

Abstract
Previously, pharmacological levels of insulin have been shown to stimulate the synthesis of normal corneal stromal collagen and proteoglycans by bovine keratocytes in culture. Here we compared insulin to physiological levels of IGF-I and found that IGF-I also stimulated the synthesis of these extracellular matrix components, but less than that of insulin. Keratocytes in monolayer culture secreted most of the collagen synthesized into the media in the form of procollagen, a precursor of collagen. We found that an overlay of 3% agarose on the keratocytes in culture enhanced the conversion of procollagen to collagen and increased the deposition of collagen and proteoglycans into the cell layer. The extracellular matrix associated with the keratocytes cultured under agarose exhibited a corneal stromal-like architecture. These results suggest that enhancing the conversion of procollagen to collagen is a key step in the formation of extracellular matrix by keratocytes in vitro. Agarose overlay of insulin-activated keratocytes in culture is a useful model for studying corneal stromal extracellular matrix assembly in vitro.
Introduction
The corneal stroma contains an extensive and transparent extracellular matrix that is interspersed with keratocytes. Transparency of this matrix requires uniformly small diameter collagen fibrils with constant inter-fibril spacing. The matrix is composed primarily of the fibrillar collagen types I and V (Birk et al. 1986), with lesser amounts of types VI (Cintron and Hong 1988), III (Cintron et al. 1988) and XII (Oh et al. 1993), and four leucine-rich type proteoglycans: three with keratan sulfate chains, lumican (Blochberger et al. 1992), keratocan (Corpuz et al. 1996), and osteoglycin/mimecan (Funderburgh et al. 1997), and one with a chondroitin sulfate chain, decorin (Li et al. 1992). In vitro studies have shown that collagen type V (Birk 2001) and the core proteins of the leucine-rich proteoglycans (Birk and Lande 1981; Silver and Birk 1983; Rada et al. 1993) can act to regulate collagen fibril growth and these findings have been confirmed in vivo in the Col5a1 haploinsufficent mouse (Segev et al. 2006; Wenstrup et al. 2006), in the lumican null mouse (Chakravarti et al. 1998; Chakravarti et al. 2006) and in the keratocan null mouse (Liu et al. 2003; Meek et al. 2003).
The keratocytes in the corneal stroma are responsible for making, maintaining and repairing the matrix. Keratocytes can be isolated from the stroma of rabbit and bovine corneas by collagenase digestion and cultured in serum free media where they maintain their normal dendritic morphology as well as other in vivo characteristics (Jester et al. 1996; Beales et al. 1999). The media supplement “ITS” and high levels of insulin, a component of “ITS”, have been shown to stimulate the proliferation of bovine keratocytes while maintaining their dendritic morphology and keratan sulfate proteoglycan synthesis (Musselmann et al. 2005). Furthermore, in the presence of ascorbic acid, a cofactor necessary for collagen triple helix stability, insulin also has been shown to increase the synthesis of collagen by 11-fold and lumican/keratocan by 2–3 fold (Musselmann et al. 2006). These studies suggest that insulin may act to maintain the normal keratocyte phenotype and that proteoglycan stability may be linked to collagen stability.
Insulin has a high affinity for its own receptor, but because it also has a low affinity for the IGF-I receptor, high levels of insulin also would activate keratocytes through IGF-IR as well [for review see (Lelbach et al. 2005)]. High levels of insulin would not normally be present in the corneal stroma, however IGF-II, another ligand for IGF-IR, is in the bovine corneal stroma and it causes bovine keratocytes to proliferate and maintain their normal phenotype in vitro (Musselmann et al. 2008). IGF-I has previously been shown to stimulate the proliferation of rabbit keratocytes in vitro while maintaining their dendritic morphology (Jester and Ho-Chang 2003).
In this study, we compare the effect of high levels of insulin and low levels of IGF-I on keratocyte proliferation and on the synthesis of collagen and proteoglycans by bovine keratocytes in culture. In addition, we also evaluate the effect of an agarose overlay on the processing of procollagen to collagen required for fibril formation and extracellular matrix assembly.
Materials and Methods
Reagents
All chemicals and growth factors were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise indicated. CyQuant kits, polyacrylamide gels, electrophoresis solutions, nitrocellulose, and DMEM/F12 were obtained from Invitrogen (Carlsbad, CA), Costar cell culture plates from Fisher Scientific (Suwanee, GA), Amicon 10,000 MWCO spin concentrators from Millipore Corporation (Bedford, MA), endo-β-galactosidase and chondroitinase ABC from Associates of Cape Cod (East Falmouth, MA) and ECL Western blotting analysis system from GE Healthcare (Piscataway, NJ)
Isolation and culture of keratocytes
Freshly harvested eyes from 1–2 year old cows were purchased from Pel Freeze (Rogers, AR) and shipped on wet ice by overnight delivery. The corneas were removed and the keratocytes isolated from the corneas by using two sequential collagenase digestions as previously described (Berryhill et al. 2001). The culture medium used throughout was DMEM/ F12 supplemented with antibiotics and 1 mM 2-phospho-L-ascorbic acid (DMEM/F12). Media was adjusted to contain 100,000 cells/ml and 2 mls were plated/well on day zero in each of 4 wells of a 6 well plate (~20,000-cells/cm2), except where specified when 2 mls were plated in each well of a 24 well plate (~100,000-cells/cm2). Plates were incubated overnight at 37 °C in a humidified atmosphere containing 5% CO2 to allow the cells to attach. The medium was changed on day 1 to DMEM/F12 or to DMEM/F12 containing either 10 ng IGF-I, 10 ng IGF-II or 10 μg insulin/ml. The media was also supplemented with 5 g of dextran (MR~40,000, Fluka, 31389)/100ml of media in one set of experiments. Media was removed and replaced with fresh media on days 4, 7, and 10 except where specified. Cultures were harvested for analysis on days 1, 4, 7, 10 and 13. Medium from 4 wells was removed and combined. The media and plates were stored at −80 °C.
Media containing 3% agarose (low melting-type, Type VII, Sigma, A9045) also was prepared and over-layered on keratocytes that previously had been plated in a 24 well plate and cultured in media containing insulin for days 1 through 4. Agarose (6g) was dissolved in 100 ml of distilled water by autoclaving, cooled to 37 °C. in a water bath and mixed with an equal volume of 2X DMEM containing 20 μg of insulin/ml that had been warmed to 37 °C. The resulting media containing 3% agarose was maintained at 37 °C. Media was removed from the 24 well plate containing keratocytes and 0.2 ml of the 3% agarose was layered on top of the cells in each well. The agarose was allowed to solidify at room temperature, 2 mls of media containing insulin was added to the well and the plates returned to the incubator. Cultures were harvested, without intervening media changes, on days 7, 10, and 13 or on days 8 and 13.
DNA measurements
The DNA content of the cells in each well was determined using a CyQuant kit and following the vendor’s instructions. Calf thymus DNA was used as a standard.
Pepsin digestion of media and cell layers
Harvested media combined from 4 wells (8 ml) was cooled on ice and adjusted to 0.5 M acetic acid by the addition of 230 μl of glacial acetic acid followed by the addition of 100 μl of a pepsin stock solution (4 mg pepsin/ml 0.5 M acetic acid). Cold 0.5 M acetic acid (2ml) containing 0.05 mg pepsin/ml was added to each well of 4 wells containing cells. Digestion of the media and cell layers was continued overnight at 4 °C. The digests of the cell layers were combined and any insoluble material was removed by centrifugation. The digests were adjusted to neutrality with NaOH, dialyzed against deionized water, lyophilized and reconstituted in 100 μl of sample buffer (Invitrogen).
Extraction of cell layers
Two mls of 0.5 M acetic acid or 250 μl of 4 M guanidine-HCl was added to each well containing cells and the cell layer extracted over night at 4 °C. The wells were scraped; the extracts from 4 wells were combined and any insoluble material pelleted by centrifugation. The supernatant was dialyzed against deionized water. The acetic acid extracts and the guanidine extracts were used in Western blots to detect procollagen type I and proteoglycans, respectively.
Concentration of media
Media samples were used directly for detection of procollagen type I by Western blot. Media samples used for SDS/PAGE Simply Blue staining and for detection of core proteins were first concentrated to 1/10th volume using spin concentrators from Amicon. Aliquots of media and guanidine extracts of the cell layers were digested with endo-β-galactosidase or chondroitinase ABC according to the vendor’s instructions for detection of lumican and keratocan core proteins or decorin core protein, respectively, by Western blot.
SDS/PAGE and Western Blot
The size of the aliquots taken from the media, the cell layer extracts and the pepsin digests for electrophoresis on polyacrylamide gels was normalized to the DNA content of those cultures or to the DNA content of parallel cultures. This made it possible to directly compare the amount of a particular protein produced by the same number of cells. All samples were reduced prior to electrophoresis and were electrophoresed according to the vendor’s instructions. Gels were stained for proteins using Simply Blue and Western blots were performed using the ECL Western blotting analysis system according to the vendor’s instructions. Primary antibodies included: rabbit anti-sera to bovine lumican and to bovine keratocan (Berryhill et al. 2001), mouse monoclonal anti-decorin (DS1, Hybridoma Bank, U. of Iowa) and mouse monoclonal anti-procollagen type I (SP1. D8, Hybridoma Bank, U. of Iowa).
Electron Microscopy
Cultured keratocytes were analyzed by transmission electron microscopy. Briefly, monolayers were fixed in 4% paraformaldehyde, 2.5% glutaraldehyde, 0.1 M sodium cacodylate pH 7.4, with 8.0 mM CaCl2 (Birk and Trelstad 1984), post-fixed with 1% osmium tetroxide and dehydrated in a graded ethanol series. The agarose layer was removed before the 100% ethanol step. After dehydration in an ethanol series, the cells were infiltrated and embedded in a mixture of EMbed 812, nadic methyl anhydride, dodecenyl succinic anhydride and DMP-30 (Electron Microscopy Sciences, Hatfield, PA.). Thin sections (90nm) were cut using a Reichert UCT ultramicrotome and post-stained with 1% aqueous uranyl acetate and 1% phosphotungstic acid, pH 3.2. The sections were analyzed using a JEOL 1400 transmission electron microscope at 80 kV and digital images were captured with a Gatan Orius camera.
Statistics
Statistical analysis was performed with four determined values for each point using Statview (SAS Institute, Cary, NC). Data are expressed as the mean + standard error. Significant differences were determined by paired t-test.
Results
The ability of insulin and IGF-1 to stimulate the proliferation of keratocytes in culture was evaluated over a 13-day period. Keratocytes were isolated from corneas and plated in DMEM/F12 on the same day. The media was changed the following day (day1) to media supplemented with 10 μg insulin/ml, 10 ng IGF-1/ml or un-supplemented media (control) and the DNA content of the cultures was determined on days 1, 4, 7, 10, and 13 of culture. Compared to controls, cultures supplemented with insulin or with IGF-1 contained significantly higher levels of DNA by day 4 of culture and essentially remained at those levels for the duration of the culture period (Figure 1).
Fig 1.

DNA content of keratocyte cultures. Keratocytes were cultured in media alone (Control/diamonds) or in media containing either IGF-I (IG/squares) or insulin (IN/triangles) and harvested on day 1, 4, 7, 10, and 13 of culture. Compared to control, cultures in media containing IGF-I or insulin contained significantly more DNA at the p< 0.016 and p< 0.005 levels respectively, for all time points.
Media from these cultures was changed every 3 days. The media harvested on days 7, 10, and 13 of culture was digested either with endo-β-galactosidase or with chondroitinase ABC and was evaluated either for keratocan and lumican content or for decorin content, respectively, by western blot using antibodies to the core proteins of these proteoglycans. The volume of media analyzed was normalized to the DNA content. The media from the IGF-1 cultures contained substantially higher levels of these proteoglycans than media from control cultures and, in most all instances, the media from the insulin cultures contained even higher levels than the media from IGF-1 cultures (Figure 2).
Fig 2.
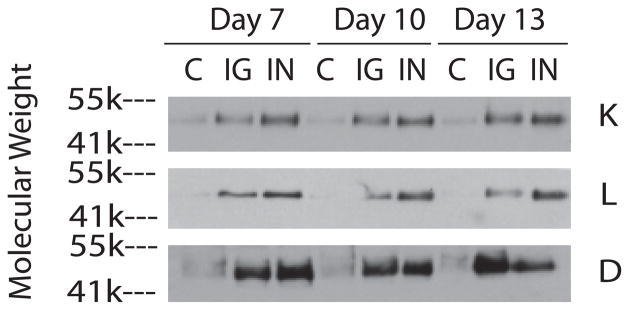
SDS/PAGE/Western blot of media from keratocyte cultures using antibodies to keratocan (K), lumican (L), and decorin (D). Keratocytes were cultured in media alone (C) or in media containing either IGF-I (IG) or insulin (IN). Media was harvested on days 7, 10, and 13 and then digested with either endo-β-galactosidase, for lumican and keratocan, or chondroinase ABC, for decorin, prior to electrophoresis.
Pepsin readily degrades globular proteins, but the triple helical domains of collagen are resistant to this protease. Aliquots of pepsin digested media and cell layers, normalized for DNA, harvested from the day 7, 10, and 13 time points were fractionated on SDS/PAGE and the collagenous proteins visualized by simply blue staining (Figure 3). Similar to that seen for the proteoglycans, the media (M) from the IGF-1 cultures contained substantially higher amounts of the α1(I), α2(I) and α1(V) chains of collagen than the control cultures and media from the days 10 and 13 insulin cultures contained slightly higher levels of these collagens than the IGF-1 cultures. Collagenous protein was not detected in the cell layer (CL) until day 10 and there was considerably less collagenous protein in the cell layer compared to that in the media, but by day 13 the same trend seen in the media was apparent in the cell layer: that is, more collagen in the IGF-1 cultures than controls and even more in the insulin cultures. These results demonstrate that most of the collagenous proteins synthesized were deposited in the media.
Fig 3.

SDS/PAGE/Blue stain of pepsin digests from keratocyte cultures. Keratocytes were cultured in media alone (C) or in media containing either IGF-I (IG) or insulin (IN). Cultures were harvested on day 7, 10, and 17. The media (M) and cell layers (CL) were digested with pepsin prior to electrophoresis.
We next determined if deposition of collagen with the cell layer could be enhanced by plating keratocytes at a higher density. The same number of cells (400,000) were plated per well in 6 and in 24 well plates to produce a five fold difference in plating density; ~20,000-cells/cm2 in 6 well plates (low density cultures) and ~100,000-cells/cm2 in 24 well plates(high density cultures). It was possible, however, to use the same amount (2mls) of media per well during subsequent culture for both cell densities because of the depth of the wells. One day after plating (day 1), there was 27% less DNA/well in the high density cultures than in the low density cultures which suggests that keratocytes may have higher attachment efficiencies at the lower plating density (Figure 4). The DNA content per well increased significantly during the subsequent 12 days of culture in both plates; ~2.1 fold increase in the low density cultures and ~1.7 fold increase in the high density cultures. Thus, during days 7–10 of culture, the cell density in the high density cultures was only ~2.6 fold higher than in the low density cultures. SDS/PAGE analyses of aliquots of pepsin digests from the cell layers, normalized to DNA content, demonstrated that the levels of collagenous proteins were slightly greater in cells at the higher density at all 3 time points (Figure 5).
Fig 4.

DNA content of keratocyte cultures. Keratocytes were plated at low density (6 well) or high density (24 well), cultured in media containing insulin starting on day 1 and harvested on days 1, 7, 10, and 13. Compared to day 1 cultures, the DNA content of cultures significantly increased for all subsequent time points whether plated at low density (p< 0.016) or high density (p< 0.003)
Fig 5.

SDS/PAGE/Blue stain of pepsin digested cell layers from keratocyte cultures. Keratocytes were plated at low density (Low) or high density (High), cultured in media containing insulin and harvested on days 7, 10, and 13. The cell layers were digested with pepsin prior to electrophoresis.
Type I collagen is initially synthesized as procollagen with N- and C- terminal globular domains. The procollagen globular domains are cleaved by specific proteases during post-translational processing and this allows the triple helical regions of collagen to laterally associate into fibrils [for recent review see (Canty and Kadler 2005)]. The processing of type I procollagen and subsequent assembly of collagen fibrils occurs quickly and completely in vivo. In contrast, cultured fibroblasts incompletely process procollagen to collagen and because procollagen cannot efficiently form normal fibrils, it accumulates in the culture medium (Goldberg et al. 1972). Consequently, we analyzed culture medium, normalized for DNA, from keratocytes harvested on day 10 for type I procollagen using a monoclonal antibody (Foellmer et al. 1983) specific for the N-terminal globular domain of α1(I) by western blot (Figure 6, lanes WB). Compared to controls, media from IGF-II cultures contained higher levels of type I procollagen. Furthermore, media from IGF-I cultures and from insulin cultures contained increasingly higher levels of type I procollagen, respectively. Staining SDS/PAGE of media from insulin cultures for protein showed that the major protein band in the media co-migrated with the major band detected by the antibody to the proα1(I) (Figure 6, lane CB).
Fig 6.

SDS/PAGE of media from keratocyte cultures. Keratocytes were cultured in media alone (C) or in media containing IGF-II (II), IGF-I (IG) or insulin (IN) and cultures were harvested on day 10. SDS/PAGE gels of the media were examined by Western blots with an antibody to procollagen type I (WB) or stained directly with blue stain (CB).
The addition of neutral polymers, such as dextran and polyethylene glycol, to the culture medium has been shown to increase the processing of procollagen to collagen by fibroblasts in culture and to cause an increase in the amount of collagen associated with the cell layer (Bateman et al. 1986; Bateman et al. 1987). We cultured insulin activated keratocytes in media supplemented with 5% dextran 40 and found that they deposited substantially lower levels of procollagen in the media and demonstrated increased levels of collagen associated with the cell layer (data not shown). However, the dextran treated keratocytes contained numerous large cytoplasmic vacuoles that were the predominant cytoplasmic feature (Figure 7a). These vacuoles were large and and found in large clusters. In contrast, the keratocytes cultured in the absence of dextran contained occasional vacuoles (Figure 7b). Organ culture of human corneas in media containing dextran also has been shown to produce vacuoles in keratocytes (Muller et al. 2001). However, these vacuoles were comparable in size, morphology and distribution to those seen in the untreated control keratocytes.
Fig 7.

Transmission electron micrographs of day 13 cultures treated with and without dextran. Large clusters of cytoplasmic vacuoles (v) were a characteristic feature of the dextran–treated cultures (a). The untreated cultures (b) contained occasional cytoplasmic vacuoles (v). (N) Keratocyte nucleus; (v) cytoplasmic vacuoles.
Encapsulation of collagenase isolated chondrocytes by suspension in media containing 2% agarose has been shown to enhance the deposition of pericellular matrix by the chondrocyte during subsequent culture (Buschmann et al. 1992; Chang et al. 1997; Dimicco et al. 2007). The pericellular matrix of chondrocytes contains collagen. Therefore, we hypothesized that culture of keratocytes in agarose would enhance procollagen processing and deposition in the cell layer. However, instead of encapsulation, keratocytes were first plated at high density (100,000-cells/cm2), cultured in media containing insulin for days 1–4 and then, on day 4 of culture, the media was removed and media containing molten 3% agarose was layered on top of the attached cell layer. After the agarose has set, 2 mls of media was added to the well above the agarose layer. Cultures were harvested on days 7, 10, and 13 of culture without intervening media replacement.
Cultures with and without an agarose overlay were evaluated for collagen content in the 2 different compartments. SDS/PAGE/Western blots with antibodies to procollagen type I, of DNA- normalized aliquots from the media and acetic acid extracts of the cell layers showed that the media of agarose over-layered cultures contained substantially less procollagen type I than the media of cultures without agarose (Figure 8). In contrast, the levels of type I procollagen in the cell layer extracts were far less than that in the media, although the extracts of cell layers over-layered with agarose contained higher levels of type I procollagen than the extracts of cell layers without agarose. SDS/PAGE analysis of DNA-normalized aliquots of pepsin digests of media and cell layers showed that compared to cultures without agarose, the agarose over-layered cultures contained substantially less collagenous protein in the media and also contained more collagenous protein in the cell layer at all 3 time points (Figure 9). Together, the results in Figures 8 and 9 indicate that the collagenous proteins detected in the media are procollagen while the collagenous proteins detected in the cell layer are collagen. These results also indicate that the agarose overlay enhances the processing of procollagen to collagen resulting in fibril formation and association with the cell layer.
Fig 8.

SDS/PAGE/western blots using antibodies to procollagen type I. Keratocytes were cultured in media containing insulin and were either overlayered with agarose (+) or were not (−). Cultures were harvested on days 7, 10 and 13. The media (M) and extracts of the cell layers (CL) from the cultures were evaluated for procollagen type I content by Western blot.
Fig 9.
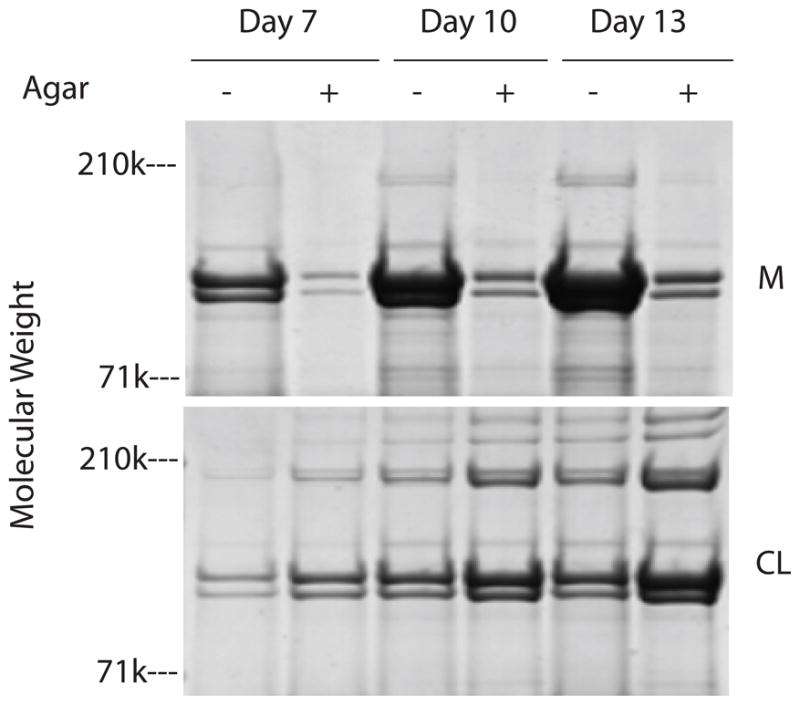
SDS/PAGE/Blue stain of pepsin digests of keratocyte cultures. Keratocytes were cultured in media containing insulin and were either overlayed with agarose (+) or were not (−). Cultures were harvested on days 7, 10, and 13. The media and cell layers were digested with pepsin prior to electrophoresis.
Cultures with and without an agarose overlay were harvested on days 8 and 13 and guanidine extracts of the cell layers were evaluated for lumican and keratocan content. DNA-normalized aliquots of the extracts were digested with endo-β-galactosidase and core protein levels were determined by western blot using antibodies to lumican and keratocan (Figure 10). Compared to cultures without agarose, cultures with agarose contained higher levels of lumican on day 13 and higher levels of keratocan on both days 8 and 13. These data indicate that the agarose overlay also enhances the incorporation of proteoglycans into the extracellular matrix.
Fig 10.

SDS/PAGE/Western blots of cell layer extracts of keratocyte cultures using antibodies to lumican (L) and keratocan (K). Keratocytes were cultured in media containing insulin and were either overlayered with agarose (+) or were not (−). Cultures were harvested on days 8 and 13. Cell layer extracts were digested with endo-β-galactosidase prior to electrophoresis
Cultured keratocytes both with and without an agarose overlay were fixed at day 13 for transmission electron microscopy (Figure 11). The keratocytes without an agarose overlay were arranged as monolayers. Keratocytes were associated with single and small groups of collagen fibrils. In contrast, the keratocyte cultures with the agarose overlay had a significant enhancement of collagen fibril deposition and also demonstrated stratification of the keratocytes. This was a major feature in large regions of the cultures with 2–4 layers of keratocytes. The regional nature coupled with the absence of keratocyte proliferation suggests the stratification resulted from reorganization and migration of keratocytes in response to the extracellular matrix deposited in the agarose overlay cultures. In addition, these cultures exhibited a more complex arrangement of keratocytes. The cells formed parallel spaced layers with numerous cytoplasmic processes delineating intercellular spaces. These spaces were filled with collagen fibrils displaying an orderly deposition of fibrils mimicking the cornea stroma-specific architecture, i.e., homogeneous small diameter fibrils with regular packing.
Fig. 11.

Transmission electron micrographs of day 13 cultures with and without an agarose overlay. Keratocyte were grown without agarose (a, b) or overlayed with agarose (c, d). Normal processing of procollagen in agarose overlay cultures results in corneal stroma-like matrix deposition. Keratocytes without agarose form monolayers with isolated collagen fibrils or small groups of fibrils (arrows). These are observed primarily associated with the keratocyte cell body. Keratocytes grown with an agarose overlay demonstrate stratification and the development of intercellular spaces defined by a series of complex cell processes. Keratocytes formed 2–4 layers separating lamellae-like structures containing abundant organized collagen fibrils. These spaces are filled with collagen fibrils that have small homogenous diameters and regular packing like that seen in the corneal stroma. The collagen fibrils are in both cross (wide arrows) and longitudinal (narrows arrows) comparable to that seen in the orthogonal stromal lamellae. Keratocytes without an agarose overlay appeared as sparse monolayers consistant with a dendritic morphology but with little matrix deposition and no stratification. (KN) keratocyte nucleus; (KP) keratocyte process.
Discussion
The results of this study show that pharmacological levels of insulin and physiological levels of IGF-I stimulated similar levels of cell proliferation, proteoglycan production and collagen synthesis by keratocytes in culture. The stimulation by insulin was, in most cases, slightly more than that of IGF-I. Insulin can bind to the receptors for IGF-I, but with much lower affinity (Lelbach et al. 2005). This suggests that the major pharmacological action of insulin could be through IGF-IR, but because the stimulation by insulin was greater than IGF-I, there may be additional actions of high levels of insulin on keratocytes, such as also activating the receptors for insulin. These data also indicate that like shown for pharmacological levels of insulin (Musselmann et al. 2005), physiological levels of IGF-I can stimulate proliferation and the synthesis of normal matrix components by keratocytes.
While insulin substantially increased collagen synthesis by keratocytes, as shown here and in a previous report (Musselmann et al. 2006), very little of the collagen made was deposited with the cell layer, i.e., assembled into fibrils. Increasing the length of time in culture and the initial plating density of the cells did, however, increase deposition of collagen with the cell layer. Most of the collagen was secreted into the media as procollagen, a precursor of collagen. The processing of procollagen to collagen by fibroblasts in culture was shown to be incomplete (Goldberg et al. 1972) and it is now apparent, from the data presented in this report, that keratocytes in culture also have this deficiency. We found, however, that culturing keratocytes under a layer of agarose enhanced the processing of procollagen to collagen, the assembly of collagen into fibrils and the deposition of fibrils and proteoglycans with the cell layer. We propose that the agarose provided a semi-permeable barrier that kept the secreted procollagen as well as the processing enzymes at or near the keratocytes surface leading to efficient processing to collagen and subsequent fibril and matrix assembly.
The processing of procollagen with subsequent fibril and matrix assembly in the cell layer associated with culture under an agarose overlay generated a corneal stroma-like architecture. We can speculate that the processing of procollagen and matrix deposition resulted in a substrate with which the keratocytes could interact. These keratocytes-matrix interactions favored the keratocytes phenotype and drove cornea-like organization. After the deposition of an extracellular matrix, the keratocytes became stratified as parallel oriented layers. It is probable that this keratocyte organization is due to the deposition of a stromal extracellular matrix that drives cell migration and organization. The intercellular spaces between the layers of keratocytes contain a well organized matrix. The collagen fibrils have small homogeneous diameters and are regularly packed. We suggest that this stroma-like architecture is generated by regulated assembly. This begins with normal procollagen processing and fibril formation involving collagen types I and V, and includes the interaction of the assembling fibrils with decorin, lumican and keratocan. These interactions would regulate the later stages in fibril formation as well as packing.
Chondrocytes in cell culture without agarose encapsulation readily synthesize procollagen type II and deposit it into the culture media (Govindraj et al. 2006). Encapsulation of chondrocytes in agarose was originally used to enhance the differentiation of chondrocytes in culture (Benya and Shaffer 1982) and was later shown to also promote the formation of the chondrocyte pericellular matrix, which would contain collagen (Buschmann et al. 1992; Chang et al. 1997; Dimicco et al. 2007). This would suggest that agarose encapsulation might be enhancing procollagen type II processing to collagen by chondrocytes in culture as well.
Agarose overlay differs from agarose encapsulation. The encapsulation method is done on unattached cells or clumps of cells in suspension and results in the formation of tiny spheres containing cells and their associated matrix. The overlay method described here is done on cells that are attached to a substrate. The keratocytes in this report are attached to the bottom of a culture dish and the overlay results in the formation of a sheet of cells and their associated matrix. Furthermore, with the overlay method, the thickness and diameter of the resulting sheet could be varied by the initial plating density and size of the culture dish.
The processing of procollagen to collagen is thought to take place on the plasma membrane or cell surface [for review see (Canty and Kadler 2005)]. Dextran and other high molecular weight neutral polymers have been shown to increase the processing of procollagen and its deposition with the cell layer in cell culture [this report and (Bateman et al. 1986; Bateman et al. 1987)]. These agents are thought to act by a volume exclusion mechanism to increase the concentration of macromolecules in a solution (Atha and Ingham 1981). Their presence would reduce the space that is available for large molecules, such as procollagen, but not for small molecules, such as salts and amino acids. Thus, for the processing of procollagen, the presence of these crowding agents in the media would increase the concentration of procollagen on the surface of the cells where the proteases are that clip off its globular domains. Agarose has not been considered to be a crowding agent but it is routinely used as a molecular sieve to separate proteins and nucleic acids according to their size. A layer of agarose over cells in culture would therefore act as a semi-permeable membrane to retard the diffusion of secreted macromolecules, such as procollagen, into the media and, as a result, the concentration of procollagen would increase on the surface of the cells where collagen processing takes place. The difference between an agarose overlay and dextran in solution is that the agarose, because it is insoluble, is a physical presence that would put some degree of confinement on the cell layer that could alter cell-cell interactions or cell movement.
Collagen binds to fibronectin and collagen fibril formation is facilitated by the presence of the integrin receptors for fibronectin, the presence of fibronectin and the formation of fibronectin fibrils [for review see (Canty and Kadler 2005)]. The agarose overlay also would act to increase the concentration of fibronectin on the cell surface and this could increase its assembly into fibrils as well as its binding to collagen and to its receptor. Type V collagen has been shown to nucleate collagen fibril formation and absence of type V collagen results in an embryonic lethal phenotype due to lack of fibril formation in the presence of normal type I collagen concentrations (Wenstrup et al. 2004). It was suggested that this nucleation involves a regulatory interaction at or near the cell surface involving a molecular complex. Integrin frequency and distribution on the plasma membrane of the cell may play a role in determining the architectural arrangement of an extracellular matrix by positioning these regulatory complexes. This agarose overlay method will likely be a useful tool for studying cell-specific extracellular matrix assembly in vitro.
Acknowledgments
Supported by Grants EY08104 and EY05129 from the National Institutes of Health
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Atha DH, Ingham KC. Mechanism of precipitation of proteins by polyethylene glycols. Analysis in terms of excluded volume. J Biol Chem. 1981;256:12108–12117. [PubMed] [Google Scholar]
- Bateman JF, Cole WG, Pillow JJ, Ramshaw JA. Induction of procollagen processing in fibroblast cultures by neutral polymers. J Biol Chem. 1986;261:4198–4203. [PubMed] [Google Scholar]
- Bateman JF, Pillow JJ, Mascara T, Medvedec S, Ramshaw JA, Cole WG. Cell-layer-associated proteolytic cleavage of the telopeptides of type I collagen in fibroblast culture. Biochem J. 1987;245:677–682. doi: 10.1042/bj2450677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beales MP, Funderburgh JL, Jester JV, Hassell JR. Proteoglycan synthesis by bovine keratocytes and corneal fibroblasts: maintenance of the keratocyte phenotype in culture. Invest Ophthalmol Vis Sci. 1999;40:1658–1663. [PubMed] [Google Scholar]
- Benya PD, Shaffer JD. Dedifferentiated chondrocytes reexpress the differentiated collagen phenotype when cultured in agarose gels. Cell. 1982;30:215–224. doi: 10.1016/0092-8674(82)90027-7. [DOI] [PubMed] [Google Scholar]
- Berryhill BL, Beales MP, Hassell JR. Production of prostaglandin D synthase as a keratan sulfate proteoglycan by cultured bovine keratocytes. Invest Ophthalmol Vis Sci. 2001;42:1201–1207. [PubMed] [Google Scholar]
- Birk DE. Type V collagen: heterotypic type I/V collagen interactions in the regulation of fibril assembly. Micron. 2001;32:223–237. doi: 10.1016/s0968-4328(00)00043-3. [DOI] [PubMed] [Google Scholar]
- Birk DE, Fitch JM, Linsenmayer TF. Organization of collagen types I and V in the embryonic chicken cornea. Invest Ophthalmol Vis Sci. 1986;27:1470–1477. [PubMed] [Google Scholar]
- Birk DE, Lande MA. Corneal and scleral collagen fiber formation in vitro. Biochim Biophys Acta. 1981;670:362–369. doi: 10.1016/0005-2795(81)90108-2. [DOI] [PubMed] [Google Scholar]
- Birk DE, Trelstad RL. Extracellular compartments in matrix morphogenesis: collagen fibril, bundle, and lamellar formation by corneal fibroblasts. J Cell Biol. 1984;99:2024–2033. doi: 10.1083/jcb.99.6.2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blochberger TC, Vergnes JP, Hempel J, Hassell JR. cDNA to chick lumican (corneal keratan sulfate proteoglycan) reveals homology to the small interstitial proteoglycan gene family and expression in muscle and intestine. J Biol Chem. 1992;267:347–352. [PubMed] [Google Scholar]
- Buschmann MD, Gluzband YA, Grodzinsky AJ, Kimura JH, Hunziker EB. Chondrocytes in agarose culture synthesize a mechanically functional extracellular matrix. J Orthop Res. 1992;10:745–758. doi: 10.1002/jor.1100100602. [DOI] [PubMed] [Google Scholar]
- Canty EG, Kadler KE. Procollagen trafficking, processing and fibrillogenesis. J Cell Sci. 2005;118:1341–1353. doi: 10.1242/jcs.01731. [DOI] [PubMed] [Google Scholar]
- Chakravarti S, Magnuson T, Lass JH, Jepsen KJ, LaMantia C, Carroll H. Lumican regulates collagen fibril assembly: skin fragility and corneal opacity in the absence of lumican. J Cell Biol. 1998;141:1277–1286. doi: 10.1083/jcb.141.5.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarti S, Zhang G, Chervoneva I, Roberts L, Birk DE. Collagen fibril assembly during postnatal development and dysfunctional regulation in the lumican-deficient murine cornea. Dev Dyn. 2006;235:2493–2506. doi: 10.1002/dvdy.20868. [DOI] [PubMed] [Google Scholar]
- Chang J, Nakajima H, Poole CA. Structural colocalisation of type VI collagen and fibronectin in agarose cultured chondrocytes and isolated chondrons extracted from adult canine tibial cartilage. J Anat. 1997;190 (Pt 4):523–532. doi: 10.1046/j.1469-7580.1997.19040523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cintron C, Hong BS. Heterogeneity of collagens in rabbit cornea: type VI collagen. Invest Ophthalmol Vis Sci. 1988;29:760–766. [PubMed] [Google Scholar]
- Cintron C, Hong BS, Covington HI, Macarak EJ. Heterogeneity of collagens in rabbit cornea: type III collagen. Invest Ophthalmol Vis Sci. 1988;29:767–775. [PubMed] [Google Scholar]
- Corpuz LM, Funderburgh JL, Funderburgh ML, Bottomley GS, Prakash S, Conrad GW. Molecular cloning and tissue distribution of keratocan. Bovine corneal keratan sulfate proteoglycan 37A. J Biol Chem. 1996;271:9759–9763. doi: 10.1074/jbc.271.16.9759. [DOI] [PubMed] [Google Scholar]
- Dimicco MA, Kisiday JD, Gong H, Grodzinsky AJ. Structure of pericellular matrix around agarose-embedded chondrocytes. Osteoarthritis Cartilage. 2007;15:1207–1216. doi: 10.1016/j.joca.2007.03.023. [DOI] [PubMed] [Google Scholar]
- Foellmer HG, Kawahara K, Madri JA, Furthmayr H, Timpl R, Tuderman L. A monoclonal antibody specific for the amino terminal cleavage site of procollagen type I. Eur J Biochem. 1983;134:183–189. doi: 10.1111/j.1432-1033.1983.tb07549.x. [DOI] [PubMed] [Google Scholar]
- Funderburgh JL, Corpuz LM, Roth MR, Funderburgh ML, Tasheva ES, Conrad GW. Mimecan, the 25-kDa corneal keratan sulfate proteoglycan, is a product of the gene producing osteoglycin. J Biol Chem. 1997;272:28089–28095. doi: 10.1074/jbc.272.44.28089. [DOI] [PubMed] [Google Scholar]
- Goldberg B, Epstein EH, Jr, Sherr CJ. Precursors of collagen secreted by cultured human fibroblasts. Proc Natl Acad Sci U S A. 1972;69:3655–3659. doi: 10.1073/pnas.69.12.3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindraj P, West L, Smith S, Hassell JR. Modulation of FGF-2 binding to chondrocytes from the developing growth plate by perlecan. Matrix Biol. 2006;25:232–239. doi: 10.1016/j.matbio.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Jester JV, Barry-Lane PA, Cavanagh HD, Petroll WM. Induction of alpha-smooth muscle actin expression and myofibroblast transformation in cultured corneal keratocytes. Cornea. 1996;15:505–516. [PubMed] [Google Scholar]
- Jester JV, Ho-Chang J. Modulation of cultured corneal keratocyte phenotype by growth factors/cytokines control in vitro contractility and extracellular matrix contraction. Exp Eye Res. 2003;77:581–592. doi: 10.1016/s0014-4835(03)00188-x. [DOI] [PubMed] [Google Scholar]
- Lelbach A, Muzes G, Feher J. The insulin-like growth factor system: IGFs, IGF-binding proteins and IGFBP-proteases. Acta Physiol Hung. 2005;92:97–107. doi: 10.1556/APhysiol.92.2005.2.1. [DOI] [PubMed] [Google Scholar]
- Li W, Vergnes JP, Cornuet PK, Hassell JR. cDNA clone to chick corneal chondroitin/dermatan sulfate proteoglycan reveals identity to decorin. Arch Biochem Biophys. 1992;296:190–197. doi: 10.1016/0003-9861(92)90562-b. [DOI] [PubMed] [Google Scholar]
- Liu CY, Birk DE, Hassell JR, Kane B, Kao WW. Keratocan-deficient mice display alterations in corneal structure. J Biol Chem. 2003;278:21672–21677. doi: 10.1074/jbc.M301169200. [DOI] [PubMed] [Google Scholar]
- Meek KM, Quantock AJ, Boote C, Liu CY, Kao WW. An X-ray scattering investigation of corneal structure in keratocan-deficient mice. Matrix Biol. 2003;22:467–475. doi: 10.1016/s0945-053x(03)00081-7. [DOI] [PubMed] [Google Scholar]
- Muller LJ, Pels E, Vrensen GF. The effects of organ-culture on the density of keratocytes and collagen fibers in human corneas. Cornea. 2001;20:86–95. doi: 10.1097/00003226-200101000-00017. [DOI] [PubMed] [Google Scholar]
- Musselmann K, Alexandrou B, Kane B, Hassell JR. Maintenance of the keratocyte phenotype during cell proliferation stimulated by insulin. J Biol Chem. 2005;280:32634–32639. doi: 10.1074/jbc.M504724200. [DOI] [PubMed] [Google Scholar]
- Musselmann K, Kane B, Alexandrou B, Hassell JR. Stimulation of collagen synthesis by insulin and proteoglycan accumulation by ascorbate in bovine keratocytes in vitro. Invest Ophthalmol Vis Sci. 2006;47:5260–5266. doi: 10.1167/iovs.06-0612. [DOI] [PubMed] [Google Scholar]
- Musselmann K, Kane BP, Alexandrou B, Hassell JR. IGF-II is present in bovine corneal stroma and activates keratocytes to proliferate in vitro. Exp Eye Res. 2008;86:506–511. doi: 10.1016/j.exer.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SP, Griffith CM, Hay ED, Olsen BR. Tissue-specific expression of type XII collagen during mouse embryonic development. Dev Dyn. 1993;196:37–46. doi: 10.1002/aja.1001960105. [DOI] [PubMed] [Google Scholar]
- Rada JA, Cornuet PK, Hassell JR. Regulation of corneal collagen fibrillogenesis in vitro by corneal proteoglycan (lumican and decorin) core proteins. Exp Eye Res. 1993;56:635–648. doi: 10.1006/exer.1993.1081. [DOI] [PubMed] [Google Scholar]
- Segev F, Heon E, Cole WG, Wenstrup RJ, Young F, Slomovic AR, Rootman DS, Whitaker-Menezes D, Chervoneva I, Birk DE. Structural abnormalities of the cornea and lid resulting from collagen V mutations. Invest Ophthalmol Vis Sci. 2006;47:565–573. doi: 10.1167/iovs.05-0771. [DOI] [PubMed] [Google Scholar]
- Silver FH, Birk DE. Kinetic analysis of collagen fibrillogenesis: I. Use of turbidity--time data. Coll Relat Res. 1983;3:393–405. doi: 10.1016/s0174-173x(83)80020-x. [DOI] [PubMed] [Google Scholar]
- Wenstrup RJ, Florer JB, Brunskill EW, Bell SM, Chervoneva I, Birk DE. Type V collagen controls the initiation of collagen fibril assembly. J Biol Chem. 2004;279:53331–53337. doi: 10.1074/jbc.M409622200. [DOI] [PubMed] [Google Scholar]
- Wenstrup RJ, Florer JB, Davidson JM, Phillips CL, Pfeiffer BJ, Menezes DW, Chervoneva I, Birk DE. Murine model of the Ehlers-Danlos syndrome. col5a1 haploinsufficiency disrupts collagen fibril assembly at multiple stages. J Biol Chem. 2006;281:12888–12895. doi: 10.1074/jbc.M511528200. [DOI] [PubMed] [Google Scholar]