 1994; Lovell et al., 1998; Syme et al., 2004; Tougu et al., 2011), and overexpression of Aβ42 in Drosophila brains may accumulate oxidative stress and cause damage in neuronal nuclei and mitochondrial DNA (Rival et al., 2009). Therefore, controlling copper status from youth may be an effective way to slow or inhibit the progress of AD. Recently, a Phase 2 clinical trial of Cu supplementation based on the “copper deficiency theory” was unsuccessful (Kessler et al., 2008) and the chelation method, such as by using clioquiniol, has effects at the organismal level and is generally harmful. Using a genetic method to manipulate copper specific transporters may be a potentially valuable strategy for treating AD. Through knocking down Ctr1C in the nervous system of Drosophila, we found oxidative stress was significantly reduced, and brain neurodegeneration was much improved and lifespan was prolonged.
In summary, our study suggests that brain copper level is vital to AD, genetically manipulating copper importer or exporter to lower copper accumulation early in life may markedly ameliorate the Aβ42-induced neurodegeneration. Because metal ions function differently in AD progress, we may need to combine diverse strategies in order to develop a better type of therapy.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
We thank Bloomington Stock Center for providing Drosophila materials for this study, and Dan Chen and Sumin Guo for technical assistance with the Drosophila experiments. We thank Dr. Michael Kanost (Kansas State University) for his generosity in providing us some valuable resources, and Dr. Ashley I. Bush (University of Melbourne, Australia) for his comments on our experiments. This work was supported by the National Basic Research Program of China (#2011CB910900), China Postdoctoral Science Foundation (#20060390479), National Institutes of Health (Ro1AI 07864), National Basic Research Project (973 program) of the Ministry of Science and Technology of China (2009CB941301).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure statement
The authors declare no competing financial interests.
References
- Adlard PA, Bush AI. Metals and Alzheimer's disease. J Alzheimers Dis. 2006;10(2–3):145–163. doi: 10.3233/jad-2006-102-303. [DOI] [PubMed] [Google Scholar]
- Arking R, Buck S, Berrios A, Dwyer S, Baker GT. Elevated Paraquat Resistance Can Be Used as a Bioassay for Longevity in a Genetically Based Long-Lived Strain of Drosophila. Dev Genet. 1991;12(5):362–370. doi: 10.1002/dvg.1020120505. [DOI] [PubMed] [Google Scholar]
- Atwood CS, Moir RD, Huang XD, Scarpa RC, Bacarra NME, Romano DM, Hartshorn MK, Tanzi RE, Bush AI. Dramatic aggregation of Alzheimer A beta by Cu(II) is induced by conditions representing physiological acidosis. J Biol Chem. 1998;273(21):12817–12826. doi: 10.1074/jbc.273.21.12817. [DOI] [PubMed] [Google Scholar]
- Barnham KJ, Bush AI. Metals in Alzheimer's and Parkinson's diseases. Curr Opin Chem Biol. 2008;12(2):222–228. doi: 10.1016/j.cbpa.2008.02.019. [DOI] [PubMed] [Google Scholar]
- Bharathi, Indi SS, Rao KSJ. Copper- and iron-induced differential fibril formation in alpha-synuclein: TEM study. Neurosci Lett. 2007;424(2):78–82. doi: 10.1016/j.neulet.2007.06.052. [DOI] [PubMed] [Google Scholar]
- Bieler S, Soto C. beta-sheet breakers for Alzheimer's disease therapy. Curr Drug Targets. 2004;5(6):553–558. doi: 10.2174/1389450043345290. [DOI] [PubMed] [Google Scholar]
- Bitan G, Kirkitadze MD, Lomakin A, Vollers SS, Benedek GB, Teplow DB. Amyloid beta-protein (A beta) assembly: A beta 40 and A beta 42 oligomerize through distinct pathways. P Natl Acad Sci USA. 2003;100(1):330–335. doi: 10.1073/pnas.222681699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleackley MR, MacGillivray RTA. Transition metal homeostasis: from yeast to human disease. Biometals. 2011;24(5):785–809. doi: 10.1007/s10534-011-9451-4. [DOI] [PubMed] [Google Scholar]
- Brewer GJ. Copper excess, zinc deficiency, and cognition loss in Alzheimer's disease. Biofactors. 2012;38(2):107–113. doi: 10.1002/biof.1005. [DOI] [PubMed] [Google Scholar]
- Bush AI, Pettingell WH, Multhaup G, Paradis MD, Vonsattel JP, Gusella JF, Beyreuther K, Masters CL, Tanzi RE. Rapid Induction of Alzheimer a-Beta Amyloid Formation by Zinc. Science. 1994;265(5177):1464–1467. doi: 10.1126/science.8073293. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Howard B, Yatin S, Koppal T, Drake J, Hensley K, Aksenov M, Aksenova M, Subramaniam R, Varadarajan S, Harris-White ME, Pedigo NW, Carney JM. Elevated oxidative stress in models of normal brain aging and Alzheimer's disease. Life Sci. 1999;65(18–19):1883–1892. doi: 10.1016/s0024-3205(99)00442-7. [DOI] [PubMed] [Google Scholar]
- Carty N, Nash KR, Brownlow M, Cruite D, Wilcock D, Selenica ML, Lee DC, Gordon MN, Morgan D. Intracranial Injection of AAV Expressing NEP but Not IDE Reduces Amyloid Pathology in APP+PS1 Transgenic Mice. PloS one. 2013;8(3):e59626. doi: 10.1371/journal.pone.0059626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherny RA, Legg JT, McLean CA, Fairlie DP, Huang XD, Atwood CS, Beyreuther K, Tanzi RE, Masters CL, Bush AI. Aqueous dissolution of Alzheimer's disease A beta amyloid deposits by biometal depletion. J Biol Chem. 1999;274(33):23223–23228. doi: 10.1074/jbc.274.33.23223. [DOI] [PubMed] [Google Scholar]
- Chiang HC, Wang L, Xie ZL, Yau A, Zhong Y. PI3 kinase signaling is involved in A beta-induced memory loss in Drosophila. P Natl Acad Sci USA. 2010;107(15):7060–7065. doi: 10.1073/pnas.0909314107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias-Santagata D, Fulga TA, Duttaroy A, Feany MB. Oxidative stress mediates tau-induced neurodegeneration in Drosophila. J Clin Invest. 2007;117(1):236–245. doi: 10.1172/JCI28769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Atwood CS, Anderson VE, Siedlak SL, Smith MA, Perry G, Carey PR. Metal binding and oxidation of amyloid-beta within isolated senile plaque cores: Raman microscopic evidence. Biochemistry-Us. 2003;42(10):2768–2773. doi: 10.1021/bi0272151. [DOI] [PubMed] [Google Scholar]
- Duce JA, Bush AI. Biological metals and Alzheimer's disease: Implications for therapeutics and diagnostics. Prog Neurobiol. 2010;92(1):1–18. doi: 10.1016/j.pneurobio.2010.04.003. [DOI] [PubMed] [Google Scholar]
- Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, Eckman CB, Tanzi RE, Selkoe DJ, Guenette S. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. P Natl Acad Sci USA. 2003;100(7):4162–4167. doi: 10.1073/pnas.0230450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finelli A, Kelkar A, Song HJ, Yang HD, Konsolaki M. A model for studying Alzheimer's A beta 42-induced toxicity in Drosophila melanogaster. Mol Cell Neurosci. 2004;26(3):365–375. doi: 10.1016/j.mcn.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Geng J, Li M, Wu L, Ren J, Qu X. Liberation of copper from amyloid plaques: making a risk factor useful for Alzheimer's disease treatment. Journal of medicinal chemistry. 2012;55(21):9146–9155. doi: 10.1021/jm3003813. [DOI] [PubMed] [Google Scholar]
- Golde TE, Dickson D, Hutton M. Filling the gaps in the abeta cascade hypothesis of Alzheimer's disease. Curr Alzheimer Res. 2006;3(5):421–430. doi: 10.2174/156720506779025189. [DOI] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nature reviews Molecular cell biology. 2007;8(2):101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JMC. Role of Free-Radicals and Catalytic Metal-Ions in Human-Disease - an Overview. Method Enzymol. 1990;186:1–85. doi: 10.1016/0076-6879(90)86093-b. [DOI] [PubMed] [Google Scholar]
- Horikoshi Y, Mori T, Maeda M, Kinoshita N, Sato K, Yamaguchi H. A beta N-terminal-end specific antibody reduced beta-amyloid in Alzheimer-model mice. Biochem Bioph Res Co. 2004;325(2):384–387. doi: 10.1016/j.bbrc.2004.10.039. [DOI] [PubMed] [Google Scholar]
- Huang XD, Cuajungco MP, Atwood CS, Hartshorn MA, Tyndall JDA, Hanson GR, Stokes KC, Leopold M, Multhaup G, Goldstein LE, Scarpa RC, Saunders AJ, Lim J, Moir RD, Glabe C, Bowden EF, Masters CL, Fairlie DP, Tanzi RE, Bush AI. Cu(II) potentiation of Alzheimer A beta neurotoxicity -Correlation with cell-free hydrogen peroxide production and metal reduction. J Biol Chem. 1999;274(52):37111–37116. doi: 10.1074/jbc.274.52.37111. [DOI] [PubMed] [Google Scholar]
- Iijima K, Chiang HC, Hearn SA, Hakker I, Gatt A, Shenton C, Granger L, Leung A, Iijima-Ando K, Zhong Y. A beta 42 Mutants with Different Aggregation Profiles Induce Distinct Pathologies in Drosophila. Plos One. 2008;3(2) doi: 10.1371/journal.pone.0001703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima K, Liu HP, Chiang AS, Hearn SA, Konsolaki M, Zhong Y. Dissecting the pathological effects of human A beta 40 and A beta 42 in Drosophila: A potential model for Alzheimer's disease. P Natl Acad Sci USA. 2004;101(17):6623–6628. doi: 10.1073/pnas.0400895101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata N, Tsubuki S, Takaki Y, Watanabe K, Sekiguchi M, Hosoki E, Kawashima-Morishima M, Lee HJ, Hama E, Sekine-Aizawa Y, Saido TC. Identification of the major A beta(1–42)-degrading catabolic pathway in brain parenchyma: Suppression leads to biochemical and pathological deposition. Nat Med. 2000;6(2):143–150. doi: 10.1038/72237. [DOI] [PubMed] [Google Scholar]
- James SA, Volitakis I, Adlard PA, Duce JA, Masters CL, Cherny RA, Bush AI. Elevated labile Cu is associated with oxidative pathology in Alzheimer disease. Free Radical Bio Med. 2012;52(2):298–302. doi: 10.1016/j.freeradbiomed.2011.10.446. [DOI] [PubMed] [Google Scholar]
- Jones CE, Abdelraheim SR, Brown DR, Viles JH. Preferential Cu2+ coordination by His(96) and His(111) induces beta-sheet formation in the unstructured amyloidogenic region of the prion protein. J Biol Chem. 2004;279(31):32018–32027. doi: 10.1074/jbc.M403467200. [DOI] [PubMed] [Google Scholar]
- Keller JN, Schmitt FA, Scheff SW, Ding Q, Chen Q, Butterfield DA, Markesbery WR. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology. 2005;64(7):1152–1156. doi: 10.1212/01.WNL.0000156156.13641.BA. [DOI] [PubMed] [Google Scholar]
- Kessler H, Bayer TA, Bach D, Schneider-Axmann T, Supprian T, Herrmann W, Haber M, Multhaup G, Falkai P, Pajonk FG. Intake of copper has no effect on cognition in patients with mild Alzheimer's disease: a pilot phase 2 clinical trial. J Neural Transm. 2008;115(8):1181–1187. doi: 10.1007/s00702-008-0080-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang ML, Wang L, Fan QW, Xiao GR, Wang XX, Zhong Y, Zhou B. Genetic Inhibition of Solute-Linked Carrier 39 Family Transporter 1 Ameliorates A beta Pathology in a Drosophila Model of Alzheimer's Disease. Plos Genet. 2012;8(4):623–639. doi: 10.1371/journal.pgen.1002683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR. Copper, iron and zinc in Alzheimer's disease senile plaques. J Neurol Sci. 1998;158(1):47–52. doi: 10.1016/s0022-510x(98)00092-6. [DOI] [PubMed] [Google Scholar]
- Lutsenko S, Petris NJ. Function and regulation of the mammalian coppertransporting ATPases: Insights from biochemical and cell biological approaches. J Membrane Biol. 2003;191(1):1–12. doi: 10.1007/s00232-002-1040-6. [DOI] [PubMed] [Google Scholar]
- Maynard CJ, Cappai R, Volitakis I, Cherny RA, White AR, Beyreuther K, Masters CL, Bush AI, Li QX. Overexpression of Alzheimer's disease amyloid-beta opposes the age-dependent elevations of brain copper and iron. J Biol Chem. 2002;277(47):44670–44676. doi: 10.1074/jbc.M204379200. [DOI] [PubMed] [Google Scholar]
- Miller LM, Wang Q, Telivala TP, Smith RJ, Lanzirotti A, Miklossy J. Synchrotron-based infrared and X-ray imaging shows focalized accumulation of Cu and Zn co-localized with beta-amyloid deposits in Alzheimer's disease. J Struct Biol. 2006;155(1):30–37. doi: 10.1016/j.jsb.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Muqit MMK, Feany MB. Modelling neurodegenerative diseases in Drosophila: a fruitful approach? Nat Rev Neurosci. 2002;3(3):237–243. doi: 10.1038/nrn751. [DOI] [PubMed] [Google Scholar]
- Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA. Oxidative damage is the earliest event in Alzheimer disease. J Neuropath Exp Neur. 2001;60(8):759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- Reddy PH. Amyloid precursor protein-mediated free radicals and oxidative damage: Implications for the development and progression of Alzheimer's disease. J Neurochem. 2006;96(1):1–13. doi: 10.1111/j.1471-4159.2005.03530.x. [DOI] [PubMed] [Google Scholar]
- Rival T, Page RM, Chandraratna DS, Sendall TJ, Ryder E, Liu B, Lewis H, Rosahl T, Hider R, Camargo LM, Shearman MS, Crowther DC, Lomas DA. Fenton chemistry and oxidative stress mediate the toxicity of the beta-amyloid peptide in a Drosophila model of Alzheimer's disease. Eur J Neurosci. 2009;29(7):1335–1347. doi: 10.1111/j.1460-9568.2009.06701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini N, Oelhafen S, Hua HQ, Georgiev O, Schaffner W, Bueler H. Extended lifespan of Drosophila parkin mutants through sequestration of redox-active metals and enhancement of anti-oxidative pathways. Neurobiol Dis. 2010;40(1):82–92. doi: 10.1016/j.nbd.2010.05.011. [DOI] [PubMed] [Google Scholar]
- Sharma AK, Pavlova ST, Kim J, Finkelstein D, Hawco NJ, Rath NP, Mirica LM. Bifunctional compounds for controlling metal-mediated aggregation of the abeta42 peptide. Journal of the American Chemical Society. 2012;134(15):6625–6636. doi: 10.1021/ja210588m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgourakis NG, Yan YL, McCallum SA, Wang CY, Garcia AE. The Alzheimer's peptides A beta 40 and 42 adopt distinct conformations in water: A combined MD/NMR study. J Mol Biol. 2007;368(5):1448–1457. doi: 10.1016/j.jmb.2007.02.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southon A, Palstra N, Veldhuis N, Gaeth A, Robin C, Burke R, Camakaris J. Conservation of copper-transporting P(IB)-type ATPase function. Biometals. 2010;23(4):681–694. doi: 10.1007/s10534-010-9332-2. [DOI] [PubMed] [Google Scholar]
- Steiger D, Fetchko M, Vardanyan A, Atanesyan L, Steiner K, Turski ML, Thiele DJ, Georgiev O, Schaffner W. The Drosophila Copper Transporter Ctr1C Functions in Male Fertility. J Biol Chem. 2010;285(22):17089–17097. doi: 10.1074/jbc.M109.090282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strozyk D, Launer LJ, Adlard PA, Cherny RA, Tsatsanis A, Volitakis I, Blennow K, Petrovitch H, White LR, Bush AI. Zinc and copper modulate Alzheimer A beta levels in human cerebrospinal fluid. Neurobiol Aging. 2009;30(7):1069–1077. doi: 10.1016/j.neurobiolaging.2007.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudoh S, Frosch MP, Wolf BA. Differential effects of proteases involved in intracellular degradation of amyloid beta-protein between detergent-soluble and -insoluble pools in CHO-695 cells. Biochemistry. 2002;41(4):1091–1099. doi: 10.1021/bi011193l. [DOI] [PubMed] [Google Scholar]
- Syme CD, Nadal RC, Rigby SEJ, Viles JH. Copper binding to the amyloid-beta (A beta) peptide associated with Alzheimer's disease – Folding, coordination geometry, pH dependence, stoichiometry, and affinity of A beta-(1–28): Insights from a range of complementary spectroscopic techniques. J Biol Chem. 2004;279(18):18169–18177. doi: 10.1074/jbc.M313572200. [DOI] [PubMed] [Google Scholar]
- Tougu V, Tiiman A, Palumaa P. Interactions of Zn(II) and Cu(II) ions with Alzheimer's amyloid-beta peptide. Metal ion binding, contribution to fibrillization and toxicity. Metallomics. 2011;3(3):250–261. doi: 10.1039/c0mt00073f. [DOI] [PubMed] [Google Scholar]
- Tully T, Cambiazo V, Kruse L. Memory through Metamorphosis in Normal and Mutant Drosophila. J Neurosci. 1994;14(1):68–74. doi: 10.1523/JNEUROSCI.14-01-00068.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully T, Quinn WG. Classical-Conditioning and Retention in Normal and Mutant Drosophila-Melanogaster. J Comp Physiol A. 1985;157(2):263–277. doi: 10.1007/BF01350033. [DOI] [PubMed] [Google Scholar]
- Turski ML, Thiele DJ. Drosophila Ctr1A functions as a copper transporter essential for development. J Biol Chem. 2007;282(33):24017–24026. doi: 10.1074/jbc.M703792200. [DOI] [PubMed] [Google Scholar]
- White AR, Multhaup G, Maher F, Bellingham S, Camakaris J, Zheng H, Bush AI, Beyreuther K, Masters CL, Cappai R. The Alzheimer's disease amyloid precursor protein modulates copper-induced toxicity and oxidative stress in primary neuronal cultures. J Neurosci. 1999;19(21):9170–9179. doi: 10.1523/JNEUROSCI.19-21-09170.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing YM, Higuchi K. Amyloid fibril proteins. Mech Ageing Dev. 2002;123(12):1625–1636. doi: 10.1016/s0047-6374(02)00098-2. [DOI] [PubMed] [Google Scholar]
- Yamamoto N, Arima H, Naruse K, Kasahara R, Taniura H, Hirate H, Sugiura T, Suzuki K, Sobue K. Ketamine reduces amyloid beta-protein degradation by suppressing neprilysin expression in primary cultured astrocytes. Neuroscience letters. 2013 doi: 10.1016/j.neulet.2013.04.016. [DOI] [PubMed] [Google Scholar]
- Yatin SM, Varadarajan S, Link CD, Butterfield DA. In vitro and in vivo oxidative stress associated with Alzheimer's amyloid beta-peptide (1–42) Neurobiol Aging. 1999;20(3):325–330. doi: 10.1016/s0197-4580(99)00056-1. [DOI] [PubMed] [Google Scholar]
- Yin H, Barnet RC, Miller RR. 2nd-Order Conditioning and Pavlovian Conditioned Inhibition - Operational Similarities and Differences. J Exp Psychol Anim B. 1994;20(4):419–428. [PubMed] [Google Scholar]
- Zhou H, Cadigan KM, Thiele DJ. A copper-regulated transporter required for copper acquisition, pigmentation, and specific stages of development in Drosophila melanogaster. J Biol Chem. 2003;278(48):48210–48218. doi: 10.1074/jbc.M309820200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.