

Abstract
Macrophages are key players in the inflammatory response. In this study, we tested the hypothesis that although all macrophage subpopulations in tumor hosts are affected by the disease, it is the close proximity to the tumor that induces major alterations in these cells. We compared tumor-associated macrophages (TAMs) with peritoneal macrophages from mice bearing D1-DMBA-3 mammary tumors (T-PEMs). Our results show that TAMs downregulate IL-12p70 but upregulate IL-12p40, IL-23, IL-6 and IL-10. Some NFκB and C/EBP transcription factors family members are decreased in TAMs; however NFκBp50 homodimers, STAT1/pSTAT1 and STAT3/pSTAT3 are overexpressed. Furthermore, while TAMs block T-cell proliferation and are more prone to apoptosis compared to T-PEMs, both types of macrophages have an impaired phagocytic capacity. Moreover, TAMs constitutively express iNOS and produce nitric oxide but do not express arginase and are Gr-1high and CD11blow. Collectively, our analysis of two spatially distinct macrophage subpopulations in tumor-bearing mice revealed that the tumor modulates them differently into two molecularly and functionally dissimilar macrophage subpopulations.
Keywords: Peritoneal and tumor-associated, macrophages, Tumor microenvironment, Inflammation, Immunosuppression
1. Introduction
Tumors are dynamic microenvironments consisting of neoplastic cells surrounded by connective tissue, newly developed blood vessels and a number of recruited immune and non-immune cells, all of which interact with each other and produce factors that have downstream effects on the immune system [1]. The cells, vessels and molecules that surround the tumor cells, influence how these grow and metastasize, yet the non-tumor components of the tumor microenvironment can be also modulated by the tumor cells in an active interplay that generally contributes to tumor survival and development. The critical role the tumor microenvironment plays in cancer has been fully recognized in recent years [2].
Chronic inflammation is intimately associated with cancerogenesis [3–5]. Macrophages, the most common non-tumor cell type in tumor microenvironments, play central roles in inflammation and participate in tumor development [5]. Although pro-inflammatory M1 macrophages may be cytotoxic to tumor cells, persistent inflammatory responses can be detrimental and help cancer initiation and/or progression through the generation of mutation-inducing reactive oxygen and nitrogen free radicals [6]. Anti-inflammatory M2 macrophages are also involved in tumor progression by fostering invasion, extracellular matrix remodeling, angiogenesis, metastasis and suppression of anti-tumor immune responses [7–9]. A considerable body of data links the density of macrophages in solid tumors to poor prognosis [10,11]. Tumor associated macrophages (TAMs) exhibit phenotypes that significantly contribute to the tumor microenvironment’s immunosuppressive properties and to tumor progression [10,12–14]. There is scarce information in the literature on how spatially distinct macrophage subpopulations are affected in tumor hosts. A comprehensive assessment of the phenotype of different macrophage subpopulations is therefore essential to the understanding of tumor-derived signals guiding polarization of innate and adaptive immunities in tumor hosts and to the identification of molecular mechanisms that might be amenable to therapeutic intervention.
In this study we aimed to analyze two spatially distant subpopulations of macrophages isolated from mammary tumor-bearing mice, i.e.: peripheral macrophages elicited to the peritoneal cavity (T-PEMs) and tumor-associated macrophages (TAMs). The physical isolation of these macrophage subpopulations from their specific locations enabled us the characterization of their distinct phenotypes and functions, yet we also studied TAMs in their in situ locations in the tumors without tumor disruption using histology and immunohistochemistry. We tested the hypothesis that all macrophage subpopulations in tumor hosts are affected by the disease, but that the proximity to the tumor microenvironment induces major alterations in macrophages. We confirmed that the presence of a tumor does affect different macrophage subpopulations in tumor hosts in very different ways. Our work shows that the local changes induced by the tumor microenvironment are by far more profound than the changes induced by the distant effects of tumor factors in peripheral locations. The role of macrophages in tumor development has been recognized and has received a great deal of interest, but the reciprocal way in which tumors modify macrophages has been less examined. Given the importance of macrophages as inflammatory and phagocytic cells central in innate immunity, understanding how immune suppression may be induced in tumor hosts by tumor factors impairing macrophage functions is highly relevant. Our study clearly demonstrates that macrophages at the tumor microenvironment (TAMs) and at the periphery (T-PEMs) are two molecularly and functionally distinct macrophage subpopulations.
2. Materials and methods
2.1. Animals and tumors
Female BALB/c mice of 10–14 weeks of age purchased from NCI-Frederick (MD) were used. The D1-DMBA-3 mammary adenocarcinoma is a transplantable tumor derived from a nonviral, non-carcinogen-induced preneoplastic alveolar nodule in a BALB/c mouse treated with 7,12-dimethylbenzanthracene [15]. The immunogenic tumor is routinely transplanted by s.c. injection of 1 × 106 tumor cells. Our institutional animal care and use committee approved the animal experiments.
2.2. Isolation of cells
Four-week D1-DMBA3 tumors were dissected, washed, cut to 1–2 mm2 pieces and enzyme digested as previously described [16]. Indirect labeling using biotinilated antibodies (F4/80 for TAMs isolation or CD3 for CD3+ cells isolation), both from eBioscience (San Diego, CA), followed by anti-biotin Miltenyi magnetic beads (Miltenyi Biotec, Auburn, CA) were used to isolate TAMs or CD3+ cells, respectively; T splenocytes and peritoneal macrophages from normal (N-PEMs) and tumor-bearing (T-PEMs) mice were isolated as previously described [17,18].
2.3. Western blot
Macrophages (107) were adhered to plastic tissue culture dishes, whole cell extracts were obtained, and Western blots were performed as previously described [18]. Rabbit α-mouse polyclonal antibodies (NFκBp50, p65, c-rel, iNOS2, arginase, p53, Bcl3, C/EBPα, C/EBPβ), all from Santa Cruz Biotechnologies (Santa Cruz, CA), were used. Anti–Bcl-x (1:2000) [19] was kindly provided by Dr. Larry Boise (Emory University), and staurosporine was from EMD/Calbiochem (San Diego, CA). Rabbit α-mouse STAT1/pSTAT1, STAT3/pSTAT3, MMP9 and α-caspase 3 antibodies were from Cell Signaling Technologies (Boston, MA). Rabbit α-mouse actin polyclonal antibody was obtained from Sigma–Aldrich (St. Louis, MO). Goat α-rabbit IgG-HRP (Santa Cruz, CA) was used as the secondary antibody.
2.4. Quantitative real-time PCR (qRT-PCR)
107 cells from individual samples of N-PEMs, T-PEMs and TAMs were cultured in complete medium (RPMI with 10% FBS), constitutively and with 10 μg/mL LPS for 2 h. Total RNA was isolated and cDNA synthesis was performed using Invitrogen reagents. qRT-PCR was done using TaqMan Gene Expression Assays (Applied Biosystems, Carlsbad, CA) for IL-12p35, IL-12p40, IL-23p19 and GAPDH 20X on a 7500 Fast Real-time PCR system (Applied Biosystems, Carlsbad, CA).
2.5. Cytokine ELISAs
eBioscience (San Diego, CA) kits for detection of mouse IL-6, IL-23, IL-10, IL-12 p40 and IL-12 p70 were used as previously described [18]; TNFα was detected using a kit from Biosource (Grand Island, NY).
2.6. Nitric oxide (NO) production
Macrophages were plated (3 × 105) and incubated at 37 °C with and without 10 μg/mL LPS in complete media for 48 h. Cell supernatant nitrite ( ) concentration served as a reflection of NO production and was measured using the colorimetric Griess reaction [20].
2.7. Flow cytometry
N-PEMs, T-PEMs, TAMs and CD3+ cells were added at 106cells/tube. For macrophages, Fc receptors were blocked for 5 min using mouse CD16/32 antibody (eBioscience, San Diego, CA) and cells were stained with CD11b-FITC, CD115-PE, Gr-1-APC antibodies (eBioscience, San Diego, CA) and F4/80-PE Cy7 (BioLegend); intracellular staining for CD68-AF700 (AbD/Serotec, Raleigh, North Carolina) was performed as previously described [21]. To assess viability, macrophages were stained with 7-amino-actinomycin-D (7-AAD; BD-PharMingen, San Jose, CA), which was added 15 min prior to flow cytometry. CD3+ T cells were stained using CD4-FITC and CD25-PE (eBioscience, San Diego, CA); intracellular staining for FoxP-3-APC (eBioscience, San Diego, CA) was performed using a Fixation and Permeabilization kit (eBioscience, San Diego, CA). For intracellular IL-17 detection, isolated CD3+ cells were incubated with PMA and Ionomycin at 10 μM (Calbiochem, Billerica, MA), Brefeldin-A (eBioscience, San Diego, CA, 3 μg/ml), and Monensin (BD-Biosciences, San Jose, CA, 0.67 μg/ml) for 4 h; staining was carried out using IL-17-AF647 antibody (eBioscience, San Diego, CA). Samples were acquired from an LSRII cytometer (BD-Biosciences, San Diego, CA) and analyzed with BDFACS Diva and FlowJo Softwares (BD-Bioscience, San Jose, CA).
2.8. T-cell proliferation
T splenocytes from normal mice were isolated using CD 90.2 Microbeads (Milteny, Cambridge, MA) and activated with αCD3/αCD28 (BD Bioscience, San Jose, CA) with or without mouse recombinant IL-2 (10 ng/ml, Peprotech, NJ) for 48 h. T splenocytes previously stimulated with αCD3/αCD28 and IL-2 were incubated with adherent N-PEMs, T-PEMs and TAMs (1:5) for 120 h at 37°C. Fluorescence intensity was measured at 485–530 nm using a 1420 Victor Multi-Label Counter (Perkin Elmer, CA) following the instructions of CyQuant Cell Proliferation Assay (Invitrogen, Grand Island, NY).
2.9. Histology and immunohistochemistry (IHC)
Two, three and four-week tumors were removed from the animals and fixed in 10% formalin for 48 h, processed and embedded in paraffin. 4-μm paraffin sections were cut and antigen retrieval was carried out in citrate buffer (Vector Labs, CA) for 20 min at 95 °C. Endogenous peroxidase activity in tissues was blocked in 3% peroxidase solution (Sigma–Aldrich, St. Louis, MO) and primary antibodies were added for 1 h at RT [rat anti-mouse F4/80 (Abcam, MA) and the rat anti-mouse CD3 (Cell Marque, Rocklin, CA)]. Sections were incubated with biotinylated Rabbit Anti-Rat IgG (secondary antibody, Vector Labs, Burlingame, CA) for 1 h at RT and then with HRP-Streptavidin for 20 min at RT. Hematoxylin was used as a counterstain; sections were also stained with Hematoxylin and Eosin (H&E) to reveal the histology of the tumors.
2.10. Phagocytosis assay
Fluorescent Zymosan A BioParticles (Alexa Fluor 59, Invitrogen, Grand Island, NY) were added to 5 × 105 adherent N-PEMs, T-PEMs and TAMs at a ratio of 1:10 (cells: bioparticles), were incubated for 30 min at 37 °C and were washed with PBS and quenched by trypan blue (1.25 mg/ml, Thermo Scientific, Kalamazoo, MI) for 1 min at RT. Samples were acquired using the Zeiss ApoTome Axiovert and analyzed using the Zeiss AxioVision software. For inhibition of phagocytosis, macrophages were first incubated for 30 min with Cytochalasin-D (1 μg/ml; Invitrogen, Grand Island, NY) and then incubated with Zymosan-A Bioparticles.
2.11. Functional arginase assay
3 × 105 N-PEMs, T-PEMs and TAMs were incubated in complete media with or without 10 μg/mL of LPS for 48 h and arginase activity was determined in the cell lysates as previously described [22].
2.12. Statistical analysis
Prism software (GraphPad, La Jolla, CA) was used for all statistical analyses. Student’s paired-t-test was used to analyze statistical significance between experimental groups; p < 0.05 was considered to be statistically significant and error bars represent standard error of the mean (SEM).
3. Results
3.1. TAMs and T-PEMs exhibit differential expression of cytokines
Cytokines are known to affect leukocyte populations both functionally and phenotypically. In this study, the differential expression of proinflammatory (IL-12p40, IL-12p70, IL-6, TNFα and IL-23) and immunosuppressive cytokines (IL-10) was examined in TAMs and T-PEMs. We previously showed that ex-vivo LPS-stimulated T-PEMs from advanced mammary tumor-bearing mice exhibit diminished expression of IL-12p70 compared to N-PEMs [18]. To determine if the immunosuppressive tumor microenvironment affects IL12p70 production, the expression of this molecule was investigated in TAMs. There was no detectable expression of IL-12p70 in either resting or LPS-stimulated TAMs (Fig. 1A). Surprisingly, significantly elevated levels of IL-12p40 protein were seen in TAMs compared to both N-PEMs and T-PEMs (either constitutively or upon LPS-stimulation). However, although neither N-PEMs nor T-PEMs produce IL-12p40 constitutively, the levels of IL-12p40 in LPS-activated N-PEMs and T-PEMs are much lower than in TAMs (Fig. 1B). Given that the IL-12p40 chain is shared by the proinflammatory cytokines IL-12p70 and IL-23, we examined the expression of IL-23 protein (associated with tumor progression [23]) in the three macrophage subpopulations. Our results (Fig. 1C) demonstrate that while resting TAMs display elevated IL-23 production, these levels decrease upon LPS activation. Interestingly there was no detectable production of IL-23 in resting or activated N-PEMs or T-PEMs. We also assessed the expression of two additional proinflammatory cytokines associated with tumorigenesis, IL-6 [24] and TNF-α [25]. Our results reveal that the patterns of constitutive or LPS-induced expression of IL-6 and TNF-α are totally different within TAMs. These cells express high levels of IL-6, both constitutively and higher after LPS-induced (as is the case with their IL-10 expression) but do not show constitutive or LPS-induced expression of TNF-α at all, following a similar pattern to their IL-12p70 expression (Fig. 1D and E). We previously reported that there were no differences in the production of the anti-inflammatory cytokine IL-10 between T-PEMs and N-PEMs [18]. In this study we show that the expression of IL-10 in TAMs was significantly higher in both resting and LPS-activated conditions compared to either N-PEMs or T-PEMs (Fig. 1F).
Fig. 1.

TAMs do not express IL-12p70 yet they constitutively upregulate IL-12p40, IL-23, IL-6 and IL-10 cytokines. ELISAs of IL-12p70 (A) IL-12p40 (B) IL-23 (C) IL-6 (D) TNFα (E) and IL-10 (F) measured protein concentration in 24-h supernatants of N-PEMs, T-PEMs and TAMs cultured with and without LPS (10 μg/mL). Columns: mean of four different experiments with similar results; bars, SEM.
To verify that the differences in cytokine expression between T-PEMs and TAMs were not due to the different isolation methods used, we treated T-PEMs with the enzyme mix, biotinilated F4/80 antibody and anti-biotin Miltenyi beads that were used to isolate TAMs, and the results were similar (data not shown). It is important to mention that TAMs purity was assessed by flow cytometry in every batch of isolated TAMs (not shown) and our results show that they were always >90% pure F4/80+ cells. Taken together, our data reveal that TAMs and T-PEMs exhibit totally different protein expression patterns of these molecules, with local tumor microenvironment favoring constitutive upregulation of the pro-inflammatory cytokines IL-23, IL-6 and IL-12p40 and of the immunesuppresive IL-10, yet total blockade of the anti-tumor [26] cytokine IL-12p70.
3.2. TAMs and T-PEMs display different mechanisms of regulation of IL-12 and IL-23 expression
IL-12p70 heterodimer is comprised of IL-12p40 and IL-12p35 chains, whereas IL-23 encompasses IL-12p40 and IL-23p19. To elucidate the mechanisms involved in decreased expression of IL-12p70 and enhanced production of IL-23 in TAMs, two proinflammatory cytokines with opposite functions in cancer development, we examined the IL-12p40, IL-12p35 and IL-23p19 transcripts in the three macrophage subpopulations. Using qRT-PCR, we confirmed our previous data obtained by Northern blot analysis [18] showing downregulation of IL-12p35 and IL-12p40 mRNAs in LPS-activated T-PEMs, as compared with N-PEMs (Fig. 2). We found that TAMs do not express measurable levels of IL-12p35 transcripts, both constitutively and upon LPS stimulation. In contrast, there was an overexpression of IL-12p40 mRNA in both resting and in LPS-activated TAMs. Negligible levels of IL-23p19 mRNA transcripts were observed in resting or LPS-activated N-PEMs and T-PEMs. However resting TAMs showed enhanced expression of IL-23p19 mRNA transcript which decreased upon LPS-activation. Thus, the lack of IL-12p70 expression in TAMs despite their upregulation of IL-12p40 could be due to a profound decline in IL-12p35 mRNA. Overall, our results demonstrate the existence of dissimilar transcriptional patterns of IL-12p40, IL-12p35 and IL-23p19 gene expression in these two macrophage subpopulations.
Fig. 2.
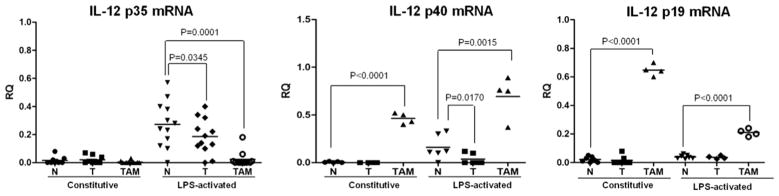
TAMs and T-PEMs differently regulate the expression of IL-12p70 and IL-23. IL-12p35, IL-12p40 and IL-23p19 mRNA levels were determined by qRT-PCR using GAPDH for normalization in constitutive and LPS-activated individual samples of N-PEMs, T-PEMs and TAMs. Mean reflects results of individual animals in one of three different experiments with similar outcomes.
3.3. Differential expression of proinflammatory transcription factors in TAMs and T-PEMs
Resting T-PEMs have been shown to exhibit impaired function of inflammation-associated transcription factors NFκB, C/EBP and STAT1, related to their decreased expression in these cells [18,21]. Examination of the same transcription factors in resting TAMs revealed that NFκBp65 and c-rel are downregulated in these cells to lower levels than those exhibited by T-PEMs. However, NFκBp50 was signficantly upregulated in TAMs compared to T-PEMs (Fig. 3A). Moreover, our data with the C/EBP transcription factor family demonstrate that resting TAMs display a more profound downregulation in C/EBPβ expression than the one shown by T-PEMs. Analysis of STAT1 revealed that TAMs show a substantial enhancement particularly in the non-phosphorylated but also in the phosphorylated forms of STAT1 as compared with T-PEMs.
Fig. 3.
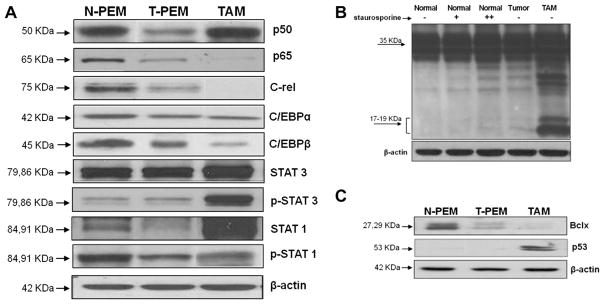
TAMs and T-PEMs differently express proinflammatory transcription factors and show different susceptibility to apoptosis. Western blots experiments comparing N-PEMs, T-PEMs and TAMs in different conditions are presented. (A) Constitutive expression patterns of several transcription factors in the three macrophage subgroups. (B) Activated levels of caspase 3 in N-PEMs, T-PEMs and TAMs; first three lanes represent N-PEMs cultured with 0, 10, and 100 nmol, respectively, of staurosporine for 2 h; lane 4, untreated T-PEMs and lane 5, untreated TAMs cultured for the same amount of time. (C) Constitutive Bcl-x and p53 expression in N-PEMs, T-PEMs and TAMs. Figures represent one of three different experiments with similar results.
We have also demonstrated no differences on the STAT3 expression in resting N-PEMs and T-PEMs [21]. However, our present results demonstrate a significant enhancement in expression of both forms of STAT3 in resting TAMs compared with T-PEMs, particularly the phosphorylated form (Fig. 3A). Altogether, our data demonstrate that there is a gradual alteration, either down or upregulation, in the expression of all these different transcription factors in T-PEMs and TAMs, with the more significant changes observed in TAMs.
3.4. TAMs are more susceptible to apoptosis than T-PEMs
We previously showed that NFκB-impaired T-PEMs exhibit diminished expression of anti-apoptotic Bcl-x and higher amounts of the 19-kD-activated form of caspase 3 than N-PEMs, which could explain elevated levels of apoptosis observed in T-PEMs [21]. In this study we assessed if TAMs were likewise prone to apoptosis. Thus, we compared the expression of activated caspase 3, Bcl-x and the pro-apoptotic p53 in the three macrophage subpopulations. We observed that resting TAMs express significantly higher levels of activated caspase 3 (Fig. 3B) with augmented expression of p53 yet lower levels of Bcl-x compared with T-PEMs (Fig. 3C). Overall, these results indicate that TAMs are more prone to apoptosis than T-PEMs.
3.5. TAMs exhibit a more immature phenotype than T-PEMs
T-PEMs are less differentiated than N-PEMs, with downregulated levels of myeloid differentiation markers F4/80, CD11b, CD68 and CD115, as previously described [21]. Current analysis of the expression of the above markers in TAMs revealed that these cells are less differentiated compared to T-PEMs based on the diminished expression of these markers (Fig. 4A). Interestingly, TAMs upregulate the granulocytic myeloid marker Gr1 at much higher levels than T-PEMs, compared with N-PEMs. Because macrophages from tumor bearers are more predisposed to apoptosis, 7-AAD viability marker was included as a control in our flow cytometric experiments. Our results with 7-AADneg (viable) cells are similar to these ones obtained with total cells (data not shown). Therefore, our studies suggest that TAMs are less differentiated than T-PEMs, yet they are not phenotypically MDSC either, since they are not CD11bhi/Gr1hi.
Fig. 4.

TAMs exhibit a more immature phenotype than T-PEMs and do not express the classical MDSCs phenotype. (A) Flow cytometric analysis of CD11b, F4/80, CD68, CD115, and Gr-1 in resting N-PEMs, T-PEMs and TAMs (MFI values). (B) Western blot analysis shows constitutive expression of iNOS, arginase and MMP9 by N-PEMs, T-PEMs and TAMs. (C) iNOS activity was detected as nitrite concentration levels in 48-h supernatants of constitutive or LPS-stimulated N-PEMs, T-PEMs and TAMs, and (D) Arginase activity measured as urea concentration levels in 48-h supernatants of constitutive and LPS-activated N-PEMs, T-PEMs and TAMs. Figures represent one of three different experiments with similar results. MFI, Mean fluorescence intensity.
3.6. Expression of iNOS, arginase and MMP-9 differs in T-PEMs and TAMs
Inducible nitric oxide synthase (iNOS), arginase and MMP-9 are three enzymes critically involved in the pro-tumor activities of myeloid cells such as TAMs and myeloid-derived suppressor cells (MDSC) [21,27]. MDSC characteristically exhibit enhanced expression/ activity of iNOS and arginase (both involved in the metabolism of arginine [28]), whereas elevated levels of MMP-9 implicated in extracellular matrix remodeling contributes to tumor metastasis [29]. Analysis of the constitutive protein expression of these three enzymes in the macrophage subpopulations examined (Fig 4B) demonstrates that TAMs overexpress iNOS and MMP-9 but significantly downregulate arginase expression compared to T-PEMs and N-PEMs. Moreover, the enzymatic activities of iNOS and arginase were examined for the production of NO and urea, respectively, in the different macrophage subpopulations under resting and LPS-activated conditions. As was seen with the iNOS protein expression, N-PEMs and T-PEMs do not produce NO constitutively, whereas TAMs do (Fig 4C). However, after activation, N-PEMs produce high levels of NO while T-PEMs are suppressed in their production of this mediator even more so than TAMs. Interestingly, and in parallel with its expression, arginase’s constitutive activity is exhibited by N-PEMs and T-PEMs at similar high levels, but the activity of this enzyme is significantly lower in TAMs (Fig. 4D). Further, LPS does not seem to regulate arginase activity in the different macrophage subpopulations. Overall, our results show that TAMs and T-PEMs display opposite expression and function of these pro-tumor/suppressor markers, but neither subpopulation corresponds to the classical functional pattern of suppressor MDSC, which simultaneously express iNOS and arginase [27].
3.7. Macrophage function is differentially altered in TAMs compared to T-PEMs
Macrophages as professional antigen presenting cells contribute to T lymphocyte activation and proliferation. Tumor-derived factors are known to alter various functions in macrophages including their ability to stimulate T cell proliferation and phagocytose particles. In this study, we compared N-PEMs, T-PEMs and TAMs in their ability to modulate cytokine-induced T cell proliferation. Thus, we analyzed ex vivo the rate of proliferation of CD3/CD28/IL-2-activated T splenocytes from normal mice in the absence or presence of N-PEMs, T-PEMs and TAMs, using a fluorescent T cell proliferation assay. N-PEMs do not significantly contribute to CD3/CD28/IL-2-activated T cell proliferation, but macrophages from tumor-bearing mice block this proliferation with TAMs exerting a much more powerful inhibitory activity on T cell proliferation than T-PEMs (Fig. 5A). Our studies demonstrate that tumor presence induces macrophages to inhibit T cell proliferation, with TAMs displaying a much stronger inhibitory capability.
Fig. 5.

TAMs and T-PEMs differ in their regulation of T cell proliferation and their phagocytosis capabilities. (A) Analysis of ex vivo proliferation of αCD3/αCD28/IL-2-activated T splenocytes from normal mice in the absence or the presence of N-PEMs, T-PEMs and TAMs; columns represent mean of four different experiments with similar results. (B) Analysis of the ability of N-PEMs, T-PEMs and TAMs to internalize fluorescent Zymosan particles using fluorescence microscopy, with and without pre-treatment with phagocytosis-interfering Cytochalasin-D; columns show mean of four different experiments with similar results.
To determine if the localization of macrophages in tumor bearers affect their ability to phagocytose particles, we analyzed the internalization of fluorescent Zymosan particles in N-PEMs, T-PEMs and TAMs using fluorescence microscopy. Our results indicate that N-PEMs are fully functional in phagocytosing these particles, whereas T-PEMs and TAMs show a similarly impaired capability to do so (Fig. 5B). Moreover, when the three macrophage subpopulations were pre-treated with phagocytosis-interfering Cytochalasin D prior to Zymosan exposure, there was approximately 50% reduction in phagocytosis in all cell subtypes with TAMs showing significantly higher phagocytic capacity than T-PEMs. In conclusion, our results indicate that macrophages from tumor bearers have in general an impaired ability to phagocytose compared with macrophages from normal mice, but that in the presence of an extrinsic inhibitor of phagocytosis, conditions in the periphery have a stronger modulating effect in blocking phagocytosis by macrophages than those in the tumor.
3.8. Numbers of TAMs and CD3+ cells increase with tumor progression
To study if tumor development has any influence in the degree of colonization by TAMs, tumors at different stages of progression were analyzed histologically. Secretion of IL-23 and IL-6 by TAMs into the microenvironment could result in the recruitment/differentiation of proinflammatory Th17 cells. Moreover, IL-6 and TGFβ (secreted by this tumor [30]) might favor recruitment of other immunosuppressive T cells such as Tregs. We investigated the presence of CD3+ cells into these mammary tumors. Towards this, mammary tumors at 2, 3 and 4-weeks post-tumor implantation were examined by IHC to detect the density of F4/80+ macrophages (TAMs) and CD3+ lymphocytes. Fig. 6A shows the H&Es and IHC of D1-DMBA3 tumors with increasing progression. Our results demonstrate that increased numbers of TAMs and CD3+ cells correlate with tumor progression. Of note, both TAMs and CD3+ cells colocalized in the tumor stroma of 3-week and increasingly in 4-week tumors. Additionally, peritumoral presence of macrophages and CD3+ cells was noticed in 4-week tumors. These results clearly indicate that TAMs and CD3+ cells accumulate and co-localize in higher numbers preferentially in advanced tumors.
Fig. 6.

TAMs coexist with CD3+ T cells, Tregs and IL-17-producing cells in the mammary tumor microenvironment. (A) Histologies (H&E) and IHC results showing F4/80+ macrophages and CD3+ lymphocytes colonizing D1-DMBA3 tumors with increasing degrees of progression. (B) One representative experiment showing flow cytometry analysis of CD4+CD25+Foxp3+ Tregs in spleens from normal mice and spleens and tumors from tumor-bearing mice. (C) Histogram showing percentages of Tregs in the three locations: data for the bar graphs was obtained from different experiments where a total of 18 normal and 18 tumor-bearing mice were used; NS (normal spleen), TS (tumor spleen), T (tumor). (D–E) Histograms corresponding to flow cytometry analysis (not shown) demonstrating presence of IL-17-producing cells (6D: percentages and 6E: MFI) in spleens from normal mice and spleens and tumors from tumor hosts (same numbers of mice as in C were used).
3.9. Tregs and IL-17-producing cells are more numerous in tumor microenvironment compared to periphery
To investigate the nature of the CD3+ lymphocytes that colonize these tumors, CD3+ cells were isolated from spleens of normal mice and spleens and tumors of tumor-bearing mice using Miltenyi beads and subjected to flow cytometric analysis. The percentages of CD4+CD25+Foxp3+ Tregs were lower in the spleens of normal mice, but were significantly increased in the spleens of tumor bearers and especially in their tumors (Fig. 6B and C). Flow cytometric analyses to detect the presence of IL-17-producing cells (not necessarily Th17) revealed that similar to T regs, IL-17-producing cells increase in the spleen of tumor bearers and significantly in their tumors (Fig. 6D and E). Our data indicate that Tregs and IL-17-producing CD3+ T cells accumulate at significant levels in the tumor microenvironment.
4. Discussion
Our comprehensive comparison of T-PEMs and TAMs revealed that these two macrophage subpopulations are very different in several of the crucial molecules they express, the functions they perform and the mechanisms they use to regulate them.
We studied these two macrophage populations from tumor bearers in their resting states and upon activation with LPS, a TLR4 ligand. The existence of other TLR4 ligands present in tumors, such as heat shock proteins and hyaluronan fragments [31] justifies the relevance of studying not only constitutive but also TLR4-activated (LPS) macrophages. It is important to mention that the differences found between TAMs and T-PEMs were not due to the methods used to isolate them, because T-PEMs were also subjected to the same treatments used to collect TAMs and the differences remained. Our studies indicate that the conditions at which macrophages are exposed in the tumor microenvironment and the periphery of tumor hosts are qualitatively distinct, and that the tumor microenvironment, compared to peripheral effects of the tumor, profoundly modifies macrophages phenotypes and functions. Thus, from the immune effect standpoint TAMs are less differentiated, more prone to apoptosis and show increased inhibitory activity against T cell proliferation, although display similarly impaired phagocytic capacity than macrophages in the periphery of tumor bearers (T-PEMs). Importantly, TAMs do not exhibit -either phenotypically or functionally- classical characteristics of MDSC.
Macrophages can be activated following two different profiles: they can be “classically” activated (or M1) when they are proinflammatory whereas they can become “alternatively” activated (or M2) when they become immune suppressive. In mice, upregulation of the proinflammatory cytokine IL-12 and downregulation of the immune suppressive cytokine IL-10 characterize M1 macrophages, and the opposite is characteristic of M2 macrophages. In this study we show that first, from the aspect of cytokine expression, TAMs are neither M2 nor M1, yet clearly exhibit phenotypes and functions that foster tumor promotion. These cells are characteristic for their decreased pro-inflammatory/anti-tumor IL-12p70, diminished pro-inflammatory/pro-tumor TNFα and high immunosuppressive IL-10, but substantially elevated levels of pro-inflammatory/ pro-tumor IL-23 and IL-6, high NO (and iNOS) and lack of arginase expression. Although TAMs have been identified in the vast majority of studies as M2 macrophages [32], it is likely that both M1 and M2 populations may coexist within tumors, depending on the cytokine micro-milieus that may be encountered in the microenvironment of different tumors [33]. Movahedi et al. [34] showed that more M2-like TAMs were enriched in hypoxic tumor areas, whereas M1-like TAMs were found in the rest of the tumors. It has been hypothesized that the dynamic changes of the tumor microenvironment may occur during the transition from early neoplastic events toward advanced tumor stages [35]. These events would drive an M1 toward M2 switch of TAM functions or could allow coexistence of both M1 and M2 phenotypes. Phenotypes of TAMs can differ in various tumor models and stages of cancer progression [36,37]. It is possible that the isolation techniques used in disrupting TAMs from the tumor could have resulted in a loss of their in situ distribution pattern in different areas which could explain our mixed M1/M2 results. In future work it will be important to determine by IHC whether M1 and M2 macrophages coexist within the tumor microenvironment, or as reported [38] individual TAMs simultaneously express M1 and M2 markers.
We previously published that several tumor-derived factors and cytokines were associated with IL-12p70 downregulation in T-PEMs, which was mainly due to decreased IL-12p40 transcription [30,39]. However, we now demonstrate that TAMs upregulate IL-12p40 mRNA and protein, as well as IL-23. Second, from the aspect of signaling pathway, we have previously shown that TGFβ and PGE2 produced by the D1-DMBA3 mammary tumor, acting either alone or synergistically, are associated with the impaired expression of NFκB and C/EBP in peripheral T-PEMs [21]. We now demonstrate that NF-kB is regulated in TAMs through decreased expression of p65 and c-rel but upregulation of p50 homodimers. TAMs with increased NFκBp50 inhibitory homodimers previously reported in a murine fibrosarcoma model and in human ovarian carcinoma have been associated with an inability to promote an effective M1 anti-tumor response [40]. These studies suggest that the roles that NFκB may play in TAMs are different depending on the types of tumors and their stages. Studies in mouse models of inflammation-associated cancers show a tumor-promoting role for NFκB activation in TAMs [41,42] while our present studies and those of others [40] using transplantable tumors demonstrate that TAMs exhibit NFκB defective function. Moreover, while our studies using mammary tumors indicate a defective NFκB function associated with a non-M1 and non-M2 phenotype, an M2 phenotype has been shown in a murine fibrosarcoma model to be also associated with NFκB impairment (41). Interestingly, these authors linked overexpression of p50 homodimers to the transcription inhibition of pro-inflammatory NFκB-dependent genes such as TNFα and IL-12p40. The augmented expression of IL-12p40 in TAMs in our model may be attributed in part to their increased STAT1 activation that is critical for IL-12p40 and iNOS2 transcription [43]. Diminished baseline NFκB and increased STAT1 have also been described in TAMs by others. While Kusmartsev and Gabrilovich [44] reported that elevated STAT1 in TAMs was associated with an up-regulation of iNOS and arginase activity in these cells, Biswas et al. [45] described a unique transcriptional program expressed by TAMs with defective NF-κB and enhanced IRF-3/STAT1 activation. Our results concur with studies [46] showing STAT3 signaling within the tumor microenvironment induces pro-carcinogenic IL-23 expression via direct transcriptional activation of the IL-23/p19 gene, while inhibiting anti-carcinogenic IL-12p70 in TAMs. These authors demonstrated that STAT3 inhibits NF-κB c-rel -dependent IL-12/p35 gene expression in tumor-associated dendritic cells as we have shown in our TAMs. In preliminary gene microarray experiments (data not shown) we observed that TAMs and T-PEMs down-regulate several genes encoding for chemokines and receptors involved in the recruitment of effector cells to the site of inflammation. By down-regulating these molecules, T-PEMs and TAMs may help induce an immune-impaired milieu both at the tumor microenvironment and systemically. In contrast, we also found that LPS-activated TAMs exhibit significantly increased levels of CXCL10, a Th1 cell-attracting chemokine expressed by M1 macrophages [32] but downregulated in T-PEMS [21].
The high levels of TGFβ, IL-6 and IL-23 existing in D1-DMBA-3 tumors (mostly contributed by TAMs) have been related with induction of Tregs and Th17 cells, two cell types associated with tumor progression [47–49]. Interestingly, IL-17 also produced by “altered” tumor-infiltrating Tregs in colorectal tumors is associated with a more aggressive tumor behavior [50]. Detection of Tregs, IL-17-producing CD3+ cells and TAMS co-localizing within the mammary tumor microenvironment suggests the possibility of a crosstalk between all these cell types in advanced tumors.
Our unpublished work with other animal tumor models shows that even within the same tumor location (i.e., mammary cancer), different tumor models may exhibit different tumor microenvironment compositions and cellular and biochemical interactions, allowing TAMs to show different characteristics. Along these lines, we have observed that TAMs isolated from 4T1 mammary tumors (ER+) do not show IL-23 production at all, as compared to the presently studied ER− D1-DMBA-3 tumor, which are high producers of IL-23.
Overall, our results show that the tumor microenvironment is the location that most alters macrophage activity, due to the intimate contact with tumor cells and other cells and tumor factors. We found that these local modifications are more profound that the changes exhibited by macrophages residing in non-tumor, peripheral locations, to which lower tumor factor gradients reach. The complex mixture of cellular components, tumor-derived factors and cytokines/chemokines within the tumor microenvironment may induce TAMs to express a new subset of molecules that are not expressed by macrophages in the periphery of tumor bearers. This could lead to altered macrophage functions that may in turn promote metastatic traits in tumor cells and facilitate tumor progression. Deleting TAMs, re-educating them in anti-tumor responses or blocking their recruitment into tumors are some of the strategies proposed to reverse the deleterious effects of these cells in cancer development.
Acknowledgments
We thank Dr. Diana Lopez (University of Miami) for critical discussion and review of this manuscript. This study was financed by the National Institutes of Health (R21 CA153172 and KO1 CA101926 from MTK).
Abbreviations
- MDSC
myeloid derived suppressor cell
- NPEM
normal peritoneal elicited macrophage
- TAM
tumor associated macrophage
- TPEM
tumor peritoneal elicited macrophage
- Treg
regulatory T cell
References
- 1.Witz IP. Yin-yang activities and vicious cycles in the tumor microenvironment. Cancer Res. 2008;68:9–13. doi: 10.1158/0008-5472.CAN-07-2917. [DOI] [PubMed] [Google Scholar]
- 2.Albini A, Sporn MB. The tumour microenvironment as a target for chemoprevention. Nat Rev Cancer. 2007;7:139–147. doi: 10.1038/nrc2067. [DOI] [PubMed] [Google Scholar]
- 3.Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7:211–217. doi: 10.1016/j.ccr.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 4.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 5.Rodriguez-Vita J, Lawrence T. The resolution of inflammation and cancer. Cytokine Growth Factor Rev. 2010;21:61–65. doi: 10.1016/j.cytogfr.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 6.Meira LB, Bugni JM, Green SL, Lee CW, Pang B, Borenshtein D, Rickman BH, Rogers AB, Moroski-Erkul CA, McFaline JL, Schauer DB, Dedon PC, Fox JG, Samson LD. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J Clin Invest. 2008;118:2516–2525. doi: 10.1172/JCI35073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol. 2010;22:231–237. doi: 10.1016/j.coi.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 8.Mantovani A, Sica A, Locati M. New vistas on macrophage differentiation and activation. Eur J Immunol. 2007;37:14–16. doi: 10.1002/eji.200636910. [DOI] [PubMed] [Google Scholar]
- 9.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–555. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 10.Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol. 2002;196:254–265. doi: 10.1002/path.1027. [DOI] [PubMed] [Google Scholar]
- 11.Ryder M, Ghossein RA, Ricarte-Filho JC, Knauf JA, Fagin JA. Increased density of tumor-associated macrophages is associated with decreased survival in advanced thyroid cancer. Endocr Relat Cancer. 2008;15:1069–1074. doi: 10.1677/ERC-08-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4:71–78. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 13.Pollard JW. Macrophages define the invasive microenvironment in breast cancer. J Leukoc Biol. 2008;84:623–630. doi: 10.1189/jlb.1107762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ostrand-Rosenberg S. Immune surveillance. A balance between protumor and antitumor immunity. Curr Opin Genet Dev. 2008;18:11–18. doi: 10.1016/j.gde.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Medina D, DeOme KB. Response of hyperplastic alveolar nodule outgrowth-line D1 to mammary tumor virus, nodule-inducing virus, and prolonged hormonal stimulation acting singly and in combination. J Natl Cancer Inst. 1969;42:303–310. [PubMed] [Google Scholar]
- 16.Owen JL, Lopez DM, Grosso JF, Guthrie KM, Herbert LM, Torroella-Kouri M, Iragavarapu-Charyulu V. The expression of CCL2 by T lymphocytes of mammary tumor bearers: Role of tumor-derived factors. Cell Immunol. 2005;235:122–135. doi: 10.1016/j.cellimm.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 17.Torroella-Kouri M, Herbert L, Perry G, Lopez DM. Altered IL-12 Signaling Pathways Contribute to the Deficient IFN-gamma Production by T Splenocytes from Tumor-Bearing Mice. Cancer Genomics Proteomics. 2004;1:345–354. [PubMed] [Google Scholar]
- 18.Torroella-Kouri M, Ma X, Perry G, Ivanova M, Cejas PJ, Owen JL, Iragavarapu-Charyulu V, Lopez DM. Diminished expression of transcription factors nuclear factor kappaB and CCAAT/enhancer binding protein underlies a novel tumor evasion mechanism affecting macrophages of mammary tumor-bearing mice. Cancer Res. 2005;65:10578–10584. doi: 10.1158/0008-5472.CAN-05-0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boise LH, Minn AJ, Noel PJ, June CH, Accavitti MA, Lindsten T, Thompson CB. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity. 1995;3:87–98. doi: 10.1016/1074-7613(95)90161-2. [DOI] [PubMed] [Google Scholar]
- 20.Calderon CL, Torroella-Kouri M, Dinapoli MR, Lopez DM. Involvement of protein kinase C and not of NF kappa B in the modulation of macrophage nitric oxide synthase by tumor-derived phosphatidyl serine. Int J Oncol. 2008;32:713–721. [PubMed] [Google Scholar]
- 21.Torroella-Kouri M, Silvera R, Rodriguez D, Caso R, Shatry A, Opiela S, Ilkovitch D, Schwendener RA, Iragavarapu-Charyulu V, Cardentey Y, Strbo N, Lopez DM. Identification of a subpopulation of macrophages in mammary tumor-bearing mice that are neither M1 nor M2 and are less differentiated. Cancer Res. 2009;69:4800–4809. doi: 10.1158/0008-5472.CAN-08-3427. [DOI] [PubMed] [Google Scholar]
- 22.Caso R, Silvera R, Carrio R, Iragavarapu-Charyulu V, Gonzalez-Perez RR, Torroella-Kouri M. Blood monocytes from mammary tumor-bearing mice. early targets of tumor-inducedimmune suppression? Int J Oncol. 2010;37:891–900. doi: 10.3892/ijo_00000740. [DOI] [PubMed] [Google Scholar]
- 23.Langowski JL, Zhang X, Wu L, Mattson JD, Chen T, Smith K, Basham B, McClanahan T, Kastelein RA, Oft M. IL-23 promotes tumour incidence and growth. Nature. 2006;442:461–465. doi: 10.1038/nature04808. [DOI] [PubMed] [Google Scholar]
- 24.Huang WL, Yeh HH, Lin CC, Lai WW, Chang JY, Chang WT, Su WC. Signal transducer and activator of transcription 3 activation up-regulates interleukin-6 autocrine production: a biochemical and genetic study of established cancer cell lines and clinical isolated human cancer cells. Mol Cancer. 2010;9:309. doi: 10.1186/1476-4598-9-309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Balkwill F. TNF-alpha in promotion and progression of cancer. Cancer Metastasis Rev. 2006;25:409–416. doi: 10.1007/s10555-006-9005-3. [DOI] [PubMed] [Google Scholar]
- 26.Portielje JE, Gratama JW, van Ojik HH, Stoter G, Kruit WH. IL-12: a promising adjuvant for cancer vaccination. Cancer Immunol Immunother. 2003;52:133–144. doi: 10.1007/s00262-002-0356-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kusmartsev S, Gabrilovich DI. Role of immature myeloid cells in mechanisms of immune evasion in cancer. Cancer Immunol Immunother. 2006;55:237–245. doi: 10.1007/s00262-005-0048-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rodriguez PC, Ernstoff MS, Hernandez C, Atkins M, Zabaleta J, Sierra R, Ochoa AC. Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res. 2009;69:1553–1560. doi: 10.1158/0008-5472.CAN-08-1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Z, Li L, Yang Z, Luo W, Li X, Yang H, Yao K, Wu B, Fang W. Increased expression of MMP9 is correlated with poor prognosis of nasopharyngeal carcinoma. BMC Cancer. 2010;10:270. doi: 10.1186/1471-2407-10-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Torroella-Kouri M, Lopez DM. Mammary tumor derived TGFb- 1 impairs crucial innate immune response in tumor hosts. J Immunol Immunopathol. 2003;5:31–38. [Google Scholar]
- 31.Tsan MF, Gao B. Endogenous ligands of Toll-like receptors. J Leukoc Biol. 2004;76:514–519. doi: 10.1189/jlb.0304127. [DOI] [PubMed] [Google Scholar]
- 32.Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889–896. doi: 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- 33.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Movahedi K, Laoui D, Gysemans C, Baeten M, Stange G, Van den Bossche J, Mack M, Pipeleers D, In’t Veld P, De Baetselier P, Van Ginderachter JA. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010;70:5728–5739. doi: 10.1158/0008-5472.CAN-09-4672. [DOI] [PubMed] [Google Scholar]
- 35.Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest. 2007;117:1155–1166. doi: 10.1172/JCI31422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Biswas SK, Sica A, Lewis CE. Plasticity of macrophage function during tumor progression: regulation by distinct molecular mechanisms. J Immunol. 2008;180:2011–2017. doi: 10.4049/jimmunol.180.4.2011. [DOI] [PubMed] [Google Scholar]
- 37.Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006;66:605–612. doi: 10.1158/0008-5472.CAN-05-4005. [DOI] [PubMed] [Google Scholar]
- 38.Pettersen JS, Fuentes-Duculan J, Suarez-Farinas M, Pierson KC, Pitts-Kiefer A, Fan L, Belkin DA, Wang CQ, Bhuvanendran S, Johnson-Huang LM, Bluth MJ, Krueger JG, Lowes MA, Carucci JA. Tumor-associated macrophages in the cutaneous SCC microenvironment are heterogeneously activated. J Invest Dermatol. 2011;131:1322–1330. doi: 10.103/jid.2011.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Torroella-Kouri M, Keith JC, Ivanova M, Lopez DM. IL-11-induced reduction of C/EBP transcription factor binding may contribute to the IL-12 downregulation in tumor-bearing mice. Int J Oncol. 2003;22:439–448. [PubMed] [Google Scholar]
- 40.Saccani A, Schioppa T, Porta C, Biswas SK, Nebuloni M, Vago L, Bottazzi B, Colombo MP, Mantovani A, Sica A. P50 nuclear factor-kappaB overexpression in tumor-associated macrophages inhibits M1 inflammatory responses and antitumor resistance. Cancer Res. 2006;66:11432–11440. doi: 10.1158/0008-5472.CAN-06-1867. [DOI] [PubMed] [Google Scholar]
- 41.Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 42.Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 43.Mancino A, Lawrence T. Nuclear factor-kappaB and tumor-associated macrophages. Clin Cancer Res. 2010;16:784–789. doi: 10.1158/1078-0432.CCR-09-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kusmartsev S, Gabrilovich DI. STAT1 signaling regulates tumor-associated macrophage-mediated T cell deletion. J Immunol. 2005;174:4880–4891. doi: 10.4049/jimmunol.174.8.4880. [DOI] [PubMed] [Google Scholar]
- 45.Biswas SK, Gangi L, Paul S, Schioppa T, Saccani A, Sironi M, Bottazzi B, Doni A, Vincenzo B, Pasqualini F, Vago L, Nebuloni M, Mantovani A, Sica A. A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-kappaB and enhanced IRF-3/STAT1 activation) Blood. 2006;107:2112–2122. doi: 10.1182/blood-2005-01-0428. [DOI] [PubMed] [Google Scholar]
- 46.Kortylewski M, Xin H, Kujawski M, Lee H, Liu Y, Harris T, Drake C, Pardoll D, Yu H. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell. 2009;15:114–123. doi: 10.1016/j.ccr.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, Yu H. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med. 2009;206:1457–1464. doi: 10.1084/jem.20090207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang JP, Yan J, Xu J, Pang XH, Chen MS, Li L, Wu C, Li SP, Zheng L. Increased intratumoral IL-17-producing cells correlate with poor survival in hepatocellular carcinoma patients. J Hepatol. 2009;50:980–989. doi: 10.1016/j.jhep.2008.12.033. [DOI] [PubMed] [Google Scholar]
- 49.Mougiakakos D, Choudhury A, Lladser A, Kiessling R, Johansson CC. Regulatory T cells in cancer. Adv Cancer Res. 2010;107:57–117. doi: 10.1016/S0065-230X(10)07003-X. [DOI] [PubMed] [Google Scholar]
- 50.Blatner NR, Bonertz A, Beckhove P, Cheon EC, Krantz SB, Strouch M, Weitz J, Koch M, Halverson AL, Bentrem DJ, Khazaie K. In colorectal cancer mast cells contribute to systemic regulatory T-cell dysfunction. Proc Natl Acad Sci USA. 2010;107:6430–6435. doi: 10.1073/pnas.0913683107. [DOI] [PMC free article] [PubMed] [Google Scholar]