

Bafilomycin B1 is the archetypal compound of the plecomacrolide family of natural products, which include concanamycins, viranamycins, hygrolidin, elaiophylin, and allied metabolites.1 This compound was first isolated from Streptomyces griseus sp. sulphurus (TÜ 1922) in 1984.2 The term plecomacrolide was later introduced to describe a subgroup of polyketides featured with a 16- or 18-membered macrolactone containing two conjugated diene units, and a hemiacetal side chain.1 These compounds have now been studied extensively as vacuolar ATPase (V-ATPase) inhibitors and, as such, have shown some promise as anti-osteoporetics.3,4 In addition, synergistic antifungal activity of bafilomycin A1 with the calcineurin inhibitor FK506 against the life-threatening fungal pathogen Cryptococcus neoformans,5 inhibition of the release of β-amyloid by bafilomycin A1,6 together with antitumor,7 antiparasitic,8 and immunosuppressant9 activities of other bafilomycins were reported. Despite continued efforts, generalized toxicity has kept these compounds from clinical application. Semi-synthetic derivatives have provided some indication that target specificity can be enhanced.10 However, extensive manipulation of the core structure for structure-activity relationship studies remains hampered by the complexity of total synthesis.11,12 Decades of work in the field of polyketide biosynthesis have yielded tools to generate subtle changes to the core macrolide via genetic manipulation.13,14 In order to utilize this knowledge to generate novel analogues, the native biosynthetic gene cluster sequence must be elucidated. When a bafilomycin-producing organism, Streptomyces lohii, was isolated during an ongoing drug discovery program in our laboratories, the characterization of the bafilomycin biosynthetic pathway was initiated.
To isolate the genes involved in bafilomycin biosynthesis, a fosmid library was constructed from S. lohii (ATCC BAA-1276, JCM 14114) genomic DNA. This library was probed for type I polyketide ketosynthase (KS) domains using a radiolabelled PCR product amplified from S. lohii gDNA using degenerate primers. Colony hybridization experiments revealed 12 KS positive fosmids. Initial restriction digestions and Southern hybridization experiments revealed two major groups of overlapping fosmids. One clone from each group was chosen for further analysis. BLAST analysis15 of the sequence from a small number of subclones suggested that one of these representatives contained only type I polyketide synthase (PKS) genes. Complete sequencing of this clone revealed the domain organization of the PKS modules to exactly match with a section of the genetic architecture predicted by the structure of bafilomycin and the typical polyketide rules of colinearity. End sequencing of the other KS positive fosmids identified those containing overlapping sequence. These were also sequenced to generate a map of the entire gene cluster.
The macrolactone core of bafilomycin is assembled by a type I PKS system. These genes are arranged as five open reading frames, bafAI – bafAV, spanning 59 kilobases (Scheme 1, Table 1). The domain organization of the PKS modules exactly follows the model suggested by the structure of the core macrolactone and the PKS pattern of colinearity. In addition, the acyltransferase (AT) domains 2 and 6 contain the signature motifs associated with the incorporation of malonate.16 Modules 5 and 11 contain the residues that have been predicted to specify the selection of methoxymalonate,17,18 while the remaining extender modules are specific for methylmalonate,16 again matching the predicted motifs of the bafilomycin structure (see Supporting Information). The AT domain of the loading module was expected to be specific for the incorporation of isobutyrate, suggested by the metabolite structure and confirmed by precursor feeding studies.19 This domain aligns most closely with the isobutyrate loading domains from the lipomycin20, 21 and tautomycin22 pathways. Downstream of the core open reading frames is bafH, a PKS type II thioesterase (TEII). Homologues of this enzyme have been found in a number of PKS clusters and are known to possess an editing role during biosynthesis.23-25
Scheme 1.

A) The organization of open reading frames in the bafilomycin biosynthetic gene cluster. B) The domain organization of the type I polyketide synthases encoded by ORFs BafAI through BafAV.
Table 1.
Predicted Functions of the Open Reading Frames Show in Scheme 1.
| Protein | Amino Acids | Putative Function | Nearest Homolog (Enzyme, Origin) | Identity/Similarity | Accession Number |
|---|---|---|---|---|---|
| BafX | 528 | acyl CoA ligase | MoeA4 [Streptomyces ghanaensis] | 72%/83% | ZP_06574361.1 |
| BafY | 514 | amide synthetase | Amide synthetase[Streptomyces tsukubaensis] | 78%/85% | ZP_10067139.1 |
| BafZ | 414 | 5-aminolevulinic acid synthase | aminolevulinate synthase [Streptomyces nodosus subsp. asukaensis] | 73%/83% | AAO62615.1 |
| BafAI | 4884 | PKS loading module + modules 1-3 | polyketide synthase [Streptomyces neyagawaensis] | 62%/70% | AAZ94387.1 |
| BafAII | 5145 | PKS modules 4-6 | BFAS2 [Streptomyces tsukubaensis] | 78%/83% | ZP_10067132.1 |
| BafAIII | 3968 | PKS modules 7,8 | BFAS2 [Streptomyces tsukubaensis] | 79%/85% | ZP_10067131.1 |
| BafAIV | 3511 | PKS modules 9,10 | polyketide synthase [Streptomyces scabiei] | 62%/73% | YP_003493865.1 |
| BafAV | 2103 | PKS module 11 + Thioesterase | polyketide synthase [Streptomyces clavuligerus] | 61%/69% | ZP_06769488.1 |
| BafB | 296 | glyceryl-ACP oxidase | methoxymalonate biosynthesis protein [Streptomyces tsukubaensis] | 72%/81% | ZP_10067126.1 |
| BafC | 93 | acyl carrier protein (ACP) | GalJ [Streptomyces galbus] | 69%/80% | ADE22334.1 |
| BafD | 363 | acyl-ACP dehydrogenase | methoxymalonate biosynthesis protein [Streptomyces tsukubaensis] | 85%/93% | ZP_10067124.1 |
| BafE | 365 | glycerate ACP ligase | methoxymalonate biosynthesis protein [Streptomyces tsukubaensis] | 83%/90% | ZP_10067123.1| |
| BafF | 220 | O-methyl transferase | O-methyltransferase mdmC [Streptomyces clavuligerus] | 73%/81% | ZP_05008783.1 |
| BafG | 609 | AfsR homolog | putative regulator [Streptomyces neyagawaensis] | 54%/64% | AAZ94408.1 |
| BafH | 253 | TEII | thioesterase [Streptomyces auratus] | 65%/76% | ZP_10551360.1 |
| ORF1 | 117 | LuxR homolog | LuxR family transcriptional regulator [Streptomyces tsukubaensis] | 65%/73% | ZP_10067119.1 |
| ORF2 | 320 | Putative Malonyl transferase | acyl-carrier-protein S-malonyltransferase [Streptomyces tsukubaensis] | 84%/90% | ZP_10067118.1 |
| ORF3 | 332 | Putative CoA ligase | hypothetical protein [Streptomyces neyagawaensis] | 74%/83% | AAZ94384.1 |
The identity of this gene cluster was established by targeted disruption of bafilomycin production using the REDIRECT system.26 When bafAIII was replaced in-frame by the apramycin resistance cassette (genotype confirmed by PCR analysis, see Supporting Information), the mutant strain of S. lohii failed to produce bafilomycins, as demonstrated by HPLC analysis of culture extracts (Figure 1).
Figure 1.
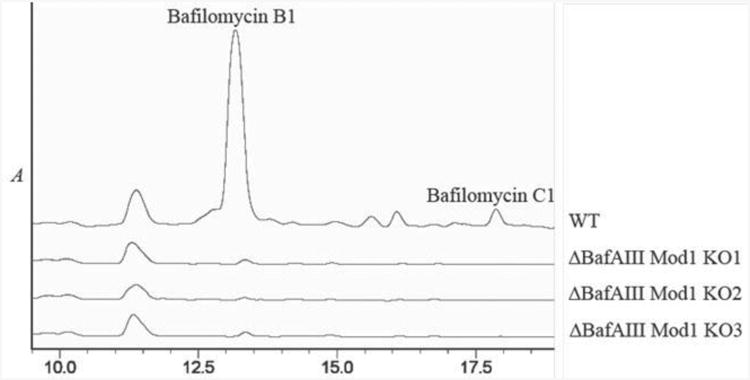
HPLC chromatograms of extracts of wild-type S. lohii compared to three separate bafAIII gene-disruption mutants.
The core macrolactone bafilomycin A1, presumably formed upon the activity of the TE domain in BafAV, acquires enhanced bioactivity by the addition of a fumarate moiety to the hydoxyl group on C21 to form bafilomycin C1.2 Further potency is provided by a 2-amino-3-hydroxy-cyclopent-2-enone (C5N) ring attached to the pendant fumaryl group via an amide bond formed with the distal carboxylate resulting in the conversion to bafilomycin B1 (Scheme 1).
Notably, this C5N moiety, present in a variety of secondary metabolites,27 often serves as a hydrogen bond donor/accepter pharmacophore. Its biosynthesis has long been proposed to be derived from 5-aminolevulinic acid (5-ALA) via an unusual cyclization process. This putative transformation was recently confirmed in the ECO-02301 pathway (Scheme 2).28 First, the pyridoxal 5′-phosphate (PLP) dependent 5-ALA synthase (ALAS) Orf34 catalyzes the condensation of succinyl-CoA and glycine to yield 5-ALA, which is subsequently activated by the acyl-CoA ligase (ACL) Orf35 to form 5-ALA-CoA. Next, Orf34 acts again to cyclize 5-ALA-CoA to afford C5N, avoiding the non-enzymatic cyclization to the 6-membered ring 2, 5-piperidinedione. Finally, the ATP-dependent amide synthetase (AMS) Orf33 is responsible for linking C5N to the carboxyl group. Interestingly, this enzyme trio is similarly encoded by a set of three genes located at the 5′ end of the bafilomycin gene cluster, wherein BafX, BafY, and BafZ are 62/72%, 40/55%, 68/80% identical/similar to Orf35, Orf33, and Orf34, respectively, at the protein level. This strongly suggests that the conversion from bafilomycin C1 to B1 is likely mediated sequentially by BafZ→BafX→BafZ→BafY (Scheme 2).
Scheme 2.

Putative pathway for 5-ALA biosynthesis and attachment.
To provide evidence for this proposed tailoring pathway, the activity of BafZ was characterized in vitro. In addition, this confirmed the proposed functionality of BafZ and validated biochemically the identity of the bafilomycin cluster, It is also expected that BafZ might be useful in industrial fermentation to overproduce 5-ALA, which has been widely used as a photodynamic medicine to treat various cancers, a selective biodegradable herbicide or insecticide, and a precursor for the production of heme-containing enzymes, porphyrin, and vitamin B12.29, 30 According to the similarity between BafZ and Orf34, the BafZ catalyzed condensation should be classified as the C4 pathway (the Shemin pathway) for 5-ALA biosynthesis, which does not exist in Escherichia coli (the normally selected industrial strain).30 Thus, the introduction of the bafZ gene into E. coli would likely supplement its native C5 pathway of 5-ALA, thereby enhancing the overall production of 5-ALA. The similar strategy using the ALAS gene hemA from Rhodobacter sphaeroides has already achieved significant improvement of 5-ALA productivity.30, 31
To characterise the in vitro activity of BafZ, its encoding gene bafZ was PCR amplified from S. lohii and cloned into the expression vector pACYCDuet-1. The protein was overexpressed in E. coli BL21 (DE3) and purified using Ni-NTA column chromatography (see Supporting Information). Upon incubation of purified BafZ (35 μM) with substrates glycine (5 mM) and succinyl-CoA (1 mM) at 28 °C for 2 h, significantly formation of a new compound with retention time and molecular weight (obs. 132.0658; calc. 132.0655) matching an authentic standard of 5-ALA was detected during HR-LCMS analysis (Figure 2).
Figure 2.
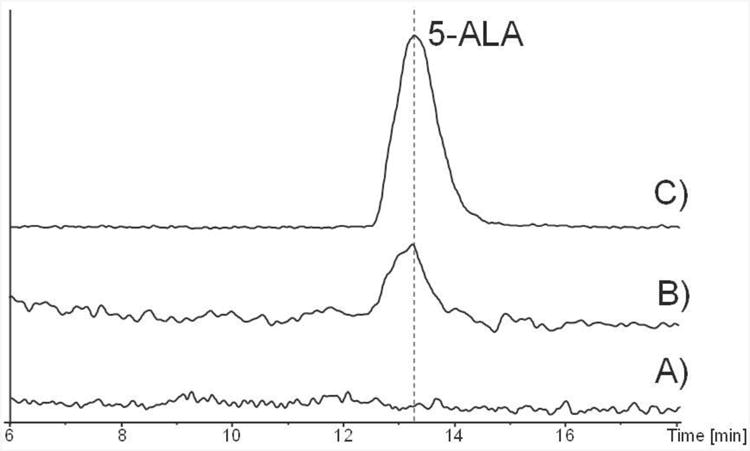
Extracted ion chromatograms (m/z=132.06). A) the negative control with boiled BafZ added to the reaction; B) the BafZ-catalyzed synthesis of 5-ALA from succinyl-CoA and glycine; C) 5-ALA authentic standard. The high resolution mass spectrum of 5-ALA is shown in Supporting Information.
In sequencing >15kb upstream and downstream of the defined baf cluster, no additional ORFs were identified to encode an enzyme catalyzing attachment of the fumarate moiety. Clustering of all genes involved in the biosynthesis of a given product is the paradigm for Streptomyces secondary metabolite production. However, separation of the 2-amino-3-hydroxy-cyclopent-2-enone production genes from those involved in the production of the core polysaccharide in the moenomycin pathway illustrates one of several exceptions to this rule.32 Therefore, this could be the case with the bafilomycin cluster in S. lohii. Alternatively, it is possible that the amide synthetase BafY also catalyzes the ester bond formation between fumarate and bafilomycin A1, which is supported by the recent report that a non-ribosomal peptide synthetase condensation enzyme SgcC5 is capable of catalyzing both amide and ester bond formation.33 To test these hypotheses, additional work will be necessary, and is currently ongoing in our laboratories.
The incorporation of glycerol-derived precursors has been inferred from feeding studies conducted more than twenty years ago.34 Recently, a number of biosynthetic gene clusters utilizing the glycerol-derived methoxymalonate extender molecule have been sequenced. Associated with these clusters is an operon responsible for the biosynthesis of this uncommon extender unit.17, 35, 36 Downstream of bafAI-AV are bafB-F, five open reading frames predicted to encode enzymes responsible for methoxymalonate biosynthesis (Table 1). BafC is a discrete acyl carrier protein that is loaded with glycerate, presumably oxidized from glycerol during primary metabolism by the enzyme BafE. The subsequent steps in the generation of methoxymalonate are each proposed to be catalyzed as a BafC bound intermediate. First, BafB is proposed to oxidize the 3-hydroxy group into an aldehyde, which is subsequently converted to a carboxylic acid. This conversion is catalyzed by BafD, an acyl-ACP dehydrogenase.37 Finally, the α-hydroxyl group is methylated by BafF, a putative O-methyltransferase. While following a genetic organization conserved within several pathways, each of these genes is most closely related to their discontinuously distributed counterparts found in the concanamycin biosynthetic gene cluster from Streptomyces neyagawaensis ATCC 27449.17
Based on the results of feeding studies, Schumann et al. have proposed that methoxymalonate is selected by specific AT domains directly from the dedicated ACP (BafC).19 Specific protein interaction was inferred due to low incorporation of an N-acetylcysteamine thioester (SNAC) of methoxymalonate. However, it is possible that methoxymalonate is transferred to coenzyme A, which then acts as the substrate for the AT. This scenario is more consistent with the classical mechanisms of PKS systems. The enzyme encoded by ORF3 could possibly fulfill this function. It is predicted to be a CoA ligase and shares high homology with an enzyme encoded as part of the concanamycin biosynthetic gene cluster.17
Downstream of the biosynthesis ORFs are a number of genes encoding putative regulatory elements. BafG is a homologue of AfsR, which has been shown to be a pleiotropic transcriptional activator involved in the regulation of secondary metabolite biosynthesis in the model organism Streptomyces coelicolor.38 Homologues of this protein are associated with a number of secondary metabolite biosynthesis pathways within the Streptomyces genus including the closely related concanamycin pathway.17, 39 Orf1 encodes a member of the LuxR family of transcriptional repressors. Repression is relieved by the binding of a quorum-sensing molecule, most often a homoserine lactone.40 LuxR family proteins are associated with a number of secondary metabolite biosynthesis clusters, including the concanamycin system.17
In summary, sequencing and analysis of the bafilomycin biosynthetic genes represents a novel 16-membered plecomacrolide pathway identified after the elaiophylin gene cluster.41 Recently, the discovery of more bafilomycin derivatives42-44 from diverse Streptomyces strains and additional putative bafilomycin biosynthetic genes45 suggests broad distribution of this biosynthetic system. Thus, the current work provides an important reference for identification and analysis of analogous gene clusters, and crucial information for the future engineered biosynthesis of bafilomycin analogues. Moreover, the in vitro characterization of BafZ expands the pool of 5-ALA synthases, which might benefit industrial production of 5-ALA.
Experimental Section
Genomic DNA was isolated from S. lohii using standard Streptomyces protocols.46 A genomic library was constructed in E. coli Epi300 using a Fosmid (Epicentre) vector. Initial candidates were selected from a library of 1152 clones by probing with a radiolabelled ketosynthase gene fragment. This probe was a heterologous gene fragment generated by PCR using degenerate KS targeting primers 4UU and 5LL (see Supporting Information) and S. lohii gDNA as the template.
Sequence was assembled using Seqman software (DNAStar). Translation analysis was carried out using the MacVector open reading frame analysis function. Functional assignment was accomplished by BLAST analysis15 against amino acid sequences contained within the NCBI and EMBL databases. Polyketide synthase domain organization was facilitated by the PKS/NRPS database maintained by the Indian National Institute of Immunology.47
Gene disruption in S. lohii was performed using a modified version of the REDIRECT system.26 A suicide knockout vector was created using fosmid 8D3 from the cluster sequencing effort by replacing the gene of interest with a selectable disruption cassette. This fosmid was transferred by interspecies conjugation from E. coli ET12567 into Streptomyces. Genetic confirmation of the disruption was accomplished by PCR amplification using genomic DNA as template.
Metabolite profiling of the genetically confirmed knock out strains and one wild-type strain was accomplished by extracting 12 ml cultures grown for 72 hours. The cultures were centrifuged to remove cells and the supernatant was extracted twice with an equal volume of dichloromethane. The organic extracts from each culture were pooled and dried in vacuo, and redissolved in DMSO. Extracts were analyzed by LC-MS on a C-18 column eluted with a gradient of acetonitrile in water with 0.1% formic acid. The mass and retention times were validated by comparison to an authentic standard purchased from Sigma Aldrich.
The bafZ gene was amplified from gDNA of S. lohii using the primers as follow: forward primer, cgcggatccgatgaaccattacctggagctcttc tca (the italic letters indicate the BamHI site); reverse primer, caatgaagctttcagccgtccgttccgggcgacccgtg (the underlined letters indicate the HindIII site), and inserted into pACYCDuet-1 (Novagen) between the restriction sites of BamHI and HindIII to afford the expression vector pACYCDuet-1-bafZ. To prepare BafZ protein, E.coli BL21 (DE3) cells carrying pACYCDuet-1-bafZ were grown at 37 °C in LB medium containing 25 μg/mL of chloramphenicol until OD600 reached ∼0.4. Then, isopropyl-β-D-thiogalactopyranoside (IPTG) was added to the final concentration of 0.1 mM to initiate the protein overexpression. The cells were cultured at 16 °C for another 16 h, collected by centrifugation (5000 g, 10 min, 4 °C), resuspended in 30 mL of lysis buffer (20 mM HEPES, pH 8.0, 0.5 M NaCl, and 5 mM imidazole), and lysed by sonication on ice. After a high-speed centrifugation (40,000 g, 30 min, 4 °C) to remove the cell debris, 1 mL Ni-NTA agarose resin (Qiagen) was added to the supernatant, and mixed at 4 °C for 1 h. Next, the resin with bound proteins was loaded onto a gravity flow column, washed with 50 mL wash buffer (20 mM HEPES, pH 8.0, 0.5 M NaCl, and 20 mM imidazole), and eluted by elution buffer (20 mM HEPES, pH 8.0, 0.5 M NaCl, and 250 mM imidazole). The purified BafZ was buffer exchanged into desalting buffer (50 mM NaH2PO4, 10% glycerol) using the PD-10 column (GE Healthcare). The final proteins were flash frozen in liquid nitrogen and stored at -80 °C for the following use.
The in vitro BafZ assay was performed by following Zhang et al.28 Briefly, 35 μM of purified BafZ was incubated with 5 mM glycine and 1 mM succinyl-CoA in a 100 μl reaction at 28 °C for 2 h; 100 μl of methanol was added to stop the reaction. After removal of the precipitated proteins by centrifugation, the supernatant was subjected to HR-LCMS analysis (Dionex Ultimate 3000 with Bruker maxis Q-TOF) using a BEH Amide column (2.1 mm*100 mm, 1.7 μm, Waters). Acetonitrile (solvent B) and water/5% acetonitrile (solvent A) were used as the mobile phases with a flow rate of 0.20 ml/min. The program was as follow: 90-70% B in A over 10 min, 70% B for 5 min, 70-90% B over 1 min, 90% B for 4 min.
Additional experimental details are available in the Supporting Information. The nucleotide sequence reported within have been deposited in GenBank under the accession number GU390405.
Supplementary Material
Acknowledgments
This work was supported by funding from “Recruitment Program of Global Experts, 2012” (S. Li) and NIH grant GM076477 (D. H. Sherman). JLF was supported in part by a NIGMS Biotechnology training grant. We thank Drs. Amy Wright and Peter McCarthy for their efforts during the early stages of this project. The authors are grateful to PNG BioNet and the University of Papua New Guinea Department of the Environment and Conservation for permission to collect research samples.
Footnotes
Supporting information for this article is available
Contributor Information
Professor David H. Sherman, Email: davidhs@umich.edu.
Professor Shengying Li, Email: lishengying@qibebt.ac.cn.
References
- 1.Bindseil KU, Zeeck A. Leibigs Ann Chem. 1994:305–312. [Google Scholar]
- 2.Werner G, Hagenmaier H, Drautz H, Baumgartner A, Zahner H. J Antibiot. 1984;37:110–117. doi: 10.7164/antibiotics.37.110. [DOI] [PubMed] [Google Scholar]
- 3.Farina C, Gagliardi S. Drug Disc Today. 1999;4:163–172. doi: 10.1016/S1359-6446(99)01321-5. [DOI] [PubMed] [Google Scholar]
- 4.Bowman EJ, Siebers A, Altendorf K. Proc Natl Acad Sci USA. 1988;85:7972–7976. doi: 10.1073/pnas.85.21.7972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Del Poeta M, Cruz MC, Cardenas ME, Perfect JR, Heitman J. Antimicrob Agents Chemother. 2000;44:739–746. doi: 10.1128/aac.44.3.739-746.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Knops J, Suomensaari S, Lee M, McCologue L, Seubert P, Sinha S. J Biol Chem. 1995;270:2419–2422. doi: 10.1074/jbc.270.6.2419. [DOI] [PubMed] [Google Scholar]
- 7.Wilton JH, Hokanson GC, French JC. J Antibiot. 1985;38:1449–1452. doi: 10.7164/antibiotics.38.1449. [DOI] [PubMed] [Google Scholar]
- 8.Goetz MA, McCormick PA, Monaghan RL, Ostlind DA. J Antibiot. 1985;38:161–168. doi: 10.7164/antibiotics.38.161. [DOI] [PubMed] [Google Scholar]
- 9.Vanek Z, Mateju J, Curdova E. Folia Microbiol. 1991;36:99–111. doi: 10.1007/BF02814487. [DOI] [PubMed] [Google Scholar]
- 10.Farina C, Gagliardi S. Curr Pharm Des. 2002;8:2033–2048. doi: 10.2174/1381612023393369. [DOI] [PubMed] [Google Scholar]
- 11.Toshima K, Jyojima T, Yamaguchi H, Noguchi Y, Yoshida T, Murase H, Nakata M, Matsumura S. J Org Chem. 1997;62:3271–3284. doi: 10.1021/jo970314d. [DOI] [PubMed] [Google Scholar]
- 12.Scheidt KA, Bannister TD, Tasaka A, Wendt MD, Savall BM, Fegley GJ, Roush WR. J Am Chem Soc. 2002;124:6981–6990. doi: 10.1021/ja017885e. [DOI] [PubMed] [Google Scholar]
- 13.Kittendorf JD, Sherman DH. Curr Opin Biotechnol. 2006;17:597–605. doi: 10.1016/j.copbio.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 14.Weissman KJ, Leadlay PF. Nat Rev Microbiol. 2005;3:925–936. doi: 10.1038/nrmicro1287. [DOI] [PubMed] [Google Scholar]
- 15.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 16.Haydock SF, Aparicio JF, Molnar I, Schwecke T, Khaw LE, Konig A, Marsden AFA, Galloway IS, Staunton J, Leadlay PF. FEBS Lett. 1995;374:246–248. doi: 10.1016/0014-5793(95)01119-y. [DOI] [PubMed] [Google Scholar]
- 17.Haydock SF, Appleyard AN, Mironenko T, Lester J, Scott N, Leadlay PF. Microbiol. 2005;151:3161–3169. doi: 10.1099/mic.0.28194-0. [DOI] [PubMed] [Google Scholar]
- 18.Del Vecchio F, Petkovic H, Kendrew SG, Low L, Wilkinson B, Lill R, Cortés J, Rudd BAM, Staunton J, Leadlay PF. J Ind Microbiol Biotechnol. 2003;30:489–494. doi: 10.1007/s10295-003-0062-0. [DOI] [PubMed] [Google Scholar]
- 19.Schumann T, Grond S. J Antibiot. 2004;57:655–661. doi: 10.7164/antibiotics.57.655. [DOI] [PubMed] [Google Scholar]
- 20.Miao V, Brost R, Chapple J, She K, Coëffet-Le Gal MF, Baltz RH. J Ind Microbiol Biotechnol. 2006;33:129–140. doi: 10.1007/s10295-005-0028-5. [DOI] [PubMed] [Google Scholar]
- 21.Bihlmaier C, Welle E, Hofmann C, Welzel K, Vente A, Breitling E, Müller M, Glaser S, Bechthold A. Antimicrob Agents Chemother. 2006;50:2113–2121. doi: 10.1128/AAC.00007-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li W, Ju J, Osada H, Shen B. J Bacteriol. 2006;188:4148–4152. doi: 10.1128/JB.00172-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heathcote ML, Staunton J, Leadlay PF. Chem Biol. 2001;8:207–220. doi: 10.1016/s1074-5521(01)00002-3. [DOI] [PubMed] [Google Scholar]
- 24.Kim BS, Cropp TA, Beck BJ, Sherman DH, Reynolds KA. J Biol Chem. 2002;277:48028–48034. doi: 10.1074/jbc.M207770200. [DOI] [PubMed] [Google Scholar]
- 25.Hu Z, Pfeifer BA, Chao E, Murli S, Kealey J, Carney JR, Ashley G, Khosla C, Hutchinson CR. Microbiol. 2003;149:2213–2225. doi: 10.1099/mic.0.26015-0. [DOI] [PubMed] [Google Scholar]
- 26.Gust B, Challis GL, Fowler K, Kieser T, Chater KF. Proc Natl Acad Sci USA. 2003;100:1541–1546. doi: 10.1073/pnas.0337542100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sattler I, Thiericke R, Zeeck A. Nat Prod Rep. 1998;15:221–240. doi: 10.1039/a815221y. [DOI] [PubMed] [Google Scholar]
- 28.Zhang W, Bolla ML, Kahne D, Walsh CT. J Am Chem Soc. 2010;121:6402–6411. doi: 10.1021/ja1002845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nishikawa S, Murooka Y. Biotechnol Genet Eng Rev. 2001;18:149–169. doi: 10.1080/02648725.2001.10648012. [DOI] [PubMed] [Google Scholar]
- 30.Kang Z, Zhang J, Zhou J, Qi Q, Du G, Chen J. Biotechnol Adv. 2012;30:1533–1542. doi: 10.1016/j.biotechadv.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 31.Fu W, Lin J, Cen P. Appl Biochem Biotechnol. 2010;160:456–466. doi: 10.1007/s12010-008-8363-4. [DOI] [PubMed] [Google Scholar]
- 32.Ostash B, Saghatelian A, Walker S. Chem Biol. 2007;14:257–267. doi: 10.1016/j.chembiol.2007.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin S, Van Lanen SG, Shen B. Proc Natl Acad Sci USA. 2009;106:4183–4188. doi: 10.1073/pnas.0808880106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walton LJ, Corre C, Challis GL. J Ind Microbiol Biotechnol. 2006;33:105–120. doi: 10.1007/s10295-005-0026-7. [DOI] [PubMed] [Google Scholar]
- 35.Carroll BJ, Moss SJ, Bai L, Kato Y, Toelzer S, Yu TW, Floss HG. J Am Chem Soc. 2002;124:4176–4177. doi: 10.1021/ja0124764. [DOI] [PubMed] [Google Scholar]
- 36.Wu K, Chung L, Revill WP, Katz L, Reeves CD. Gene. 2000;251:81–90. doi: 10.1016/s0378-1119(00)00171-2. [DOI] [PubMed] [Google Scholar]
- 37.Watanabe K, Khosla C, Stroud RM, Tsai SC. J Mol Biol. 2003;334:435–444. doi: 10.1016/j.jmb.2003.10.021. [DOI] [PubMed] [Google Scholar]
- 38.Floriano B, Bibb M. Mol Microbiol. 1996;21:385–396. doi: 10.1046/j.1365-2958.1996.6491364.x. [DOI] [PubMed] [Google Scholar]
- 39.Umeyama T, Lee PC, Horinouchi S. Appl Microbiol Biotechnol. 2002;59:419–425. doi: 10.1007/s00253-002-1045-1. [DOI] [PubMed] [Google Scholar]
- 40.Fuqua C, Winans SC, Greenberg EP. Ann Rev Microbiol. 1996;50:727–751. doi: 10.1146/annurev.micro.50.1.727. [DOI] [PubMed] [Google Scholar]
- 41.Haydock SF, Mironenko T, Ghoorahoo HI, Leadlay PF. J Biotechnol. 2004;113:55–68. doi: 10.1016/j.jbiotec.2004.03.022. [DOI] [PubMed] [Google Scholar]
- 42.Li J, Lu C, Shen Y. J Antibiot. 2010;63:595–599. doi: 10.1038/ja.2010.95. [DOI] [PubMed] [Google Scholar]
- 43.Zhang Dj, Wei G, Wang Y, Si Cc, Tian L, Tao Lm, Li Yg. J Antibiot. 2011;64:391–393. doi: 10.1038/ja.2011.12. [DOI] [PubMed] [Google Scholar]
- 44.Yu Z, Zhao Lx, Jiang Cl, Duan Y, Wong L, Carver KC, Schuler LA, Shen B. J Antibiot. 2011;64:159–162. doi: 10.1038/ja.2010.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Natsuko I, Akio O, Haruo I, Jun I, Shigeru K, Yumi W, Sanae N, Yoko K, Emi K, Machi S, Akiho A, Shigehiro F, Yoshimi H, Sachi K, Misa O, Satoshi T, Takuya N, Sueharu H, Yasuo O, Masayuki H, Tomohisa K, Akira A, Fumiki N, Hiromi M, Yoko T. F DNA Res. 2010;17:393–406. [Google Scholar]
- 46.Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. Practical Streptomyces Genetics. The John Innes Foundation; Norwich, UK: 2000. [Google Scholar]
- 47.Ansari MZ, Yadav G, Gokhale RS, Mohanty D. Nucl Acids Res. 2004;32:W405–413. doi: 10.1093/nar/gkh359. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.