

Abstract
Medullary thyroid carcinoma (MTC) is a neoplasm of the endocrine system, which originates from parafollicular C-cells of the thyroid gland. For MTC therapy, the Food and Drug Administration recently approved vandetanib and cabozantinib, multi-kinase inhibitors targeting RET and other tyrosine kinase receptors of vascular endothelial growth factor, epidermal growth factor, or hepatocyte growth factor. Nevertheless, not all patients with the progressive MTC respond to these drugs, requiring the development of additional therapeutic modalities that have distinct activity. Previously, we reported that expression of activated Ras or Raf in the human MTC cell lines, TT and MZ-CRC-1, can induce growth arrest and RET downregulation via a leukemia inhibitory factor (LIF)-mediated autocrine/paracrine loop. In this study, we aimed to evaluate bacterially-produced recombinant human LIF for its efficacy to suppress human MTC xenografts in mice. Here, we report that, consistent with its effects in vitro, locally or systemically administered recombinant LIF effectively suppressed growth of TT and MZ-CRC-1 xenografts in mice. Further, as predicted from its effects in TT and MZ-CRC-1 cell cultures in vitro, recombinant LIF activated the JAK/STAT pathway and downregulated RET and E2F1 expression in tumors in mice. These results suggest that LIF is a potent cytostatic agent for MTC cells, which regulates unique mechanisms that are not targeted by currently available therapeutic agents.
Keywords: Medullary thyroid carcinoma, RET, leukemia inhibitory factor, STAT3
1. Introduction
Medullary thyroid carcinoma (MTC) is a neoplasm of the endocrine system, which originates from parafollicular C-cells of the thyroid gland [1]. MTC occurs either sporadically or in hereditary forms, i.e., familial MTC and multiple endocrine neoplasia (MEN) type 2 syndrome. MTC progresses slowly and is relatively rare, comprising about 5% of all thyroid cancers. Nevertheless, MTC can be fatal. Currently, the only curative therapy for MTC is surgical resection, which is not effective for metastatic or recurring MTC. It is, therefore, necessary to develop additional therapeutic means, which requires identification of a mechanism exploitable to control MTC cell growth/survival. Although recent studies report the detection of additional oncogenic alterations (e.g., RasG12V) in MTC at lower frequencies [2–4], MTC is mainly caused by altered activity of the receptor tyrosine kinase, rearranged during transfection (RET). For example, various activation mutations occur in the cell surface receptor domain or the cytoplasmic kinase domain of RET in about 95% of hereditary MTC and about 50% of sporadic MTC cases (reviewed in [5]). Accordingly, RET is a key primary target for the design of a therapeutic strategy for MTC. The Food and Drug Administration recently approved vandetanib (trade name Caprelsa, AstraZeneca) and cabozantinib (Cometriq, Exelixis), the multi-kinase inhibitors targeting RET and other tyrosine kinase receptors activated by vascular endothelial growth factor, epidermal growth factor, or hepatocyte growth factor for the treatment of inoperable progressive MTC [6, 7]. Nevertheless, these drugs are not always effective, demanding additional therapeutic strategies [6–8]. In this regard, a cytokine that inhibits oncogenic RET expression in MTC cells may be useful.
It is now well understood that oncogenic stress can elicit growth inhibitory responses in different cell types, as an innate tumor defense mechanism [9–11]. Therefore, an emerging question is whether these phenomena can be exploited for therapy. Previously, we reported that expression of activated Ras or Raf in the human MTC cell lines, TT and MZ-CRC-1, can induce growth arrest and RET downregulation via a leukemia inhibitory factor (LIF)-mediated autocrine/paracrine loop [12, 13]. LIF is a multifunctional cytokine of the interleukin-6 family (reviewed in Ref. [14]). In MTC cells, LIF mediated growth inhibition via activation of the JAK/STAT pathway through the LIFR-gp130 receptor and subsequent downregulation of RET and E2F1 [12, 13] and induction of IFI16 [15]. E2F1 is a critical transcription factor involved in S-phase cell cycle progression [16] and IFI16 is a member of the interferon-inducible HIN200 nuclear protein family, which is well known for its growth inhibitory effects in cells [15, 17]. Of note, LIF sufficiently induced growth arrest of TT and MZ-CRC-1 cells in vitro, presenting an important possibility that LIF has a potential as a therapeutic agent for MTC. It is therefore necessary to test this possibility in a physiologically more relevant MTC model.
In this study, we evaluated bacterially-produced recombinant human LIF for its efficacy to suppress human MTC xenografts in mice. Consistent with its effects in vitro, recombinant LIF induced growth inhibitory responses in MTC xenografts in mice. Further, as predicted from it effects in MTC cell culture in vitro, recombinant LIF activated JAK/STAT pathway and downregulated RET and E2F1 expression in MTC xenografts in mice. These results suggest that LIF is a potent cytostatic agent for MTC cells, which regulates unique mechanisms that are not targeted by currently available therapeutic agents.
2. Materials and Methods
2.1. Cell culture and reagents
Two well characterized human MTC lines, TT and MZ-CRC-1, were used in this study. TT was obtained from ATCC and MZ-CRC-1 was provided by Dr. Robert Gagel (MD Anderson). Generation of TT-GAS3 cell line that stably expresses the STAT3 luciferase reporter construct, (GAS)3-Luc, was previously described [12]. Maintenance of these cell lines was previously described [18, 19]. Briefly, TT and TT-GAS3 were maintained in RPMI 1640 (Invitrogen, Carlsbad, CA) supplemented with 16% fetal bovine serum (FBS), 100 units of penicillin and 100 μg of streptomycin per ml. MZ-CRC-1 was maintained in high-glucose DMEM (Invitrogen) supplemented with 10% FBS in culture dishes coated with rat collagen (Sigma, St. Louis, MO). The control human LIF was obtained from EMD Millipore (Billerica, MA). Silver stain kit was purchased from Thermo Scientific (Rockford, IL).
2.2. Recombinant LIF
The recombinant human LIF used in this study was produced using the tandem hexa-histidine (His)-sumoylation (SUMO) tagging system, as previously described [20]. Briefly, LIF cDNA was cloned into a modified pQE30 vector (Qiagen, Germantown, MD) to generate a tandem hexa-His tagged SUMO-LIF fusion protein. BL21 E. Coli containing the pREP4 plasmid (for lac I expression) were transformed with this pQE30-SUMO-LIF plasmid. One liter cultures in Luria-Bertani media were grown at 37 °C to A600 of 0.7. Expression of SUMO-LIF was induced with 1 mM isopropyl-β-D-thiogalactopyranoside for 20 hours at 15 °C. Cell pellets collected by centrifugation were resuspended in 20 ml of lysis buffer (50 mM Tris-Cl, 300 mM NaCl, pH 7.8) and lysed using a French Press (1,000 psi). The supernatant containing soluble SUMO-LIF was loaded onto Ni-NTA agarose resin (Clontech), washed with 50 mM Tris-Cl/300 mM NaCl/10 mM imidazole (pH 7.8) and eluted with 50 mM Tris-Cl/300 mM NaCl/400 mM imidazole (pH 7.8). Elutions containing hexa-His tagged SUMO-LIF were digested with Ulp1 (Ubiquitin-like specific protease 1/SUMO-Protease 1) leaving the native N-terminus of mature LIF. After dialysis, the cleaved hexa-His tagged SUMO and LIF were loaded onto Ni-NTA agarose resin and the flow-through containing LIF was collected. LIF was further purified by reverse phase HPLC purification to >98% homogeneity. The purity of the final product was estimated by SDS-PAGE and silver staining. The identity of LIF was confirmed by matrix-assisted laser desorption/ionization-time-of-flight (MALDI-TOF) mass spectrometry. The in vitro biologic activity of LIF was analyzed in a reporter assay using TT-GAS3 cell line, containing the (GAS)3-Luc reporter. Luciferase activity was measured using the Luciferase Assay System (Promega, Madison, WI) and data were normalized according to the manufacturer’s instructions.
2.3. Immunoblot analysis
Cells were lysed in 62.5 mM Tris (pH 6.8)-2% SDS mixed with the protease inhibitor cocktail (Sigma) and briefly sonicated before determining the protein concentration using the BCA reagent (Pierce, Rockford, IL). 50 μg of protein was resolved by SDS-PAGE and transferred to a polyvinylidene difluoride membrane filter (Bio-Rad, Hercules, CA). Membrane filters were blocked in 0.1 M Tris (pH 7.5)-0.9% NaCl-0.05% Tween 20 with 5% nonfat dry milk, and incubated with appropriate antibodies. Antibodies were diluted as follows: ERK1/2, 1:2,500; phospho-ERK1/2 (Thr202/Tyr204), 1:1,000; p27, 1:2,000; glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 1:5,000; RET, 1:1,000; IFI16, 1:1,000 (Santa Cruz Biotechnology); phospho-RET (Tyr1062), 1:1,000 (R&D Systems); phospho-STAT3 (Tyr705), 1:2,000; phospho-STAT3 (Ser727), 1:2,000; STAT3, 1:2,000 (Cell Signaling); E2F-1 1:1,000 (NeoMarkers). The Supersignal West Femto and Pico chemiluminescence kits (Pierce) were used for visualization of the signal. Images of immunoblots were taken and processed using ChemiDoc XRS+ and Image Lab 3.0 (Bio-Rad).
2.4. Tumor xenograft studies
1 × 107 TT or 1.5 × 107 MZ-CRC-1 cells in 200 μl Hank’s Balanced Salt Solution were inoculated subcutaneously into the rear flanks of 6-week-old female athymic nude (nu/nu) mice (Charles River Laboratories). Once palpable, tumors were measured using Vernier calipers at intervals indicated in the text. Tumor volumes were calculated using the formula: length × width × height × 0.5236. Mice were sorted into groups of 4 to achieve equal distribution of tumor size in all treatment groups. Lyophilized recombinant LIF was dissolved in sterile nonpyrogenic saline at 0.2 mg/ml and administered every two days (total 10 doses). For subcutaneous (s.c.) administration, LIF-treated groups for each cell line received recombinant LIF (0.5 mg/kg per dose), and control groups received only the vehicle (sterile saline), injected s.c. in the area adjacent to tumors. For systemic administration, LIF-treated groups for each cell line received recombinant LIF (1 mg/kg per dose), and control groups received only the vehicle, sterile phosphate-buffered saline (PBS), injected into tail vein (i.v.) of each mouse. At the end of the experiments, animals were euthanized by CO2 asphyxiation. All animal studies were performed according to protocols approved by the Institutional Animal Care and Use Committee at Medical College of Wisconsin.
2.5. Analysis of tumors
Tumors were homogenized in buffer containing 20 mM Tris (pH 7.5), 150 mM NaCl, 1mM EDTA, 1mM EGTA, 1% Triton X-100 mixed with the protease inhibitor cocktail (Sigma), briefly sonicated and cleared by centrifugation. Measurement of protein concentration and immunoblot analyses were performed as described above.
2.6. Statistical analysis
Two-tailed unpaired Student’s t-test was used to assess the statistical significance of data sets. P value < 0.05 was considered statistically significant.
3. Results
3.1. Production and validation of recombinant LIF
We produced a recombinant human LIF from bacteria using the tandem hexa-histidine-sumoylation tagging system (Fig. 1A). Analysis of the final purification fraction by SDS-PAGE and silver staining showed only one protein band on the gel, indicating the purity of our recombinant LIF (Fig. 1B). Analysis of this recombinant LIF by MALDI- TOF mass spectrometry identified the purified protein as monomeric LIF (Fig. 1C). We then determined activity of the purified LIF in a reporter assay using TT-GAS3 cell line, which was stably transfected with the (GAS)3-Luc reporter. Our recombinant LIF increased the activity of the luciferase reporter activity as effectively as a commercially available LIF when used at an equivalent dose (Fig. 1D), confirming the in vitro biological activity of the purified LIF.
Figure 1. Production of recombinant LIF.

A, Schematic of SUMO-LIF. B, Silver staining of purified fractions resolved by SDS-PAGE. C, MALDI-TOF mass spectrometry analysis of purified LIF. D, The STAT3 activity reporter cell line TTGAS3 was treated with indicated doses of recombinant human LIF produced in this study (SUMO-LIF) for 48 hours before determining the luciferase activity. Equal doses of commercially available human LIF (Cont. LIF) were used for comparison. Data are mean μ SD (n=3).
3.2. Local administration of LIF effectively suppresses growth of MTC xenografts in mice
We first evaluated the potential of recombinant human LIF to suppress TT and MZ-CRC-1 xenografts in mice. These cell lines are the most thoroughly characterized human MTC lines and, thus, have been extensively used for the evaluation of different therapeutic agents [21–23]. TT contains an extracellular domain mutation (RETC634W) whereas MZ-CRC-1 contains a mutation in the kinase domain of Ret (RETM918T), representing two different mechanisms of oncogenic RET activation in MEN2A and MEN2B tumors, respectively. TT and MZ-CRC-1 cells were subcutaneously injected into the flanks of immune-compromised nude mice. After 15 days, when tumors became palpable, we began treating mice by injecting 10 μg LIF (0.5 mg/kg) or same volume of the vehicle, PBS, into the tumor sites. These treatments were performed every two days for 20 days. We found that recombinant LIF treatment substantially suppressed growth of TT and MZ-CRC-1 xenografts (Fig. 2A). When tumor weights were measured at the end of the treatment, recombinant LIF-treated TT and MZ-CRC-1 tumors showed 81% and 93% reduction in size relative to the PBS-treated control tumors, respectively (Fig. 2B and 2C). Importantly, during these treatments, we did not observe any apparent adverse effects of LIF on mice except that LIF induced mild decreases in body weight (Fig. 2D).
Figure 2. Effects of local administration of LIF on MTC tumor xenografts.

Athymic mice bearing TT and MZ-CRC-1 xenografts were treated with ten doses of LIF (0.5 mg/kg/dose). LIF was administered s.c. at the area adjacent to tumors every two days beginning at day 15 after tumor implantation. The control group was treated with the vehicle only (details in the Materials and Methods). A, Changes in tumor sizes were determined at the indicated time-points. B, Images of tumors collected at the end of the treatment. C, Weights of tumors collected at the end of the treatment. D, Body weight changes of animals at the end of treatment. Data are mean μ SEM (n=4), * p < 0.05, ** p < 0.01, t-test.
3.3. Systemic administration of LIF effectively suppresses growth of MTC xenografts in mice
Observing positive effects from the local administration experiment above, we next investigated whether TT and MZ-CRC-1 tumor growth could also be suppressed by a systemic administration of recombinant LIF. For this, when TT and MZ-CRC-1 cells developed palpable tumors in the flanks of nude mice, LIF was administered by tail vein injection at a dose of 1 mg/kg every two days for a total of 10 doses. This amount of LIF (20 μg per dose) is twice as much as that used for the local administration. Indeed, this systemic administration of LIF suppressed growth of MTC tumors very effectively (Fig. 3A). We observed that growth inhibitory effects of LIF were more significant in TT tumors than in MZ-CRC-1 tumors; the tumor relapse of TT was not observed for about three weeks after cessation of the treatment (Fig. 3A, left panel). Intriguingly, two of four TT tumors did not relapse after cessation of the treatment, as monitored for upto 40 days post-treatment (Fig. 3B). In contrast, MZ-CRC-1 tumors relapsed shortly after the treatment (Fig. 3A, right panel). When tumor suppressive effects were compared during the treatment periods (20 days), the systemic administration was as effective as the local administration (compare Fig. 3 with Fig. 2).
Figure 3. Effects of systemic administration of LIF on MTC tumor xenografts.

A, Athymic mice bearing TT and MZ-CRC-1 xenografts were treated with ten doses of LIF (1 mg/kg/dose) via tail vein injection every two days beginning at day 20 after tumor implantation. The control group was treated with the vehicle only. Mice were observed until 40 days after the termination of LIF treatment. Data are mean μ SEM (n=4). B, Individual monitoring of tumor relapse of LIF-treated TT xenografts in (A). C, Body weight changes of animals at the end of the treatment. Data are mean μ SEM (n=4).
Consistent with the local administration, the systemic administration of LIF did not induce any apparent adverse effects on mice, except that LIF induced mild decreases in body weight (Fig. 3C). Loss of body weight was previously noted as a harmless side effect of LIF [24]. These data suggest that LIF may be useful for the suppression of MTC without significant systemic toxicity.
3.4. Recombinant LIF consistently activates the JAK/STAT pathway and downregulates RET and E2F1 in MTC xenografts in mice
In TT and MZ-CRC-1 cultures in vitro, LIF mediates growth inhibition via activation of the JAK/STAT pathway, which is accompanied by downregulation of RET and E2F1, and induction of IFI16 [12, 13, 15]. To determine whether recombinant LIF consistently induces these effects in the tumor xenografts in mice, we analyzed TT and MZ-CRC-1 tumors collected after the local administration of LIF (N = 4 per group). Western blot analyses of tumor extracts revealed that LIF induced significant elevation of STAT3 phosphorylation at Tyr705 in all LIF-treated TT and MZ-CRC-1 tumors (Fig. 4A and 4C). We previously showed that STAT3 phosphorylation at Tyr705, a key mechanism for activation of STAT3 as a transcription factor [25], is necessary for LIF-mediated growth inhibition [12, 13]. Moreover, recombinant LIF significantly downregulated RET and E2F1 in all TT and MZ-CRC-1 xenografts (Fig. 4A and 4C; densitometry shown in Fig. 4B and 4D). These effects of LIF on MTC cells in an in vivo microenvironment are consistent with our previously observed in vitro effects of LIF.
Figure 4. Effects of LIF on cell signaling in MTC tumor xenografts.

TT (A and B) and MZ-CRC-1 (C and D) xenografts from seven and eight mice, respectively, were homogenized as described in the Materials and Methods. The lysates were examined by Western blotting for expression of the indicated proteins. Total cell lysates of TT and MZ- CRC-1 cultures treated with LIF (100 ng/ml, 2 days) were loaded on the two right hand side lanes for comparison. B, D, Total optical density was measured from the Western blot images of RET and E2F-1. Mean values of the control vs LIF-treated tumors were compared to calculate p-value by t-test.
Nevertheless, not all LIF effects on MTC cells were consistent between in vitro and in vivo conditions. Although LIF consistently induced STAT3 phosphorylation at Ser727 in TT tumors, it was not observed in MZ-CRC-1 tumors (Fig. 4A and 4C). Importantly, we previously demonstrated that Ser727 phosphorylation of STAT3 is not necessary for LIF to mediate growth inhibition in TT cells [12]. Although LIF induced ERK1/2 phosphorylation in MZ-CRC-1 tumors, LIF did not induce ERK1/2 phosphorylation in TT tumors (Fig. 4A and 4C). Of note, inhibition of ERK1/2 phosphorylation using the MEK1/2 inhibitor, AZD6244, did not affect LIF-induced RET downregulation or STAT3 phosphorylation in MZ-CRC-1 cells in vitro (Fig. 5), indicating that MEK/ERK activation is not necessary for LIF to mediate growth inhibition. This is consistent with our previous observation in TT cells [12]. In addition, unexpectedly, LIF-mediated IFI16 regulation was not detected in both MTC tumors (Fig. 4A and 4C). These differences between in vitro and in vivo settings were not due to the different LIF treatment schedules. We observed that in vitro responses of TT and MZ-CRC-1 cells to LIF could be sustained for a prolonged time period, as determined by monitoring cells in cultures treated with 10 or 100 ng/ml of LIF every two days for 20 days (Fig. 6). These data demonstrate that, while the key mechanisms required for LIF to induce tumor suppression are conserved between in vitro and in vivo, not all MTC cell responses to LIF are conserved between in vitro and in vivo microenvironments.
Figure 5. Effects of MEK/ERK inhibition on STAT3 activation in MZ-CRC-1 cells.
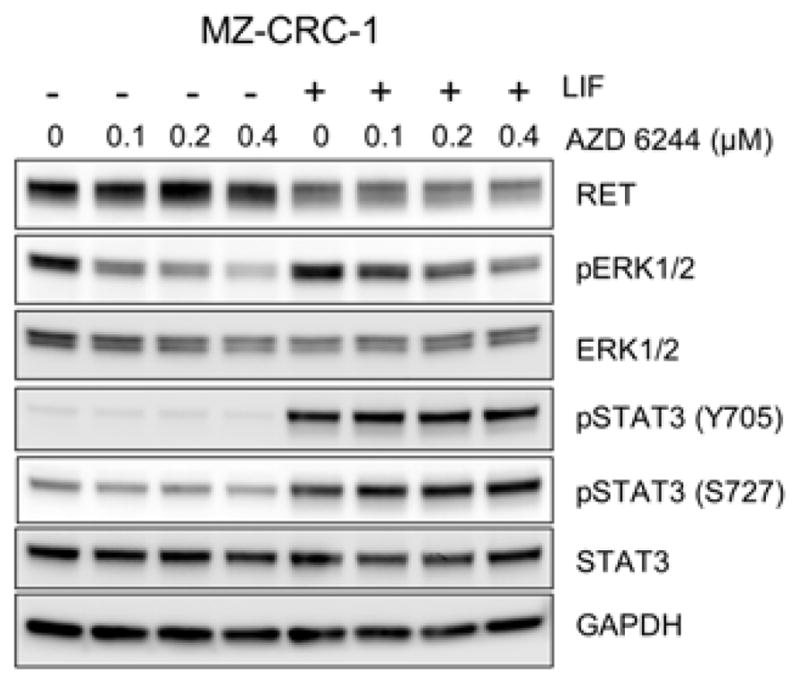
MZ-CRC-1 cells were treated with increasing doses of the MEK1/2 inhibitor, AZD 6244, for 48 hours in the absence/presence of LIF (100 ng/ml). Total cell lysates were examined by Western blotting for expression of the indicated proteins.
Figure 6. Effects of long term LIF treatment on MTC cells in vitro.
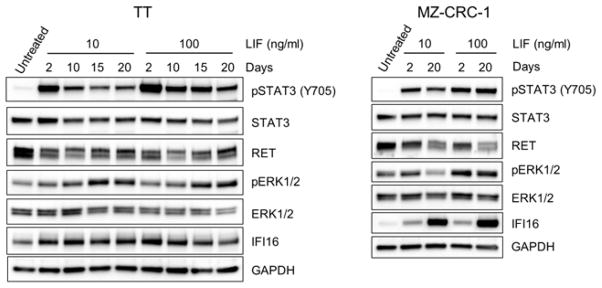
TT and MZ-CRC-1 cells were cultured for 20 days in the presence of LIF (10 or 100 ng/ml). Total cell lysates were examined by Western blot analysis for expression of the indicated proteins.
4. Discussion
This study demonstrates that administration of recombinant human LIF can suppress growth of the human MTC xenografts in mice. MTC is generally not responsive to classic chemo- or radiation-therapies. Current targets for the development of novel therapeutics for surgically incurable progressive MTC have focused on inhibition of the receptor tyrosine kinases, including RET and receptors of vascular endothelial growth factor, epidermal growth factor, or hepatocyte growth factor. Although the multi-kinase inhibitors, vandetanib and cabozantinib, have been shown to be effective in about 50% or 47% of progressive MTC cases, respectively [7, 26–28], these kinase inhibitors also cause multiple side effects. For example, significant dose reduction or discontinuation of the treatment was reported in 28% of the drug-treated patients [27]. Therefore, additional therapeutic options for MTC are required. Importantly, when compared to the effects of vandetanib that we recently reported using the same preclinical model [19], LIF treatment appears to be similarly effective as this tyrosine kinase inhibitor in suppressing TT xenografts (compare Fig. 2 and 3 with Fig. 5 of the reference 19).
Consistent with its previously shown in vitro effects [12, 13, 29], LIF induced STAT3 activation in MTC xenografts in mice. However, LIF-mediated regulation of the MEK/ERK pathway was not consistent between in vitro and in vivo experimental settings of TT and MZ-CRC-1 growth. Importantly, our previous data [12, 13, 29] and current study indicates that MEK/ERK activation is not necessary for LIF to mediate RET downregulation in these MTC cells. Our previous and current studies characterize STAT3 as an upstream regulator of RET in MTC cells, which is contrary to a previous characterization of STAT3 as a downstream mediator of RET-MEN2A in NIH3T3 fibroblasts [30]. Along with STAT3 activation, LIF consistently induced downregulation of RET-MEN2A and RET-MEN2B in MTC xenografts in mice. These observations support the capability of LIF to induce downregulation of the RET oncogenes in MTC cells. LIF also induced downregulation of the S-phase transcription factor, E2F1, which may underlie an important mechanism for LIF-induced cytostatic effects. Currently, we do not know whether these two LIF-induced mechanisms are connected to each other. This will have to be addressed in our future study for better understating of how LIF mediates growth inhibition in MTC cells. Although STAT3 activation and RET downregulation were consistent between the in vitro and in vivo experimental settings, not all cellular responses to LIF were conserved between the two settings. For example, LIF did not consistently regulate IFI16 expression in the MTC xenografts, suggesting that there is a significant difference between MTC cell responses under in vitro conditions and in in vivo microenvironments.
There are a few interesting questions as to the effects of LIF on MTC cells. First, why does LIF induce cytostatic effects in MTC cells? Our previous studies characterized STAT3 activation in MTC cells as a key mechanism for LIF to downregulate RET and to induce cytostatic effects [12, 13, 29], which is contrasted to the characterization of STAT3 as an oncogene in different tumor types [31–33]. Of note, a recent study has demonstrated that STAT3 has a tumor suppressive role in papillary thyroid cancer [34]. In conjunction with this, our study may suggest that STAT3 has a tissue type specific role and that STAT3 activity is growth inhibitory in thyroid epithelium. Second, is LIF-induced growth inhibitory signaling specific to MTC cells? It has been reported that different cell types undergoing Ras/Raf-induced growth inhibition secrete soluble factors that can help reinforce cellular growth inhibitory responses [35–37]. These factors were cell type-specific and include insulin-like growth factor binding protein-7, plasminogen activator inhibitor-1, interleukin-8, and growth-regulated oncogene-α. We previously showed that LIF is induced upon Raf activation in different cell types, including MTC cells, a pheochromocytoma line (MPC8622), human small cell lung carcinoma cell lines (NCI-H209 and DMS53), and a human prostate carcinoma line (LNCaP) [12, 29]. Intriguingly, LIF induced growth inhibitory responses only in MTC and pheochromocytoma cell lines [12, 29]. Considering that pheochromocytoma often arises in association with MEN2B [38], one may speculate that the growth inhibitory effects of LIF are specific to certain cell types, possibly associated with MEN type 2 syndrome.
Various cytokines have been tested for their efficacy in cancer therapy [39]. Some of those factors stimulate the immune system or modulate the microenvironment of tumor cells while others (e.g., tumor necrosis factor-related apoptosis inducing ligand) target tumor cells directly and induce cell death. Although LIF has not been tested in cancer, it has been tested for other pathophysiological conditions, including a few neurological disorders (e.g., amyotrophic lateral syndrome and radiation- or chemotherapy-induced neuropathy) [40–42]. Moreover, LIF has been shown to be relatively nontoxic in a few clinical settings [43]. For example, recombinant LIF proteins (e.g., rhuLIF/emfilemin/AM424) were tested in phase 2 clinical trials at a relatively high dose (4 μg/kg) without inducing adverse effects or adverse changes in the patients’ quality of life [42]. These previous studies support the potential advantage of LIF to be easily introduced into a clinical application. In conclusion, our study suggests that LIF may have potential for MTC therapy.
Acknowledgments
We thank Dr. Robert Gagel for MZ-CRC-1 and Dr. Brain Volkman for generous support for protein purification. This work was supported by American Cancer Society (RSGM-10-189-01-TBE), National Cancer Institute (1R01CA138441), and FAMRI Young Investigator Award (062438) to J.P.
Abbreviations
- MTC
medullary thyroid carcinoma
- LIF
leukemia inhibitory factor
- MEN
multiple endocrine neoplasia
- RET
rearranged during transfection
- MALDI-TOF
matrix-assisted laser desorption/ionization-time-of-flight
- PBS
phosphate-buffered saline
Footnotes
Disclosure
The authors have nothing to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tuttle RM, Ball DW, Byrd D, Daniels GH, Dilawari RA, Doherty GM, et al. Medullary carcinoma. J Natl Compr Canc Netw. 2010;8:512–30. doi: 10.6004/jnccn.2010.0040. [DOI] [PubMed] [Google Scholar]
- 2.Agrawal N, Jiao Y, Sausen M, Leary R, Bettegowda C, Roberts NJ, et al. Exomic sequencing of medullary thyroid cancer reveals dominant and mutually exclusive oncogenic mutations in RET and RAS. J Clin Endocrinol Metab. 2013;98:E364–9. doi: 10.1210/jc.2012-2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boichard A, Croux L, Al Ghuzlan A, Broutin S, Dupuy C, Leboulleux S, et al. Somatic RAS mutations occur in a large proportion of sporadic RET-negative medullary thyroid carcinomas and extend to a previously unidentified exon. J Clin Endocrinol Metab. 2012;97:E2031–5. doi: 10.1210/jc.2012-2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ciampi R, Mian C, Fugazzola L, Cosci B, Romei C, Barollo S, et al. Evidence of a low prevalence of RAS mutations in a large medullary thyroid cancer series. Thyroid. 2013;23:50–7. doi: 10.1089/thy.2012.0207. [DOI] [PubMed] [Google Scholar]
- 5.Ichihara M, Murakumo Y, Takahashi M. RET and neuroendocrine tumors. Cancer Lett. 2004;204:197–211. doi: 10.1016/S0304-3835(03)00456-7. [DOI] [PubMed] [Google Scholar]
- 6.Degrauwe N, Sosa JA, Roman S, Deshpande HA. Vandetanib for the treatment of metastatic medullary thyroid cancer. Clin Med Insights Oncol. 2012;6:243–52. doi: 10.4137/CMO.S7999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagilla M, Brown RL, Cohen EE. Cabozantinib for the treatment of advanced medullary thyroid cancer. Adv Ther. 2012;29:925–34. doi: 10.1007/s12325-012-0060-6. [DOI] [PubMed] [Google Scholar]
- 8.Wells SA, Jr, Robinson BG, Gagel RF, Dralle H, Fagin JA, Santoro M, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol. 2012;30:134–41. doi: 10.1200/JCO.2011.35.5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 10.Zhu J, Woods D, McMahon M, Bishop JM. Senescence of human fibroblasts induced by oncogenic Raf. Genes & development. 1998;12:2997–3007. doi: 10.1101/gad.12.19.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin AW, Barradas M, Stone JC, van Aelst L, Serrano M, Lowe SW. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes & development. 1998;12:3008–19. doi: 10.1101/gad.12.19.3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park JI, Strock CJ, Ball DW, Nelkin BD. The Ras/Raf/MEK/extracellular signal-regulated kinase pathway induces autocrine-paracrine growth inhibition via the leukemia inhibitory factor/JAK/STAT pathway. Mol Cell Biol. 2003;23:543–54. doi: 10.1128/MCB.23.2.543-554.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arthan D, Hong SK, Park JI. Leukemia inhibitory factor can mediate Ras/Raf/MEK/ERK-induced growth inhibitory signaling in medullary thyroid cancer cells. Cancer Lett. 2010;297:31–41. doi: 10.1016/j.canlet.2010.04.021. [DOI] [PubMed] [Google Scholar]
- 14.Auernhammer CJ, Melmed S. Leukemia-inhibitory factor-neuroimmune modulator of endocrine function. Endocr Rev. 2000;21:313–45. doi: 10.1210/edrv.21.3.0400. [DOI] [PubMed] [Google Scholar]
- 15.Kim EJ, Park JI, Nelkin BD. IFI16 is an essential mediator of growth inhibition, but not differentiation, induced by the leukemia inhibitory factor/JAK/STAT pathway in medullary thyroid carcinoma cells. J Biol Chem. 2005;280:4913–20. doi: 10.1074/jbc.M410542200. [DOI] [PubMed] [Google Scholar]
- 16.La Thangue NB. The yin and yang of E2F-1: balancing life and death. Nat Cell Biol. 2003;5:587–9. doi: 10.1038/ncb0703-587. [DOI] [PubMed] [Google Scholar]
- 17.Choubey D, Deka R, Ho SM. Interferon-inducible IFI16 protein in human cancers and autoimmune diseases. Front Biosci. 2008;13:598–608. doi: 10.2741/2705. [DOI] [PubMed] [Google Scholar]
- 18.Park JI, Strock CJ, Ball DW, Nelkin BD. Interleukin-1beta can mediate growth arrest and differentiation via the leukemia inhibitory factor/JAK/STAT pathway in medullary thyroid carcinoma cells. Cytokine. 2005;29:125–34. doi: 10.1016/j.cyto.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 19.Starenki D, Park JI. Mitochondria-Targeted Nitroxide, Mito-CP, Suppresses Medullary Thyroid Carcinoma Cell Survival In Vitro and In Vivo. J Clin Endocrinol Metab. 2013;98:1529–40. doi: 10.1210/jc.2012-3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Love M, Sandberg JL, Ziarek JJ, Gerarden KP, Rode RR, Jensen DR, et al. Solution structure of CCL21 and identification of a putative CCR7 binding site. Biochemistry. 2012;51:733–5. doi: 10.1021/bi201601k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Groot JW, Plaza Menacho I, Schepers H, Drenth-Diephuis LJ, Osinga J, Plukker JT, et al. Cellular effects of imatinib on medullary thyroid cancer cells harboring multiple endocrine neoplasia Type 2A and 2B associated RET mutations. Surgery. 2006;139:806–14. doi: 10.1016/j.surg.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 22.Morisi R, Celano M, Tosi E, Schenone S, Navarra M, Ferretti E, et al. Growth inhibition of medullary thyroid carcinoma cells by pyrazolo-pyrimidine derivates. J Endocrinol Invest. 2007;30:RC31–4. doi: 10.1007/BF03349220. [DOI] [PubMed] [Google Scholar]
- 23.Plaza-Menacho I, Mologni L, Sala E, Gambacorti-Passerini C, Magee AI, Links TP, et al. Sorafenib functions to potently suppress RET tyrosine kinase activity by direct enzymatic inhibition and promoting RET lysosomal degradation independent of proteasomal targeting. J Biol Chem. 2007;282:29230–40. doi: 10.1074/jbc.M703461200. [DOI] [PubMed] [Google Scholar]
- 24.Jansson JO, Moverare-Skrtic S, Berndtsson A, Wernstedt I, Carlsten H, Ohlsson C. Leukemia inhibitory factor reduces body fat mass in ovariectomized mice. Eur J Endocrinol. 2006;154:349–54. doi: 10.1530/eje.1.02082. [DOI] [PubMed] [Google Scholar]
- 25.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, et al. Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 26.Degrauwe N, Sosa JA, Roman S, Deshpande HA. Vandetanib for the treatment of metastatic medullary thyroid cancer. Clinical Medicine Insights Oncology. 2012;6:243–52. doi: 10.4137/CMO.S7999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wells SA, Jr, Robinson BG, Gagel RF, Dralle H, Fagin JA, Santoro M, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012;30:134–41. doi: 10.1200/JCO.2011.35.5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schoffski PER, Müller S, Brose MS, Shah MH, Licitra LF, Jarzab B, Medvedev V, Kreissl M, Niederle B, Cohen EEW, Wirth LJ, Ali HY, Hessel C, Yaron Y, Ball DW, Nelkin B, Sherman SI, Schlumberger M EXAM Study Group. An international, double-blind, randomized, placebo-controlled phase III trial (EXAM) of cabozantinib (XL184) in medullary thyroid carcinoma (MTC) patients (pts) with documented RECIST progression at baseline. J Clin Oncol. 2012;30:Abstract 5508. [Google Scholar]
- 29.Park JI, Powers JF, Tischler AS, Strock CJ, Ball DW, Nelkin BD. GDNF-induced leukemia inhibitory factor can mediate differentiation via the MEK/ERK pathway in pheochromocytoma cells derived from nf1-heterozygous knockout mice. Exp Cell Res. 2005;303:79–88. doi: 10.1016/j.yexcr.2004.09.028. [DOI] [PubMed] [Google Scholar]
- 30.Schuringa JJ, Wojtachnio K, Hagens W, Vellenga E, Buys CH, Hofstra R, et al. MEN2A-RET-induced cellular transformation by activation of STAT3. Oncogene. 2001;20:5350–8. doi: 10.1038/sj.onc.1204715. [DOI] [PubMed] [Google Scholar]
- 31.Garcia R, Bowman TL, Niu G, Yu H, Minton S, Muro-Cacho CA, et al. Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene. 2001;20:2499–513. doi: 10.1038/sj.onc.1204349. [DOI] [PubMed] [Google Scholar]
- 32.Song JI, Grandis JR. STAT signaling in head and neck cancer. Oncogene. 2000;19:2489–95. doi: 10.1038/sj.onc.1203483. [DOI] [PubMed] [Google Scholar]
- 33.Subramaniam A, Shanmugam MK, Perumal E, Li F, Nachiyappan A, Dai X, et al. Potential role of signal transducer and activator of transcription (STAT)3 signaling pathway in inflammation, survival, proliferation and invasion of hepatocellular carcinoma. Biochim Biophys Acta. 2013;1835:46–60. doi: 10.1016/j.bbcan.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 34.Couto JP, Daly L, Almeida A, Knauf JA, Fagin JA, Sobrinho-Simoes M, et al. STAT3 negatively regulates thyroid tumorigenesis. Proc Natl Acad Sci U S A. 2012;109:E2361–70. doi: 10.1073/pnas.1201232109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–18. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 36.Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–31. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 37.Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell. 2008;132:363–74. doi: 10.1016/j.cell.2007.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee NC, Norton JA. Multiple endocrine neoplasia type 2B--genetic basis and clinical expression. Surg Oncol. 2000;9:111–8. doi: 10.1016/s0960-7404(00)00038-4. [DOI] [PubMed] [Google Scholar]
- 39.Dranoff G. Cytokines in cancer pathogenesis and cancer therapy. Nat Rev Cancer. 2004;4:11–22. doi: 10.1038/nrc1252. [DOI] [PubMed] [Google Scholar]
- 40.Farese AM, Myers LA, MacVittie TJ. Therapeutic efficacy of recombinant human leukemia inhibitory factor in a primate model of radiation-induced marrow aplasia. Blood. 1994;84:3675–8. [PubMed] [Google Scholar]
- 41.Kurek JB, Radford AJ, Crump DE, Bower JJ, Feeney SJ, Austin L, et al. LIF (AM424), a promising growth factor for the treatment of ALS. J Neurol Sci. 1998;160 (Suppl 1):S106–13. doi: 10.1016/s0022-510x(98)00208-1. [DOI] [PubMed] [Google Scholar]
- 42.Davis ID, Kiers L, MacGregor L, Quinn M, Arezzo J, Green M, et al. A randomized, double-blinded, placebo-controlled phase II trial of recombinant human leukemia inhibitory factor (rhuLIF, emfilermin, AM424) to prevent chemotherapy-induced peripheral neuropathy. Clin Cancer Res. 2005;11:1890–8. doi: 10.1158/1078-0432.CCR-04-1655. [DOI] [PubMed] [Google Scholar]
- 43.Metcalf D. The unsolved enigmas of leukemia inhibitory factor. Stem Cells. 2003;21:5–14. doi: 10.1634/stemcells.21-1-5. [DOI] [PubMed] [Google Scholar]