

The therapeutic potential of glucagon-like peptide-1 (7–36) amide (GLP-1) receptor (GLP-1R) targeting for the treatment of type 2 diabetes (T2D) has been validated extensively through basic and clinical research.[1] GLP-1, a major incretin, has a wide variety of physiological roles centered on the postprandial homeostasis of glucose, including the potentiation of insulin secretion and the suppression of glucagon secretion.[2] The insulinotropic functions of GLP-1 are glucose-dependent, making GLP-1 an ideal treatment for hyperglycemia with minimal risk of hypoglycemia.[3] Recently documented preclinical and clinical research has also established the role of GLP-1R agonists in increasing satiety,[4] promoting modest weight loss,[5] improving β-cell function,[6] and preserving β-cell mass by inhibiting apoptosis,[6] which might help to slow or reverse the progression of T2D.[7]
GLP-1 is secreted from enteroendocrine L cells in the ileum and colon in response to orally ingested fats and carbohydrates, and it enters the systemic circulation via intestinal capillaries draining into the hepatic portal vein.[2] GLP-1 is rapidly degraded (t1/2=1–2 min) by dipeptidyl peptidase IV (DPP-IV), and only 10–15% of secreted GLP-1 survives intact to the systemic circulation.[8,9] GLP-1R has been identified on pancreatic β cells, in the hepatic portal vein, central nervous system, and on the vagal afferent fiber terminals of the gastrointestinal tract (GIT).[10]
A number of GLP-1-related therapeutics are currently available for the treatment of T2D, including a GLP-1 analogue, liraglutide, and a GLP-1R agonist, exenatide.[11] Both of these prescription therapeutics are administered by subcutaneous injections.[11] An orally deliverable GLP-1 therapeutic would have the potential to provide patients with a noninvasive therapy and mimic more closely the physiological effects of native GLP-1, specifically interactions with receptors in the intestines.[9]
Vitamin B12 (B12) is essential for the survival of all living organisms. It plays an important role in the normal functioning of the brain and nervous system, as well as the formation of red blood cells.[12] B12 is only synthesized naturally by bacteria, so humans must acquire the vitamin through their diet.[12,13] Due to the low bioavailability of B12, the body has an uptake mechanism, recently reviewed by Nexo et al., to ensure successful absorption of the vitamin from the diet.[13] Briefly, the pathway involves haptocorrin, a salivary enzyme that can protect and transport B12 through the stomach and into the small intestine. The B12 is then bound by intrinsic factor (IF) and proceeds down the small intestine where the IF–B12 complex is recognized by the CUB5–8 domain of cubilin-amnionless (cubam) receptor in the ileum.[13] The IF–B12 receptor complex then undergoes transcytosis, releasing B12 into the blood serum where it is bound to transcobalamin II (TCII) and transported to cells.[13]
The full potential of the B12 uptake pathway as a delivery system has yet to be realized.[14] A number of limitations to this delivery strategy have been identified in recent years that have hindered development, including a limited uptake capacity (nmol per dose),[15] relatively slow delivery time (in the order of hours),[16] and questions regarding protection for the transported peptide.[16] The critical components to the success of this delivery strategy are 1) the uptake capacity of the B12 pathway must meet the necessary increase in plasma peptide levels required to produce a physiological response; 2) questions regarding protection of the peptide must be addressed; 3) B12 conjugation must have minimal negative effect on the function of the peptide.
The B12 uptake pathway could be the answer to overcoming the major hurdles to oral peptide delivery. Our previous reported work with B12–insulin suggests there is a protective element to the B12 uptake pathway for peptides.[17] We have also previously demonstrated the ability to use B12 to orally delivery clinically relevant amounts of the appetite suppressant peptide YY.[18]
Although the IF-mediated endocytosis via cubam is the limiting step in the B12 uptake pathway, it is important to note that cubam recycles,[19] and as such, multiple dosing (e.g., morning and evening) could be adopted to increase the quantity of peptide absorbed throughout the day. This recycling will also be important when considering dietary B12 competition.[20]
To adapt the B12 uptake pathway for the delivery of the insulinotropic peptide, GLP-1, the biological potency of GLP-1 (EC50~3 nm)[21] must not be greatly decreased by B12 conjugation.[14] Herein, we report the synthesis, purification and characterization of a B12-GLP-1 conjugate based on a GLP-1 analogue, namely K34R-GLP-1. The modification of lysine (K) with arginine (R) at position 34 has been shown to not effect GLP-1 activity and allows targeted conjugation to the K26 residue.[22] In vitro experimentation with the B12-K34R-GLP-1 conjugate was utilized to determine the effect of B12 conjugation on the biological potency of GLP-1.
Unmodified GLP-1 has three amine groups (N terminus, K26 and K34) available for conjugation. For production of B12-K34R-GLP-1 (1), the peptide was prepared with modifications (amino acid substitution K34R, and fluorenyl-9-methoxycarbonyl (FMOC) protection of the N-terminal amine to give FMOC-K34R-GLP-1) to ensure selective conjugation at K26 (see Figure 1). The FMOC protection was removed after purification to ensure biological activity would be maintained.[22]
Figure 1.
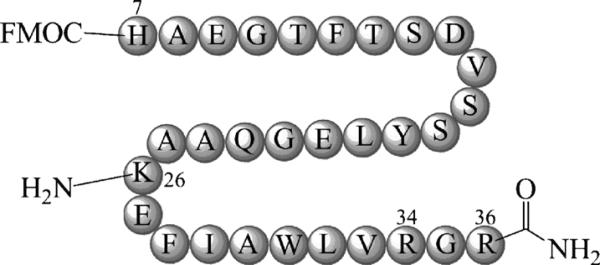
K34R-GLP-1(7-36) was purchased with N-terminal FMOC protection and K34R amino acid substitution. The C terminus was also amidated as is found for native GLP-1(7–36). The K34R modification and FMOC protection allow for site-directed B12 conjugation at lysine 26 (K26).
Modification of the 5’-hydroxy group of B12 has been repeatedly shown to maintain binding with the transport proteins vital for oral B12 uptake,[17,18,23] and this site was again chosen for this work. Reaction of B12 with 1,1’-carbonyl-di-(1,2,4-triazole) (CDT) in dry dimethyl sulfoxide (DMSO) at room temperature for five minutes furnished an activated ester at the 5’-hydroxy group. A 2.5-fold excess of activated B12 was then added to a slowly stirring solution of FMOC-K34R-GLP-1 in DMSO with 0.6% triethylamine. The reaction proceeded for two hours before a 10-fold excess of 9-fluorenylmethyl succinimidyl carbonate (FMOC-OSu) was added to help with the purification of 1, as described below.
For the purification of B12-K34R-GLP-1 (1), the reaction was first extracted by diethyl ether addition and centrifugation. The isolated pellet was re-dissolved in 25/75 acetonitrile/water with 0.1% trifluoroacetic acid (TFA), and the target product (1) was isolated on a C18 analytical column using reversed-phase (RP)-HPLC. Unreacted B12 was readily separated from FMOC-K34R-GLP-1 in this manner. Purification of unconjugated FMOC-K34R-GLP-1 and B12-conjugated FMOC-K34R-GLP-1 proved challenging due to the similarity in their hydrophobicity. The addition of excess FMOC-OSu was used to aid in purification, as previously reported for our B12 insulin conjugate.[16] Figure 2 shows that the additional FMOC moiety increases the retention time of the unconjugated (hence, unprotected) FMOC-K34R-GLP-1, allowing for separation. Following HPLC purification, the protecting groups were removed with 10% piperidine (10 min) and dialyzed against water (5 L) overnight in dialysis tubing. A B12 immunoaffinity column was used to further establish the absence of unreacted GLP-1 in subsequent in vitro assays (for details, see the Experimental Section). While exhaustive and probably poorly scalable, this route was used to ensure no free K34R-GLP-1 remained in the sample, which could otherwise contributed to a positive result in subsequent in vitro assays.
Figure 2.

Reversed-phase (RP)-HPLC chromatogram showing separation of unconjugated B12 and FMOC-K34RGLP-1 from B12-FMOC-K34R-GLP-1 with detection at 254 nm (·····) and 360 nm (—). The peak at retention time (tR) 20.7 min is B12-FMOC-K34R-GLP-1.
B12-K34R-GLP-1 (1) was identified as the peak at a retention time of 20.7 minutes (see Figure 2) by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-ToF MS) after removal of the FMOC protection as described above. The peak in Figure 3 at 4682 m/z represents a 1:1 [1–CN]+ conjugate.
Figure 3.

MALDI-ToF MS spectrum of 1 indicated by the presence of the prominent peak at 4682 m/z.
GLP-1 mediates its insulinotropic effects by binding to GLP-1R on pancreatic β cells and stimulating cAMP formation, calcium mobilization and insulin secretion.[2,8,24] The effect of B12 conjugation on GLP-1 physiological function (cAMP signaling, calcium mobilization, and insulin secretion) was studied in HEK-293 cells stably expressing human GLP-1R (HEK-GLP-1R) and in isolated human pancreatic islets.
To determine the ability of the conjugate to perform as a cAMP elevating agent, HEK-GLP-1R cells were transfected with a reporter construct containing the coding sequence for the luciferase (Luc) enzyme and a promoter incorporating a cAMP response element (CRE) originally identified within the 5’ promoter region of the rat insulin 1 gene (RIP1-CRE).[25] This cAMP-responsive Luc reporter (RIP1-CRE-Luc) can be used to determine the effect of agonist for G protein-coupled receptors positively linked to cAMP production, such as GLP-1R. The level of Luc activity was determined in whole-cell lysates by Luc-catalyzed oxidation of luciferin, which generated photons detected by a photomultiplier tube. After exposure to 1 for four hours, Luc activity was determined and shown to be concentration-dependent with an EC50 value of 4.4 nm (Figure 4). These findings were comparable to those found for the K34R-GLP-1 control with an EC50 value of 4.1 nm. These EC50 values are consistent with those reported for GLP-1 in the literature (~3 nm).[21] Figure 4 also shows that B12 did not produce any response in this assay. The EC50 value recorded for 1 suggests that B12 has little negative effect on the binding of GLP-1R by GLP-1.
Figure 4.

The fold-increase of luciferase activity was determined for B12-K34R-GLP-1 (1) and K34R-GLP-1(7–36) after HEK-GLP-1R cells were transfected with a RIP1-CRE-Luc reporter and exposed to 1, K34R-GLP-1 or free B12 for 4 h. B12-K34R-GLP-1 (1) produced a concentration-dependent response with an EC50 value of 4.4 nm, while K34R-GLP-1 exhibited an EC50 value of 4.1 nm.
We subsequently sought to determine whether an actual physiologically relevant response could be achieved with 1. The stimulatory effects of GLP-1 on pancreatic islet insulin secretion have been attributed to the capability of GLP-1 to increase levels of cytosolic [Ca2+] in β cells. In HEK cells, GLP-1 produces a rise in cytosolic [Ca2+] by facilitating the action of P2Y purinergic receptor agonists to mobilize Ca2+ from intracellular Ca2+ stores. The ability of 1 to facilitate an increase in cytosolic [Ca2+] was therefore assessed in HEK-GLP-1R cells that express endogenous P2Y purinergic receptors using a Fura-2 assay.[26] Figure 5 shows Fura-2 determinations of [Ca2+] obtained in a two-step injection protocol where 1 or native, unmodified, GLP-1 were administered to each well after baseline recording for 100 seconds. After a further 200 seconds, the P2Y receptor agonist adenosine di-phosphate (ADP) was added, and the increase in Fura-2 emission ratio was recorded. Figure 5 demonstrates that both forms of GLP-1 potentiated the action of ADP to mobilize intracellular [Ca2+] in a dose-dependent manner, with native GLP-1 only slightly more potent than 1.
Figure 5.

Fura-2 determinations of intracellular [Ca2+] in HEK-GLP-1R cells that express endogenous P2Y purinergic receptors: a) B12-K34R-GLP-1 (1) and b) native GLP-1. Note that both forms of GLP-1 potentiated the action of ADP (10 nm) to mobilize intracellular [Ca2+] and that native GLP-1 was slightly more potent than 1. Data are the mean of n=12 wells for each data point, and similar results were obtained in four independent plates with three different batches of 1.
To further document the physiologically relevant action of 1, insulin secretion was assayed under standard conditions of static incubation using human islets.[27] Human pancreatic islets are a critical tool for diabetes researchers because they provide invaluable information about the cellular biology of the pancreas and the pathology of diabetes. Furthermore, the uniqueness of human pancreatic biology is an invaluable tool for the development of therapeutics for the treatment of diabetes.
Human islets were equilibrated in Krebs-ringer buffer (KRB) containing glucose (2.8 mm), followed by a subsequent elevation of the glucose concentration to 16.7 mm with or without the added test compound. The rise in glucose concentration causes glucose stimulated insulin secretion (GSIS) in healthy insulin-producing islets, and the addition of functional GLP-1 should potentiate this effect. For three batches of human islets from three donors, the average GSIS measured in response to 16.7 mm glucose was 2.2-fold in the absence of 1 (Figure 6). GSIS was potentiated an additional 45% by 15 nm 1 (total 3.2-fold stimulation). The effect of 1 was similar to that measured when islets were instead treated with 15 nm K34R-GLP-1 (total 3.3-fold stimulation).
Figure 6.

Static incubation assays using human pancreatic islets. Human Islets (40–50 per insert) were exposed to Krebs-ringer buffer (KRB) containing 2.8 mm glucose for 30 min producing an average basal insulin secretion of 21.4 ± ng mL–1 × 30 min (1st bar). The islets were then exposed to KRB containing 16.7 mm glucose (G) and no test compound (2nd bar), 1 (denoted here as B12-GLP-1; 3rd bar), or K34R-GLP-1 (denoted here as GLP-1; 4th bar). In the absence of added peptide, glucose produced a 2.2-fold stimulation of insulin secretion. When 1 was included with 16.7 mm glucose, a 3.2-fold stimulation was measured, which is comparable to that measured for K34R-GLP-1 (3.3-fold increase).
We have reported herein the synthesis, purification and in vitro characterization of a B12-K34R-GLP-1 conjugate. Experimentation with this lead conjugate in vitro demonstrated B12 attachment has little negative effect on the insulinotropic nature of GLP-1. The data collected for B12-K34R-GLP-1 are very encouraging, and we are moving into in vivo experimentation to determine the biological activity of 1.
Experimental Section
General
Chemicals and solvents were purchased from Sigma–Aldrich or Fluka and were used without further purification. Glucagon-like peptide (7–36) amide with a K34R amino acid substitution and N-terminal FMOC protection (FMOC-K34R-GLP-1) was purchased from NEOBiolab (Cambridge, MA, USA). DMSO was dried over 4 Å molecular sieves (200–400 mesh, Sigma) under dry nitrogen gas. Dialysis tubing (2000 and 3500 Da cutoff) was purchased from Pierce. H2O was distilled and deionized to 18.2 MΩ-cm using a Barnstead Nano Diamond ultrapurification machine. Reversed-phase (RP)-HPLC purification was carried out on an Agilent 1100 system with a variety of Agilent columns discussed below. MALDI-ToF MS was performed on a Bruker Autoflex III Smartbeam machine with laser intensity ranging from 50–73%. The matrix used was 10 mg α-cyano-4-hydroxycinnamic acid (CHCA) dissolved in deionized (d)H2O/CH3CN (1:1 v/v) containing 0.1% trifluoroacetic acid (TFA). Angiotensin I was used as an internal control (1296 m/z). Electronic absorption spectroscopy (EAS) was performed on a Varian Cary 50 Biospectrometer in a 1 mL quartz cuvette (Sigma) between 200 nm and 600 nm. All centrifugation was done at 4000 rpm for 10 min using a Sorvall centrifuge with a swinging rotor (Sorvall Heraeus 75006441K). 96-Well plates were coated with rat-tail collagen from BD Biosciences prior to assays. CMRL-1066-modified culture medium was obtained from Mediatech Inc. (Manassas, VA, USA). Fetal bovine serum (FBS), Dulbecco's modified Eagle's medium (DMEM), Pluronic F-127 and Fura-2 acetoxymethyl ester (Fura-2AM) were purchased from Invitrogen Life Technologies (Carlsbad, CA, USA). A Mercodia ultrasensitive insulin ELISA kit (#10-1247-10) was used for determination of EC50 values. Measurements of intercellular [Ca2+] were performed using a FlexStation 3 microplate reader using SoftMax Pro software from Molecular Devices. A Lipofectamine 2000 kit was purchased from Invitrogen for transfections. The luciferase assay kit used was purchased from Promega (Madison, WI, USA), and the FlexStation 3 microplate reader was used to record luciferase activity.
Preparation and purification
Vitamin B12 (cyanocobalamin; 10 mg, 0.007 mmol) was activated at the 5’-hydroxy group using CDT (1.7 mg, 0.010 mmol) in dry DMSO (1 mL) under dry N2 for 5 min. The activated B12 solution (0.096 mL, 0.0007 mmol) was subsequently added to a stirring solution of FMOC-K34R-GLP-1 (1.0 mg, 0.0028 mmol) dissolved in DMSO (0.1 mL) with 0.6% Et3N (v/v). The reaction was stirred for 3 h at RT before H2O/0.1% TFA (20% v/v) was added to the reaction. A solution of FMOC-OSu (10 mg, 0.028 mmol) in DMF was added, and the reaction was allowed to proceed for an additional 1 h. The reaction was stopped by the addition of Et2O (40 mL). Purification of the FMOC-B12-K34R-GLP-1 conjugate was accomplished by RP-HPLC using an Agilent C18 column and HPLC: solvent A: dH2O/0.1% TFA; solvent B: CH3CN; 75/35→35/65% A/B over 25 min. The solution was adjusted to pH 8 with NH2HCO3 immediately following isolation by HPLC. Removal of the N-terminal FMOC protection was accomplished with the addition of 10% piperidine after 10 min. The solution was transferred to 3500 MWCO dialysis tubing and dialyzed against H2O for 18 h, changing solution every 4–6 h. 1 was collected as a light pink solution (0.10 mg, 10%). HPLC: tR=20.7 min; MALDI-ToF: m/z calcd for K34R-GLP-1-[B12-CN]+: 4680, found: 4682; Yield was calculated from EAS using ε361=27,500 m–1 cm–1. A B12 immunoaffinity column was utilized to further establish the absence of unreacted GLP-1 from a HPLC and dialyzed sample of 1. After establishing GLP-1 does not bind to the B12 immunoaffinity column and that B12-GLP-1 does, the unbound flow through was tested for biological activity in a luciferase assay producing no biological response.
Luciferase assay
HEK-GLP-1R cells were plated in a 96-well plate at a density of 55000–65000 cells per well and incubated overnight at 37°C. The next day, the cells were transfected with RIP1-CRE-Luc using Lipofectamine 2000 according to the manufacturer's instructions. The cells (HEK-GLP-1R-RIP1-CRE-Luc) were cultured in DMEM containing 10% FBS overnight and then exposed to serum-free culture medium containing 1% bovine serum albumin (BSA) and the test substance for 4 h. Cells were then lysed and assayed for luciferase-catalyzed photoemissions using a luciferase assay kit and luminometer.
Fura-2 assay
The experiment was performed using a monolayer of Fura-2-loaded HEK-GLP-1R cells grown on rat-tail-collagen-coated Costar 3904 plate. Fura-2 was loaded in a standard extracellular solution (SES) containing: 138 mm NaCl, 5.6 mm KCl, 2.6 mm CaCl2, 1.2 mm MgCl2, 10 mm HEPES (pH 7.4) and 11.1 mm glucose. The SES was also supplemented with 20 μL per mL of FBS, 1 μL per mL of Pluronic F-127, and 1 μm Fura-2AM. Spectrofluorimetry was performed using excitation light at 355/9 and 375/9 nm (center/bandpass) delivered using a 455 nm dichroic mirror. Emitted light was detected at 505/15 nm, and the ratio of emission light intensities due to excitation at 355 and 375 nm was calculated. Insulin secretion was determined by static incubation.
Insulin secretion
After overnight culture in CMRL-1066 medium, human islets were transferred to MilliCell polycarbonate film (PCF) culture plate filter inserts at a density of 40 islets per insert. The inserts were mounted within individual wells of a 24-well cell culture plate, and each well of the plate was filled with 1 mL of Krebsringer buffer (KRB) containing 24 mm aq NaHCO3 and the indicated concentration of glucose (pH 7.4). Culture plates containing islets were placed within a cell culture incubator gassed with 95% air and 5% CO2 at 37°C. Prior to the start of an experiment, islets were exposed to KRB with 2.8 mm glucose for 30 min. The inserts were then transferred between adjacent wells on the plate to measure basal and stimulated insulin secretion. For each test solution, the duration of exposure was 30 min with 16.7 mm glucose. A 200 μL fraction of each solution was then collected and assayed for insulin content using an ultrasensitive insulin ELISA kit, after appropriate dilution of samples.
Acknowledgements
R.P.D. acknowledges the W. M. Wrigley, Jr. Co. (Chicago, IL, USA) for funding this work. The authors would like to thank Dr. Christian Roth (Seattle Children's Hospital, Seattle, WA, USA) and Dr. Timothy J. Fairchild (Murdoch University, Western Australia, Australia) for helpful discussions.
References
- 1.Aroda VR, Henry RR, Han J, Huang W, DeYoung MB, Darsow T, Hoogwerf BJ. Clin. Ther. 2012;34:1247–1258. doi: 10.1016/j.clinthera.2012.04.013. [DOI] [PubMed] [Google Scholar]
- 2.Drucker DJ. Cell Metab. 2006;3:153–165. doi: 10.1016/j.cmet.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 3.Kreymann B, Ghatei MA, Williams G, Bloom SR. Lancet. 1987;330:1300–1304. doi: 10.1016/s0140-6736(87)91194-9. [DOI] [PubMed] [Google Scholar]
- 4.a Williams DL, Baskin DG, Schwartz MW. Endocrinology. 2009;150:1680–1687. doi: 10.1210/en.2008-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Renner E, Pushas N, Dobolyi A, Palkovits M. Peptides. 2012;35:14–22. doi: 10.1016/j.peptides.2012.02.018. [DOI] [PubMed] [Google Scholar]; c Kanoski SE, Fortin SM, Arnold M, Grill HJ, Hayes MR. Endocrinology. 2011;152:3103–3112. doi: 10.1210/en.2011-0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vilsbøll T, Christensen M, Junker AE, Knop FK, Gluud LL. BMJ. 2012;344:d7771. doi: 10.1136/bmj.d7771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a Tourrel C, Bailbé D, Lacorne M, Meile MJ, Kergoat M, Portha B. Diabetes. 2002;51:1443–1452. doi: 10.2337/diabetes.51.5.1443. [DOI] [PubMed] [Google Scholar]; b Talbot J, Joly E, Prentki M, Buteau J. Mol. Cell. Endocrinol. 2012;364:65–70. doi: 10.1016/j.mce.2012.08.010. [DOI] [PubMed] [Google Scholar]; c Farilla L, Hui H, Bertolotto C, Kang E, Bulotta A, Di Mario U, Perfetti R. Endocrinology. 2002;143:4397–4408. doi: 10.1210/en.2002-220405. [DOI] [PubMed] [Google Scholar]
- 7.Rutti S, Sauter NS, Bouzakri K, Prazak R, Halban PA, Donath MY. PLoS One. 2012;7:e35801. doi: 10.1371/journal.pone.0035801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holst JJ. Physiol. Rev. 2007;87:1409–39. doi: 10.1152/physrev.00034.2006. [DOI] [PubMed] [Google Scholar]
- 9.Steinert RE, Poller B, Castelli MC, Drewe J, Beglinger C. Am. J. Clin. Nutr. 2010;92:810–817. doi: 10.3945/ajcn.2010.29663. [DOI] [PubMed] [Google Scholar]
- 10.Willard FS, Sloop KW. Exp. Diabetes Res. 2012;2012:470851. doi: 10.1155/2012/470851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amori RE, Lau J, Pittas AG. JAMA J. Am. Med. Assoc. 2007;298:194–206. doi: 10.1001/jama.298.2.194. [DOI] [PubMed] [Google Scholar]
- 12.Banerjee R, editor. Chemistry and Biochemistry of B12. John Wiley & Sons; New York: 1999. [Google Scholar]
- 13.Nielsen MJ, Rasmussen MR, Andersen CB, Nexo E, Moestrup SK. Nat. Rev. Gastroenterol. Hepatol. 2012;9:345–354. doi: 10.1038/nrgastro.2012.76. [DOI] [PubMed] [Google Scholar]
- 14.Clardy SM, Allis DG, Fairchild TJ, Doyle RP. Expert Opin. Drug Delivery. 2011;8:127–140. doi: 10.1517/17425247.2011.539200. [DOI] [PubMed] [Google Scholar]
- 15.A Report of the Standing Committee on the Scientific Evaluation of Dietary Reference Intakes and its Panel on Folate, Other B Vitamins, and Choline and Subcommittee on Upper Reference Levels of Nutrients; Food and Nutrition Board; Institute of Medicine. The National Academies Press; Washington, DC (USA): 1998. [Google Scholar]
- 16.Russell-Jones GJ. Ther. Delivery. 2011;2:1575–1593. doi: 10.4155/tde.11.129. [DOI] [PubMed] [Google Scholar]
- 17.Clardy-James S, Allis DG, Fairchild TJ, Doyle RP. Med. Chem. Commun. 2012;3:1054–1058. [Google Scholar]
- 18.Fazen CH, Valentin D, Fairchild TJ, Doyle RP. J. Med. Chem. 2011;54:8707–8711. doi: 10.1021/jm2012547. [DOI] [PubMed] [Google Scholar]
- 19.Moestrup SK, Verroust PJ. Annu. Rev. Nutr. 2001;21:407–428. doi: 10.1146/annurev.nutr.21.1.407. [DOI] [PubMed] [Google Scholar]
- 20.Lildballe DL, Mutti E, Birn H, Nexo E. PLoS One. 2012;7:e46657. doi: 10.1371/journal.pone.0046657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brubaker PL, Drucker DJ. Recept. Channels. 2002;8:179–188. [PubMed] [Google Scholar]
- 22.Madsen K, Knudsen LB, Agersoe H, Nielsen PF, Thogersen H, Wilken M, Johansen N. J. Med. Chem. 2007;50:6126–6132. doi: 10.1021/jm070861j. [DOI] [PubMed] [Google Scholar]
- 23.Petrus AK, Vortherms AR, Fairchild TJ, Doyle RP. ChemMedChem. 2007;2:1717–1721. doi: 10.1002/cmdc.200700239. [DOI] [PubMed] [Google Scholar]
- 24.Leech CA, Dzhura I, Chepurny OG, Kang G, Schwede F, Genieser HG, Holz GG. Prog. Biophys. Mol. Biol. 2011;107:236–247. doi: 10.1016/j.pbiomolbio.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chepurny OC, Holz GG, Biomol J. Screening. 2007;12:740–746. doi: 10.1177/1087057107301856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Landa LR, Harbeck M, Kaihara K, Chepurny O, Kitiphongspattana K, Graf O, Nikolaev VO, Lohse MJ, Holz GG, Roe MW. J. Biol. Chem. 2005;280:31294–31302. doi: 10.1074/jbc.M505657200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chepurny OG, Kelley GG, Dzhura I, Leech CA, Roe MW, Dzhura E, Li X, Schwede F, Genieser HG, Holz GG. Am. J. Physiol. Endocrinol. Metabol. 2010;298:E622–633. doi: 10.1152/ajpendo.00630.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]